
Comparative Proteomic Analysis of Drug Trichosanthin Addition to BeWo Cell Line

 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. MTT Method
2.3. Sample Preparation
2.4. Two-Dimensional Gel Electrophoresis (2DE)
2.5. Silver Staining and Image Analysis
2.6. In-Gel Digestion and Mass Spectrometric Identification
2.7. In-Solution Trypsin Digestion and LC-MS/MS (Label-Free Method)
3. Results
3.1. MTT Method
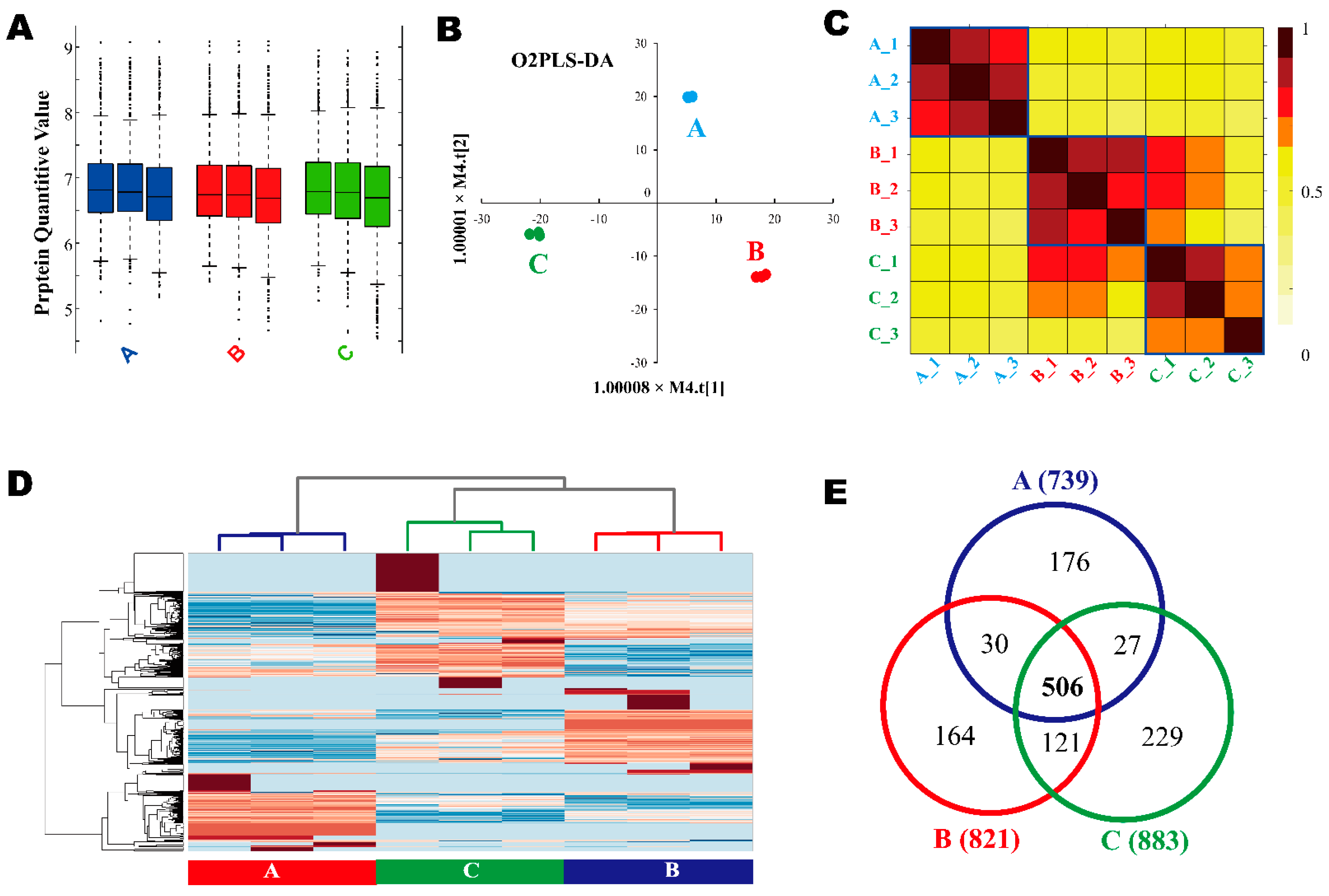
3.2. DE-Derived Proteome
3.3. DEPs Identified by MALDI-TOF/TOF MS
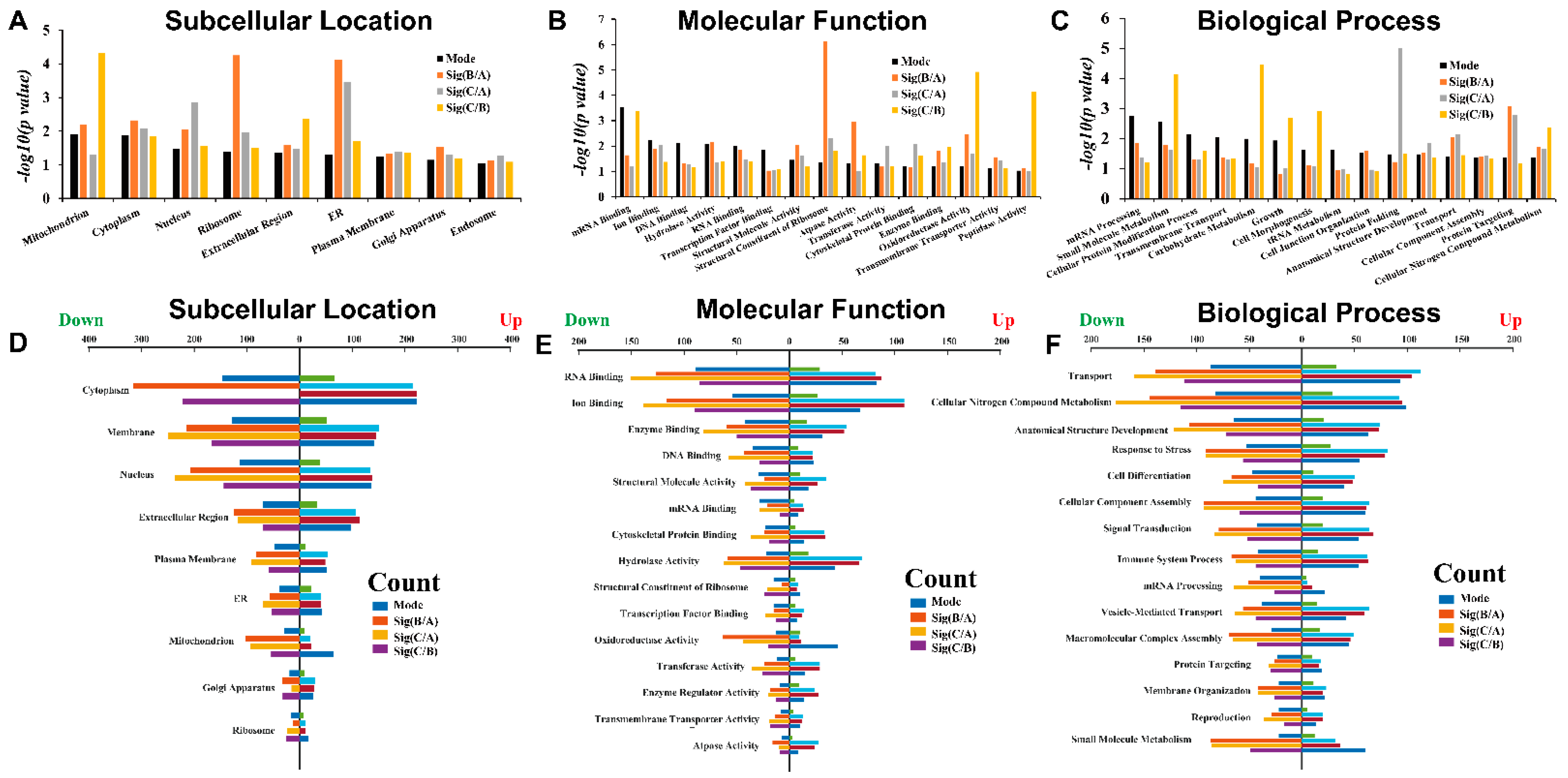
3.4. LC-MS/MS-Derived Proteome and Bioinformatics Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| DEPs | Differentially Expressed Proteins |
| LFQ | Label-Free Quantification |
| MALDI-TOF/TOF MS | Matrix-Assisted Laser Desorption Ionization/Time-Of-Flight–Time-Of-Flight Mass Spectrometry |
| MTT | 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide |
| O2PLS-DA | Orthogonal Partial Least Square Analysis-Discriminant Analysis |
| TCS | Trichosanthin |
References
- Tang, Y.S.; Liang, J.M.; Wu, A.H.; Chen, Y.Z.; Zhao, P.F.; Lin, T.L.; Zhang, M.; Xu, Q.; Wang, J.X.; Huang, Y.Z. Co-Delivery of Trichosanthin and Albendazole by Nano-Self Assembly for Overcoming Tumor Multidrug-Resistance and Metastasis. ACS Appl. Mater. Interfaces 2017, 9, 26648–26664. [Google Scholar] [CrossRef]
- Zhao, J.; Ben, L.H.; Wu, Y.L.; Hu, W.; Ling, K.; Xin, S.M.; Nie, H.L.; Ma, L.; Pei, G. Anti-HIV agent trichosanthin enhances the capabilities of chemokines to stimulate chemotaxis and G protein activation, and this is mediated through interaction of trichosanthin and chemokine receptors. J. Exp. Med. 1999, 190, 101–111. [Google Scholar] [CrossRef] [Green Version]
- Li, M.X.; Yeung, H.W.; Pan, L.P.; Chan, S.I. Trichosanthin, a potent HIV-1 inhibitor, can cleave supercoiled DNA in vitro. Nucleic Acids Res. 1991, 19, 6309–6312. [Google Scholar] [CrossRef] [Green Version]
- Too, P.H.M.; Ma, M.K.W.; Mak, A.N.S.; Wong, Y.T.; Tung, C.K.C.; Zhu, G.; Au, S.W.N.; Wong, K.B.; Shaw, P.C. The C-terminal fragment of the ribosomal P protein complexed to trichosanthin reveals the interaction between the ribosome-inactivating protein and the ribosome. Nucleic Acids Res. 2009, 37, 602–610. [Google Scholar] [CrossRef] [Green Version]
- Sha, O.; Niu, J.F.; Ng, T.B.; Cho, E.Y.P.; Fu, X.Y.; Jiang, W.Q. Anti-tumor action of trichosanthin, a type 1 ribosome-inactivating protein, employed in traditional Chinese medicine: A mini review. Cancer Chemother. Pharmacol. 2013, 71, 1387–1393. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.Z.; Li, P.F.; Li, Y.Z.; Luo, F.; Guo, C.; Lin, B.; Cao, X.W.; Zhao, J.; Wang, F.J. Enhanced anti-tumor activity of trichosanthin after combination with a human-derived cell-penetrating peptide, and a possible mechanism of activity. Fitoterapia 2016, 112, 183–190. [Google Scholar] [CrossRef]
- Wang, H.X.; Zhao, Y.X.; Wang, H.Y.; Gong, J.B.; He, H.N.; Shin, M.C.; Yang, V.C.; Huang, Y.Z. Low-molecular-weight protamine-modified PLGA nanoparticles for overcoming drug-resistant breast cancer. J. Control. Release 2014, 192, 47–56. [Google Scholar] [CrossRef]
- Wang, P.; Li, J.C. Trichosanthin-induced specific changes of cytoskeleton configuration were associated with the decreased expression level of actin and tubulin genes in apoptotic Hela cells. Life Sci. 2007, 81, 1130–1140. [Google Scholar] [CrossRef]
- Fang, E.F.; Zhang, C.Z.Y.; Zhang, L.; Wong, J.H.; Chan, Y.S.; Pan, W.L.; Dan, X.L.; Yin, C.M.; Cho, C.H.; Ng, T.B. Trichosanthin Inhibits Breast Cancer Cell Proliferation in Both Cell Lines and Nude Mice by Promotion of Apoptosis. PLoS ONE 2012, 7, e41592. [Google Scholar] [CrossRef] [Green Version]
- Tsao, S.W.; Yan, K.T.; Yeung, H.W. Selective Killing of Choriocarcinoma Cells-Invitro by Trichosanthin, a Plant Protein Purified from Root Tubers of the Chinese Medicinal Herb Trichosanthes-Kirilowii. Toxicon 1986, 24, 831–840. [Google Scholar] [CrossRef]
- An, Q.X.; Lei, Y.F.; Jia, N.; Zhang, X.Q.; Bai, Y.L.; Yi, J.; Chen, R.; Xia, A.J.; Yang, J.; Wei, S.H.; et al. Effect of site-directed PEGylation of trichosanthin on its biological activity, immunogenicity, and pharmacokinetics. Biomol. Eng. 2007, 24, 643–649. [Google Scholar] [CrossRef]
- Chen, Y.Z.; Zhang, M.; Jin, H.Y.; Tang, Y.S.; Wu, A.H.; Huang, Y.Z. Prodrug-Like, PEGylated Protein Toxin Trichosanthin for Reversal of Chemoresistance. Mol. Pharm. 2017, 14, 1429–1438. [Google Scholar] [CrossRef]
- Ficarro, S.B.; McCleland, M.L.; Stukenberg, P.T.; Burke, D.J.; Ross, M.M.; Shabanowitz, J.; Hunt, D.F.; White, F.M. Phosphoproteome analysis by mass spectrometry and its application to Saccharomyces cerevisiae. Nat. Biotechnol. 2002, 20, 301–305. [Google Scholar] [CrossRef]
- Pieper, R.; Su, Q.; Gatlin, C.L.; Huang, S.T.; Anderson, N.L.; Steiner, S. Multi-component immunoaffinity subtraction chromatography: An innovative step towards a comprehensive survey of the human plasma proteome. Proteomics 2003, 3, 422–432. [Google Scholar] [CrossRef]
- Greenough, C.; Jenkins, R.E.; Kitteringham, N.R.; Pirmohamed, M.; Park, B.K.; Pennington, S.R. A method for the rapid depletion of albumin and immunoglobulin from human plasma. Proteomics 2004, 4, 3107–3111. [Google Scholar] [CrossRef]
- Adamczyk, M.; Gebler, J.C.; Wu, J. Selective analysis of phosphopeptides within a protein mixture by chemical modification, reversible biotinylation and mass spectrometry. Rapid Commun. Mass Spectrom. 2001, 15, 1481–1488. [Google Scholar] [CrossRef]
- Butt, A.; Davison, M.D.; Smith, G.J.; Young, J.A.; Gaskell, S.J.; Oliver, S.G.; Beynon, R.J. Chromatographic separations as a prelude to two-dimensional electrophoresis in proteomics analysis. Proteomics 2001, 1, 42–53. [Google Scholar] [CrossRef]
- Van den Bergh, G.; Clerens, S.; Cnops, L.; Vandesande, F.; Arckens, L. Fluorescent two-dimensional difference gel electrophoresis and mass spectrometry identify age-related protein expression differences for the primary visual cortex of kitten and adult cat. J. Neurochem. 2003, 85, 193–205. [Google Scholar] [CrossRef] [Green Version]
- Badock, V.; Steinhusen, U.; Bommert, K.; Otto, A. Prefractionation of protein samples for proteome analysis using reversed-phase high-performance liquid chromatography. Electrophoresis 2001, 22, 2856–2864. [Google Scholar] [CrossRef]
- Gygi, S.P.; Rist, B.; Gerber, S.A.; Turecek, F.; Gelb, M.H.; Aebersold, R. Quantitative analysis of complex protein mixtures using isotope-coded affinity tags. Nat. Biotechnol. 1999, 17, 994–999. [Google Scholar] [CrossRef]
- Mann, M.; Hendrickson, R.C.; Pandey, A. Analysis of proteins and proteomes by mass spectrometry. Annu. Rev. Biochem. 2001, 70, 437–473. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Shen, S.; Rasam, S.S.; Qu, J. MS1 ion current-based quantitative proteomics: A promising solution for reliable analysis of large biological cohorts. Mass Spectrom. Rev. 2019, 38, 461–482. [Google Scholar] [CrossRef]
- Sztacho, M.; Šalovská, B.; Červenka, J.; Balaban, C.; Hoboth, P.; Hozák, P. Limited Proteolysis-Coupled Mass Spectrometry Identifies Phosphatidylinositol 4,5-Bisphosphate Effectors in Human Nuclear Proteome. Cells 2021, 10, 68. [Google Scholar] [CrossRef]
- Available online: https://www.atcc.org/resources/culture-guides/animal-cell-culture-guide (accessed on 13 May 2018).
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Denizot, F.; Lang, R. Rapid Colorimetric Assay for Cell-Growth and Survival—Modifications to the Tetrazolium Dye Procedure Giving Improved Sensitivity and Reliability. J. Immunol. Methods 1986, 89, 271–277. [Google Scholar] [CrossRef]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Nesterenko, M.V.; Tilley, M.; Upton, S.J. A simple modification of Blum’s silver stain method allows for 30 minute detection of proteins in polyacrylamide gels. J. Biochem. Biophys. Methods 1994, 28, 239–242. [Google Scholar] [CrossRef]
- Cox, J.; Hein, M.Y.; Luber, C.A.; Paron, I.; Nagaraj, N.; Mann, M. Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol. Cell. Proteom. 2014, 13, 2513–2526. [Google Scholar] [CrossRef] [Green Version]
- Ma, J.; Chen, T.; Wu, S.; Yang, C.; Bai, M.; Shu, K.; Li, K.; Zhang, G.; Jin, Z.; He, F.; et al. iProX: An integrated proteome resource. Nucleic Acids Res. 2019, 47, D1211–D1217. [Google Scholar] [CrossRef] [Green Version]
- Hetzer, M.; Mattaj, I.W. An ATP-dependent, Ran-independent mechanism for nuclear import of the U1A and U2B″ spliceosome proteins. J. Cell. Biol. 2000, 148, 293–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horst, D.; Reu, S.; Kriegl, L.; Engel, J.; Jung, A. The intratumoral distribution of nuclear beta-catenin is a prognostic marker in colon cancer. Cancer 2010, 115, 2063–2070. [Google Scholar] [CrossRef]
- Matsumoto, K.; Moriuchi, T.; Koji, T.; Nakane, P.K. Molecular cloning of cDNA coding for rat proliferating cell nuclear antigen (PCNA)/cyclin. EMBO J. 1987, 6, 637–642. [Google Scholar] [CrossRef]
- Bowman, G.D.; O’Donnell, M.; Kuriyan, J. Structural analysis of a eukaryotic sliding DNA clamp-clamp loader complex. Nature 2004, 429, 724–730. [Google Scholar] [CrossRef]
- Turan, G.; Turan, M. The Evaluation of TUNEL, PCNA and SOX2 Expressions in Lens Epithelial Cells of Cataract Patients with Pseudoexfoliation Syndrome. Curr. Eye Res. 2020, 45, 12–16. [Google Scholar] [CrossRef]
- Butera, G.; Mullappilly, N.; Masetto, F.; Palmieri, M.; Scupoli, M.T.; Pacchiana, R.; Donadelli, M. Regulation of Autophagy by Nuclear GAPDH and Its Aggregates in Cancer and Neurodegenerative Disorders. Int. J. Mol. Sci. 2019, 20, 2062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, M.R.; Garcin, E.D. D-Glyceraldehyde-3-Phosphate Dehydrogenase Structure and Function. Subcell Biochem. 2017, 83, 413–453. [Google Scholar] [PubMed]
- Kosova, A.; Khodyreva, S.N.; Lavrik, O.I. Role of Glyceraldehyde-3-Phosphate Dehydrogenase (GAPDH) in DNA Repair. Biochemistry 2017, 82, 643–654. [Google Scholar] [CrossRef] [PubMed]
- Muronetz, V.I.; Melnikova, A.K.; Barinova, K.V.; Schmalhausen, E.V. Inhibitors of Glyceraldehyde 3-Phosphate Dehydrogenase and Unexpected Effects of Its Reduced Activity. Biochemistry 2019, 84, 1268–1279. [Google Scholar] [CrossRef] [PubMed]
- Duan, X.; Zhang, J.; Liu, S.; Zhang, M.; Wang, Q.; Cheng, J. Methylation of nucleolar and coiled-body phosphoprotein 1 is associated with the mechanism of tumorigenesis in hepatocellular carcinoma. Oncol. Rep. 2013, 30, 2220–2228. [Google Scholar] [CrossRef]
- Huang, H.; Li, T.; Chen, M.; Liu, F.; Wu, H.; Wang, J.; Chen, J.; Li, X. Identification and validation of NOLC1 as a potential target for enhancing sensitivity in multidrug resistant non-small cell lung cancer cells. Cell Mol. Biol. Lett. 2018, 23, 54. [Google Scholar] [CrossRef] [Green Version]
- Nozawa, R.S.; Nagao, K.; Igami, K.T.; Shibata, S.; Shirai, N.; Nozaki, N.; Sado, T.; Kimura, H.; Obuse, C. Human inactive X chromosome is compacted through a PRC2-independent SMCHD1-HBiX1 pathway. Nat. Struct. Mol. Biol. 2013, 20, 566–573. [Google Scholar] [CrossRef] [PubMed]
- Gurzau, A.D.; Chen, K.; Xue, S.; Dai, W.; Lucet, I.S.; Ly, T.T.N.; Reversade, B.; Blewitt, M.E.; Murphy, J.M. FSHD2- and BAMS-associated mutations confer opposing effects on SMCHD1 function. J. Biol. Chem. 2018, 293, 9841–9853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coker, H.; Brockdorff, N. SMCHD1 accumulates at DNA damage sites and facilitates the repair of DNA double-strand breaks. J. Cell Sci. 2014, 127, 1869–1874. [Google Scholar] [CrossRef] [Green Version]
- Tang, M.; Li, Y.; Zhang, X.; Deng, T.; Zhou, Z.; Ma, W.; Songyang, Z. Structural maintenance of chromosomes flexible hinge domain containing 1 (SMCHD1) promotes non-homologous end joining and inhibits homologous recombination repair upon DNA damage. J. Biol. Chem. 2014, 289, 34024–34032. [Google Scholar] [CrossRef] [Green Version]
- Yan, Y.; Yin, P.; Gong, H.; Xue, Y.; Zhang, G.; Fang, B.; Chen, Z.; Li, Y.; Yang, C.; Huang, Z.; et al. Nucleosome Assembly Protein 1-Like 1 (Nap1l1) Regulates the Proliferation of Murine Induced Pluripotent Stem Cells. Cell Physiol. Biochem. 2016, 38, 340–350. [Google Scholar] [CrossRef] [PubMed]
- Okuwaki, M.; Kato, K.; Nagata, K. Functional characterization of human nucleosome assembly protein 1-like proteins as histone chaperones. Genes Cells 2010, 15, 13–27. [Google Scholar] [CrossRef]
- Huang, X.; Liew, K.; Natalang, O.; Siau, A.; Zhang, N.; Preiser, P.R. The role of serine-type serine repeat antigen in Plasmodium yoelii blood stage development. PLoS ONE 2013, 8, e60723. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Symbol | Location | Type | Human Protein Atlas Biomarker Comments |
|---|---|---|---|
| PCNA | Nucleus | Enzyme | prognosis of oropharyngeal neoplasm |
| GAPDH | Nucleus | Enzyme | diagnosis of ovarian cancer |
| CTNNA1 | Membrane | Other | diagnosis of gastric cancer. |
| CTNNB1 | Nucleus | TR | diagnosis of Colorectal Cancer, thyroid cancer, gastric cancer, Cervical Cancer, mesothelioma |
| HNRNPA1 | Nucleus | Other | |
| NOLC1 | Nucleus | Transcription Regulator | Prognostic of thyroid cancer (unfavorable) and renal cancer (unfavorable) |
| SMHD1 | Nucleus | Enzyme | Prognostic of renal cancer (unfavorable) and liver cancer (unfavorable) |
| NP1L1 | Nucleus | Other | Prognostic of renal cancer (unfavorable) and liver cancer (unfavorable) |
| PHGDH | Cytoplasm | Enzyme | Prognostic of endometrial cancer (unfavorable) and glioma (favorable) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hu, Y.; Yao, J.; Wang, Z.; Liang, H.; Li, C.; Zhou, X.; Yang, F.; Zhang, Y.; Jin, H. Comparative Proteomic Analysis of Drug Trichosanthin Addition to BeWo Cell Line. Molecules 2022, 27, 1603. https://doi.org/10.3390/molecules27051603
Hu Y, Yao J, Wang Z, Liang H, Li C, Zhou X, Yang F, Zhang Y, Jin H. Comparative Proteomic Analysis of Drug Trichosanthin Addition to BeWo Cell Line. Molecules. 2022; 27(5):1603. https://doi.org/10.3390/molecules27051603
Chicago/Turabian StyleHu, Yajun, Jun Yao, Zening Wang, Hui Liang, Cunyu Li, Xinwen Zhou, Fengying Yang, Yang Zhang, and Hong Jin. 2022. "Comparative Proteomic Analysis of Drug Trichosanthin Addition to BeWo Cell Line" Molecules 27, no. 5: 1603. https://doi.org/10.3390/molecules27051603
APA StyleHu, Y., Yao, J., Wang, Z., Liang, H., Li, C., Zhou, X., Yang, F., Zhang, Y., & Jin, H. (2022). Comparative Proteomic Analysis of Drug Trichosanthin Addition to BeWo Cell Line. Molecules, 27(5), 1603. https://doi.org/10.3390/molecules27051603