
A Benchmark Protocol for DFT Approaches and Data-Driven Models for Halide-Water Clusters


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Computational Details, Results and Discussion
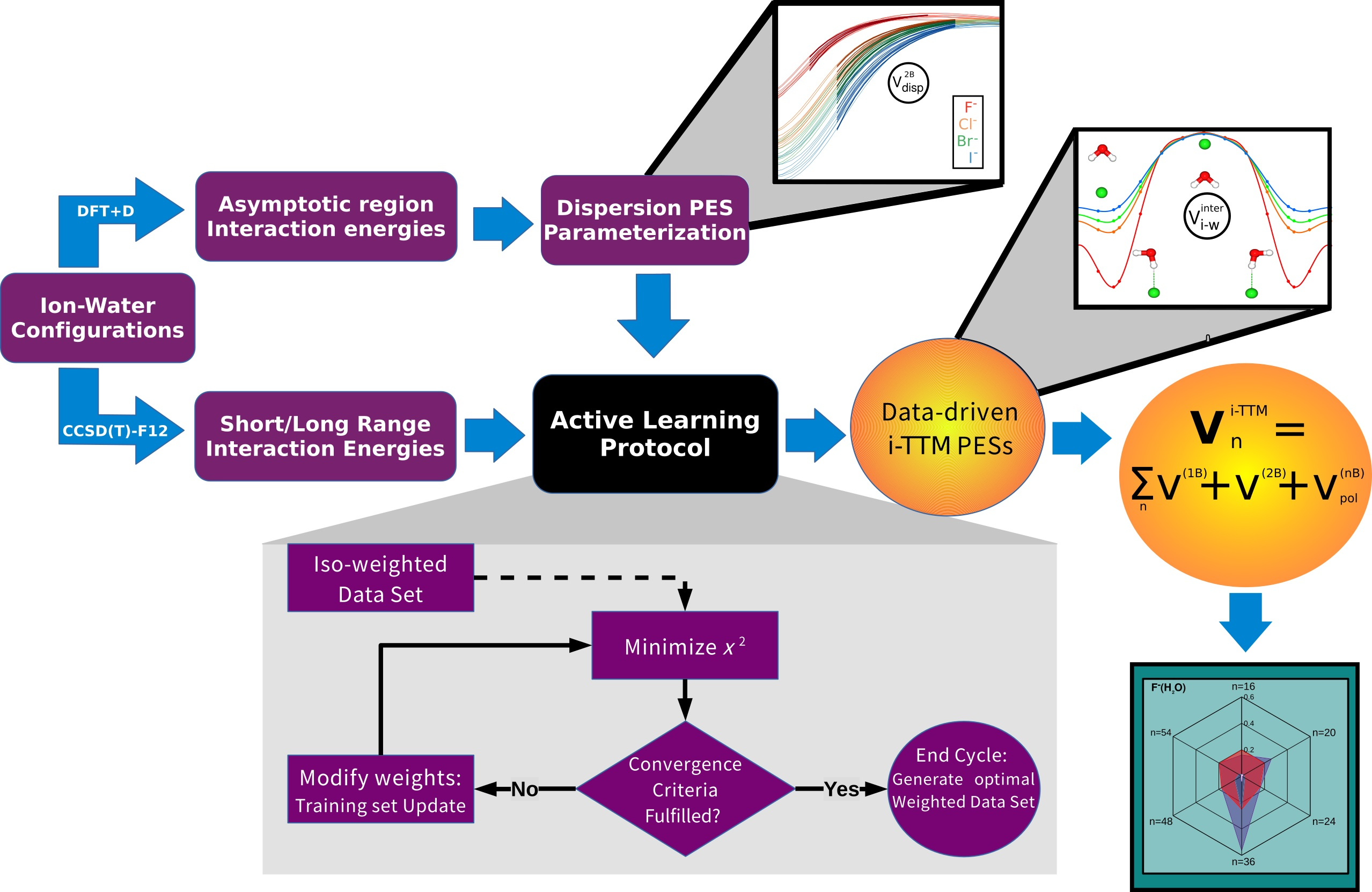
2.1. Building Up Data-Driven Interaction Models
- We start from a pool of fixed points, in our case those points are equidistant in all , and coordinates.
- For each iteration, a minimization of the parameters for the target function is performed, using a simplex procedure.
- Once the minimization has ended, we check the fulfillment of the convergence criteria, such as the maximum number of steps, minimum average error, and certain error value, along the whole curve to see if it reached convergence.
- If none of the convergence criteria have been reached, the error at each point is obtained.
- For the N points with an error larger than a predefined percentage of the maximum error value, we increase their weights in a gradual descent manner throughout the iterations.
- Minimization is performed with the new training set of weights.
- This procedure is repeated until convergence is achieved.
2.2. Evolutionary Programming Procedure and Selected Optimal Reference Structures
2.3. Electronic Structure Calculations and Reference Energy Data
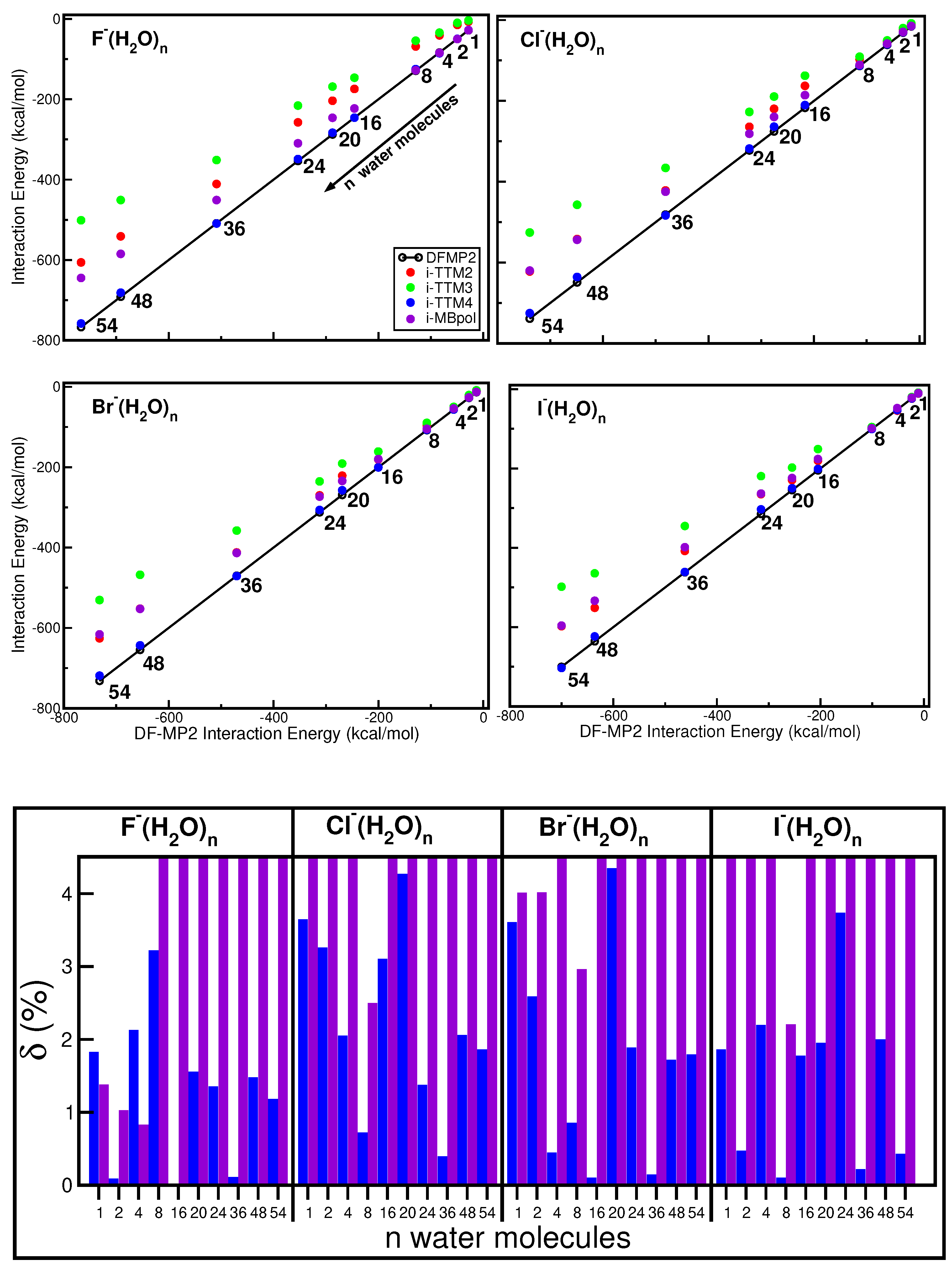
2.4. Comparative Analysis of the Different Halide–Water Potentials: Equilibrium Structures
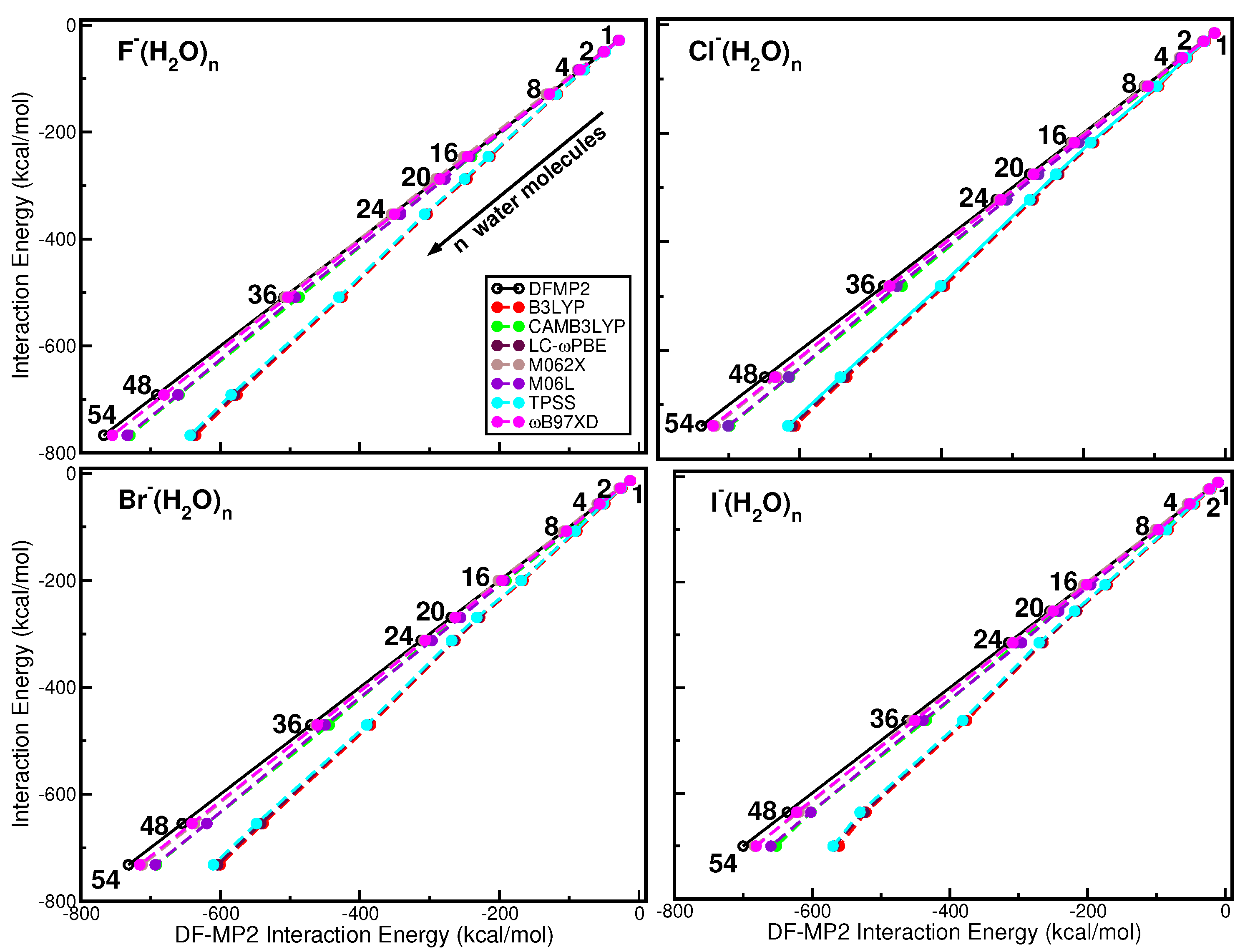
2.5. Comparative Analysis of DFT/DFT+D Approaches: Equilibrium Structures
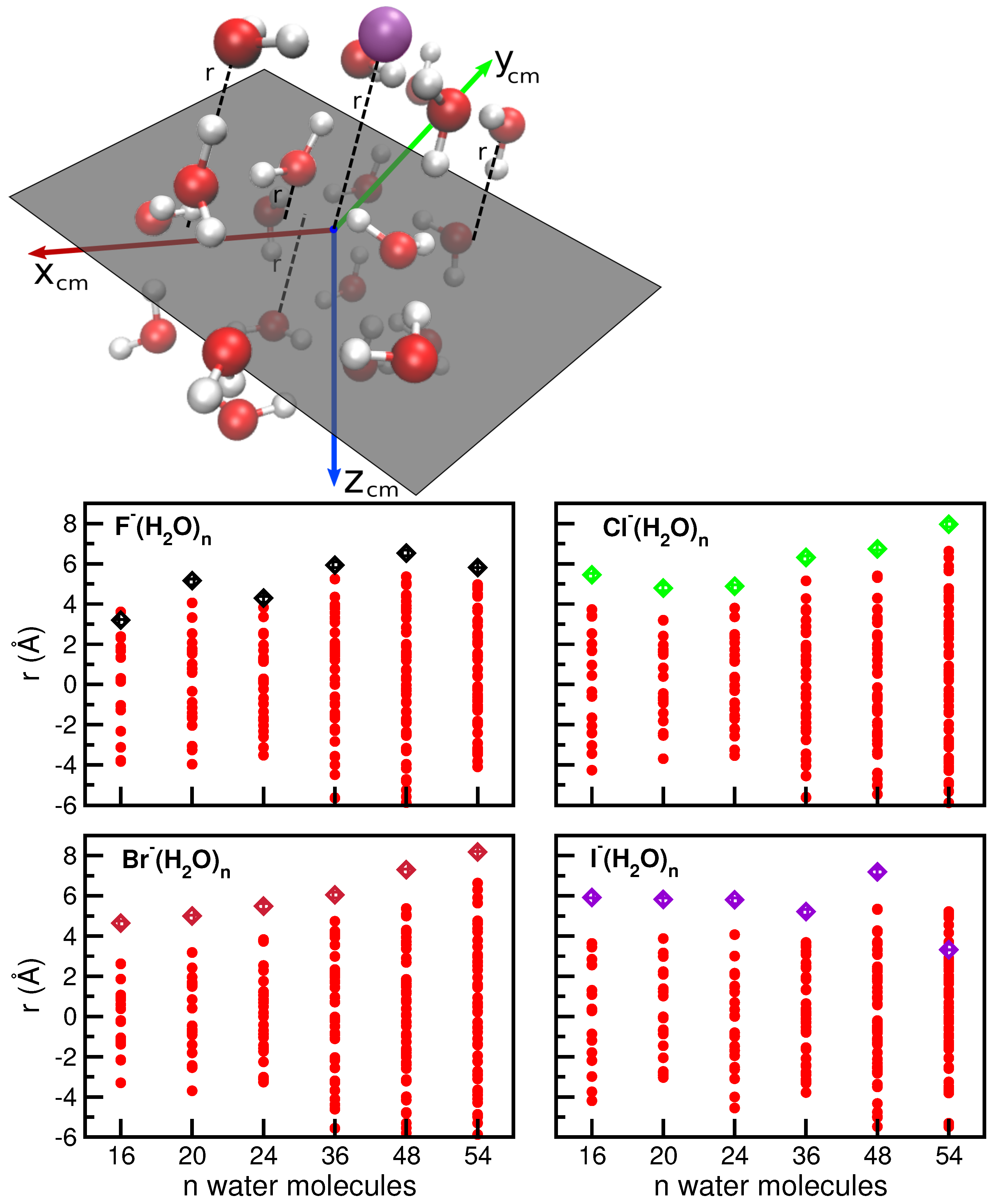
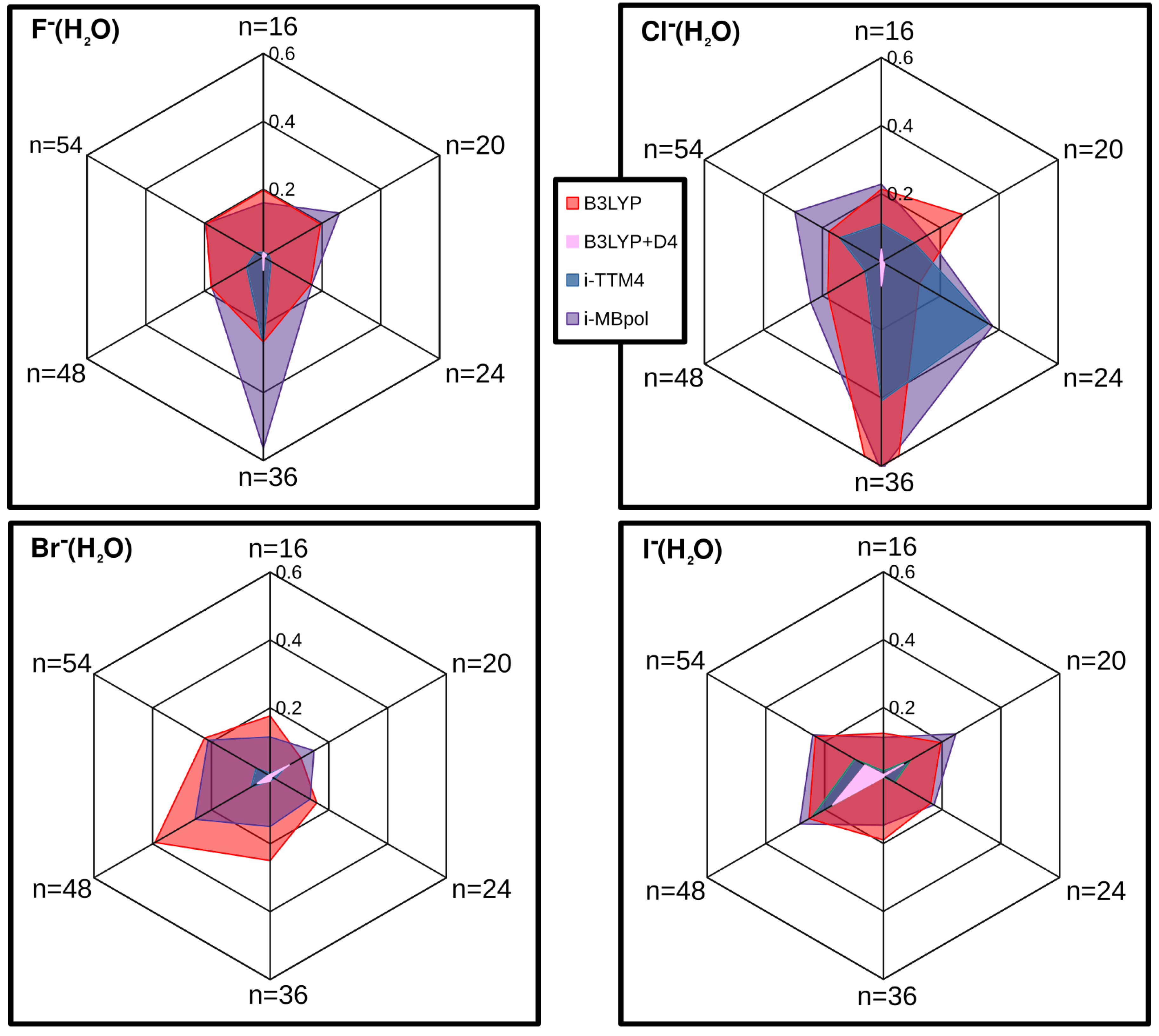
2.6. Overall Comparative Analysis: Non-Equilibrium Configurations
3. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ball, P. Life’s Matrix: A Biography of Water; University of California Press: Berkeley, CA, USA, 2001. [Google Scholar]
- Sharp, K.A. Water: Structure and Properties. In eLS; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2001. [Google Scholar]
- Brini, E.; Fennell, C.J.; Fernandez-Serra, M.; Hribar-Lee, B.; Lukšič, M.; Dill, K.A. How Water’s Properties Are Encoded in Its Molecular Structure and Energies. Chem. Rev. 2017, 117, 12385–12414. [Google Scholar] [CrossRef] [Green Version]
- Perakis, F.; Camisasca, G.; Lane, T.J.; Späh, A.; Wikfeldt, K.T.; Sellberg, J.A.; Lehmkühler, F.; Pathak, H.; Kim, K.H.; Amann-Winkel, K.; et al. Coherent X-rays reveal the influence of cage effects on ultrafast water dynamics. Nat. Commun. 2018, 9, 1917. [Google Scholar] [CrossRef]
- Markland, T.E.; Ceriotti, M. Nuclear quantum effects enter the mainstream. Nat. Rev. Chem. 2018, 2, 0109. [Google Scholar] [CrossRef] [Green Version]
- Shi, R.; Tanaka, H. Direct Evidence in the Scattering Function for the Coexistence of Two Types of Local Structures in Liquid Water. J. Am. Chem. Soc. 2020, 142, 2868–2875. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Dettori, R.; Nunes, J.P.F.; List, N.H.; Biasin, E.; Centurion, M.; Chen, Z.; Cordones, A.A.; Deponte, D.P.; Heinz, T.F.; et al. Direct observation of ultrafast hydrogen bond strengthening in liquid water. Nature 2021, 596, 531–535. [Google Scholar] [CrossRef]
- Kebarle, P.; Arshadi, M.; Scarborough, J. Hydration of Negative Ions in the Gas Phase. J. Chem. Phys. 1968, 49, 817–822. [Google Scholar] [CrossRef]
- Marcus, Y. Effect of Ions on the Structure of Water: Structure Making and Breaking. Chem. Rev. 2009, 109, 1346–1370. [Google Scholar] [CrossRef]
- Marcus, Y. Ions in Solution and Their Solvation; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2015. [Google Scholar]
- Robertson, W.H.; Johnson, M.A. Molecular Aspects of Halide Ion Hydration: The Cluster Approach. Ann. Rev. Phys. Chem. 2003, 54, 173–213. [Google Scholar] [CrossRef]
- Smith, J.D.; Saykally, R.J.; Geissler, P.L. The Effects of Dissolved Halide Anions on Hydrogen Bonding in Liquid Water. J. Am. Chem. Soc. 2007, 129, 13847–13856. [Google Scholar] [CrossRef]
- Tielrooij, K.J.; Garcia-Araez, N.; Bonn, M.; Bakker, H.J. Cooperativity in Ion Hydration. Science 2010, 328, 1006–1009. [Google Scholar] [CrossRef] [PubMed]
- Funkner, S.; Niehues, G.; Schmidt, D.A.; Heyden, M.; Schwaab, G.; Callahan, K.M.; Tobias, D.J.; Havenith, M. Watching the Low-Frequency Motions in Aqueous Salt Solutions: The Terahertz Vibrational Signatures of Hydrated Ions. J. Am. Chem. Soc. 2012, 134, 1030–1035. [Google Scholar] [CrossRef] [PubMed]
- Tobias, D.J.; Stern, A.C.; Baer, M.D.; Levin, Y.; Mundy, C.J. Simulation and Theory of Ions at Atmospherically Relevant Aqueous Liquid-Air Interfaces. Ann. Rev. Phys. Chem. 2013, 64, 339–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Migliorati, V.; Sessa, F.; Aquilanti, G.; D’Angelo, P. Unraveling halide hydration: A high dilution approach. J. Chem. Phys. 2014, 141, 044509. [Google Scholar] [CrossRef] [PubMed]
- Piatkowski, L.; Zhang, Z.; Backus, E.H.G.; Bakker, H.J.; Bonn, M. Extreme surface propensity of halide ions in water. Nat. Commun. 2014, 5, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Waluyo, I.; Nordlund, D.; Bergmann, U.; Schlesinger, D.; Pettersson, L.G.M.; Nilsson, A. A different view of structure-making and structure-breaking in alkali halide aqueous solutions through X-ray absorption spectroscopy. J. Chem. Phys. 2014, 140, 244506. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.P.; McKiernan, K.A.; Gomes, J.; Beauchamp, K.A.; Head-Gordon, T.; Rice, J.E.; Swope, W.C.; Martínez, T.J.; Pande, V.S. Building a More Predictive Protein Force Field: A Systematic and Reproducible Route to AMBER-FB15. J. Phys. Chem. B 2017, 121, 4023–4039. [Google Scholar] [CrossRef] [PubMed]
- Kistenmacher, H.; Popkie, H.; Clementi, E. Study of the structure of molecular complexes. II. Energy surfaces for a water molecule in the field of a sodium or potassium cation. J. Chem. Phys. 1973, 58, 1689–1699. [Google Scholar] [CrossRef]
- Perera, L.; Berkowitz, M.L. Structures of Cl−(H2O)n and F−(H2O)n (n = 2,3,…,15) clusters. Molecular dynamics computer simulations. J. Chem. Phys. 1994, 100, 3085–3093. [Google Scholar] [CrossRef]
- Ayotte, P.; Nielsen, S.B.; Weddle, G.H.; Johnson, M.A.; Xantheas, S.S. Spectroscopic Observation of Ion-Induced Water Dimer Dissociation in the X-.(H2O)2 (X = F, Cl, Br, I) Clusters. J. Phys. Chem. A 1999, 103, 10665–10669. [Google Scholar] [CrossRef]
- Cabarcos, O.M.; Weinheimer, C.J.; Lisy, J.M.; Xantheas, S.S. Microscopic hydration of the fluoride anion. J. Chem. Phys. 1999, 110, 5–8. [Google Scholar] [CrossRef]
- Guàrdia, E.; Skarmoutsos, I.; Masia, M. On Ion and Molecular Polarization of Halides in Water. J. Chem. Theory Comput. 2009, 5, 1449–1453. [Google Scholar] [CrossRef] [PubMed]
- Sala, J.; Guárdia, E.; Masia, M. The polarizable point dipoles method with electrostatic damping: Implementation on a model system. J. Chem. Phys. 2010, 133, 234101. [Google Scholar] [CrossRef] [PubMed]
- Horvath, S.; McCoy, A.B.; Elliott, B.M.; Weddle, G.H.; Roscioli, J.R.; Johnson, M.A. Anharmonicities and Isotopic Effects in the Vibrational Spectra of X-.H2O, .HDO, and .D2O [X = Cl, Br, and I] Binary Complexes. J. Phys. Chem. A 2010, 114, 1556–1568. [Google Scholar] [CrossRef]
- Vega, C.; Abascal, J.L.F. Simulating water with rigid non-polarizable models: A general perspective. Phys. Chem. Chem. Phys. 2011, 13, 19663–19688. [Google Scholar] [CrossRef]
- Trumm, M.; Martínez, Y.O.G.; Réal, F.; Masella, M.; Vallet, V.; Schimmelpfennig, B. Modeling the hydration of mono-atomic anions from the gas phase to the bulk phase: The case of the halide ions F−, Cl−, and Br-. J. Chem. Phys. 2012, 136, 044509. [Google Scholar] [CrossRef]
- Wolke, C.T.; Menges, F.S.; Tötsch, N.; Gorlova, O.; Fournier, J.A.; Weddle, G.H.; Johnson, M.A.; Heine, N.; Esser, T.K.; Knorke, H.; et al. Thermodynamics of Water Dimer Dissociation in the Primary Hydration Shell of the Iodide Ion with Temperature-Dependent Vibrational Predissociation Spectroscopy. J. Phys. Chem. A 2015, 119, 1859–1866. [Google Scholar] [CrossRef] [PubMed]
- Wick, C.D.; Xantheas, S.S. Computational Investigation of the First Solvation Shell Structure of Interfacial and Bulk Aqueous Chloride and Iodide Ions. J. Phys. Chem. B 2009, 113, 4141–4146. [Google Scholar] [CrossRef] [PubMed]
- Baer, M.D.; Mundy, C.J. Toward an Understanding of the Specific Ion Effect Using Density Functional Theory. J. Phys. Chem. Lett. 2011, 2, 1088–1093. [Google Scholar] [CrossRef]
- Werhahn, J.C.; Akase, D.; Xantheas, S.S. Universal scaling of potential energy functions describing intermolecular interactions. II. The halide-water and alkali metal-water interactions. J. Chem. Phys. 2014, 141, 064118. [Google Scholar] [CrossRef]
- Cisneros, G.A.; Wikfeldt, K.T.; Ojamäe, L.; Lu, J.; Xu, Y.; Torabifard, H.; Bartók, A.P.; Csányi, G.; Molinero, V.; Paesani, F. Modeling Molecular Interactions in Water: From Pairwise to Many-Body Potential Energy Functions. Chem. Rev. 2016, 116, 7501–7528. [Google Scholar] [CrossRef]
- Gillan, M.J.; Alfé, D.; Michaelides, A. Perspective: How good is DFT for water? J. Chem. Phys. 2016, 144, 130901. [Google Scholar] [CrossRef] [PubMed]
- Arismendi-Arrieta, D.J.; Riera, M.; Bajaj, P.; Prosmiti, R.; Paesani, F. i-TTM Model for Ab Initio-Based Ion—Water Interaction Potentials. 1. Halide–Water Potential Energy Functions. J. Phys. Chem. B 2016, 120, 1822–1832. [Google Scholar] [CrossRef] [PubMed]
- Dasgupta, S.; Lambros, E.; Perdew, J.P.; Paesani, F. Elevating density functional theory to chemical accuracy for water simulations through a density-corrected many-body. Nat. Commun. 2021, 12, 6359. [Google Scholar] [CrossRef]
- Joseph, P. Heindel, J.P.; Xantheas, S.S. The Many-Body Expansion for Aqueous Systems Revisited: II. Alkali Metal and Halide Ion—Water Interactions. J. Chem. Theory Comput. 2021, 17, 2200–2216. [Google Scholar]
- Amann-Winkel, K.; Bellissent-Funel, M.C.; Bove, L.E.; Loerting, T.; Nilsson, A.; Paciaroni, A.; Schlesinger, D.; Skinner, L. X-ray and Neutron Scattering of Water. Chem. Rev. 2016, 116, 7570–7589. [Google Scholar] [CrossRef] [Green Version]
- Antalek, M.; Pace, E.; Hedman, B.; Hodgson, K.O.; Chillemi, G.; Benfatto, M.; Sarangi, R.; Frank, P. Solvation structure of the halides from x-ray absorption spectroscopy. J. Chem. Phys. 2016, 145, 044318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dorsett, H.E.; Watts, R.O.; Xantheas, S.S. Probing Temperature Effects on the Hydrogen Bonding Network of the Cl−(H2O)2 Cluster. J. Phys. Chem. A 1999, 103, 3351–3355. [Google Scholar] [CrossRef]
- Kim, J.; Lee, H.M.; Suh, S.B.; Majumdar, D.; Kim, K.S. Comparative ab initio study of the structures, energetics and spectra of X−(H2O)n=1–4 [X = F,Cl,Br,I] clusters. J. Chem. Phys. 2000, 113, 5259–5272. [Google Scholar] [CrossRef]
- Prakash, M.; Gopalsamy, K.; Subramanian, V. Studies on the structure, stability, and spectral signatures of hydride ion-water clusters. J. Chem. Phys. 2011, 135, 214308. [Google Scholar] [CrossRef]
- Neogi, S.G.; Chaudhury, P. Structure and spectroscopic aspects of water-halide ion clusters: A study based on a conjunction of stochastic and quantum chemical methods. J. Comput. Chem. 2013, 34, 471–491. [Google Scholar] [CrossRef]
- Kamarchik, E.; Toffoli, D.; Christiansen, O.; Bowman, J.M. Ab initio potential energy and dipole moment surfaces of the F-(H2O) complex. Spectrochim. Acta A 2014, 119, 59–62. [Google Scholar] [CrossRef] [PubMed]
- León-Merino, I.; Rodríguez-Segundo, R.; Arismendi-Arrieta, D.J.; Prosmiti, R. Assessing Intermolecular Interactions in Guest-Free Clathrate Hydrate Systems. J. Phys. Chem. A 2018, 122, 1479–1487. [Google Scholar] [CrossRef] [PubMed]
- Bizzarro, B.B.; Egan, C.K.; Paesani, F. Nature of Halide—Water Interactions: Insights from Many-Body Representations and Density Functional Theory. J. Chem. Theory Comput. 2019, 15, 2983–2995. [Google Scholar] [CrossRef] [PubMed]
- Perera, L.; Berkowitz, M.L. Many-body effects in molecular dynamics simulations of Na+(H2O)n and Cl−(H2O)n clusters. J. Chem. Phys. 1991, 95, 1954–1963. [Google Scholar] [CrossRef]
- Burnham, C.J.; Xantheas, S.S. Development of transferable interaction models for water. IV. A flexible, all-atom polarizable potential (TTM2-F) based on geometry dependent charges derived from an ab initio monomer dipole moment surface. J. Chem. Phys. 2002, 116, 5115–5124. [Google Scholar] [CrossRef]
- Rheinecker, J.; Bowman, J.M. The calculated infrared spectrum of Cl-H2O using a new full dimensional ab initio potential surface and dipole moment surface. J. Chem. Phys. 2006, 125, 133206. [Google Scholar] [CrossRef]
- Fanourgakis, G.S.; Xantheas, S.S. The Flexible, Polarizable, Thole-Type Interaction Potential for Water (TTM2-F) Revisited. J. Phys. Chem. A 2006, 110, 4100–4106. [Google Scholar] [CrossRef]
- Fanourgakis, G.S.; Xantheas, S.S. Development of transferable interaction potentials for water. V. Extension of the flexible, polarizable, Thole-type model potential (TTM3-F, v. 3.0) to describe the vibrational spectra of water clusters and liquid water. J. Chem. Phys. 2008, 128, 074506. [Google Scholar] [CrossRef]
- Babin, V.; Leforestier, C.; Paesani, F. Development of a “First Principles” Water Potential with Flexible Monomers: Dimer Potential Energy Surface, VRT Spectrum, and Second Virial Coefficient. J. Chem. Theory Comput. 2013, 9, 5395–5403. [Google Scholar] [CrossRef]
- Kamarchik, E.; Bowman, J.M. Coupling of Low- and High-Frequency Vibrational Modes: Broadening in the Infrared Spectrum of F-(H2O)2. J. Phys. Chem. Lett. 2013, 4, 2964–2969. [Google Scholar] [CrossRef]
- Werhahn, J.C.; Miliordos, E.; Xantheas, S.S. A new variation of the Buckingham exponential-6 potential with a tunable, singularity-free short-range repulsion and an adjustable long-range attraction. Chem. Phys. Lett. 2015, 619, 133–138. [Google Scholar] [CrossRef] [Green Version]
- Metz, M.P.; Piszczatowski, K.; Szalewicz, K. Automatic Generation of Intermolecular Potential Energy Surfaces. J. Chem. Theory Comput. 2016, 12, 5895–5919. [Google Scholar] [CrossRef] [PubMed]
- Zhai, Y.; Caruso, A.; Gao, S.; Paesani, F. Active learning of many-body configuration space: Application to the Cs+—water MB-nrg potential energy function as a case study. J. Chem. Phys. 2020, 152, 144103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bilbrey, J.A.; Heindel, J.P.; Schram, M.; Bandyopadhyay, P.; Xantheas, S.S.; Choudhury, S. A look inside the black box: Using graph-theoretical descriptors to interpret a Continuous-Filter Convolutional Neural Network (CF-CNN) trained on the global and local minimum energy structures of neutral water clusters. J. Chem. Phys. 2020, 153, 024302. [Google Scholar] [CrossRef]
- Nandi, A.; Qu, C.; Houston, P.L.; Conte, R.; Yu, Q.; Bowman, J.M. A CCSD(T)-Based 4-Body Potential for Water. J. Phys. Chem. Lett. 2021, 12, 10318–10324. [Google Scholar] [CrossRef]
- Behler, J. Four Generations of High-Dimensional Neural Network Potentials. Chem. Rev. 2021, 121, 10037–10072. [Google Scholar] [CrossRef]
- Smith, J.S.; Nebgen, B.; Lubbers, N.; Isayev, O.; Roitberg, A.E. Less is more: Sampling chemical space with active learning. J. Chem. Phys. 2018, 148, 241733. [Google Scholar] [CrossRef] [Green Version]
- Westermayr, J.; Gastegger, M.; Schütt, K.T.; Maurer, R.J. Perspective on integrating machine learning into computational chemistry and materials science. J. Chem. Phys. 2021, 154, 230903. [Google Scholar] [CrossRef]
- Ceriotti, M.; Clementi, C.; Anatole von Lilienfeld, O. Machine learning meets chemical physics. J. Chem. Phys. 2021, 154, 160401. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian16 Revision C.01; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Becke, A.D.; Johnson, E.R. Exchange-hole dipole moment and the dispersion interaction revisited. J. Chem. Phys. 2007, 127, 154108. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Caldeweyher, E.; Ehlert, S.; Hansen, A.; Neugebauer, H.; Spicher, S.; Bannwarth, C.; Grimme, S. A generally applicable atomic-charge dependent London dispersion correction. J. Chem. Phys. 2019, 150, 154122. [Google Scholar] [CrossRef] [PubMed]
- Baudin, P.; Ettenhuber, P.; Reine, S.; Kristensen, K.; Kjærgaard, T. Efficient linear-scaling second-order Møller-Plesset perturbation theory: The divide—expand—consolidate RI-MP2 model. J. Chem. Phys. 2016, 144, 054102. [Google Scholar] [CrossRef] [Green Version]
- Werner, H.J.; Knowles, P.; Knizia, G.; Manby, R.; Schütz, M.; Celani, P.; Györffy, W.; Kats, D.; Korona, T.; Lindh, R.; et al. MOLPRO, Version 2012.1, a Package of ab Initio Programs. 2012. Available online: http://www.molpro.net (accessed on 28 January 2022).
- Murrell, J.N.; Carter, S.; Farantos, S.C.; Huxley, P.; Varandas, A. Molecular Potential Energy Function; Wiley: Hoboken, NJ, USA, 1984. [Google Scholar]
- Partridge, H.; Schwenke, D.W. The determination of an accurate isotope dependent potential energy surface for water from extensive ab initio calculations and experimental data. J. Chem. Phys. 1997, 106, 4618–4639. [Google Scholar] [CrossRef] [Green Version]
- Burnham, C.J.; Anick, D.J.; Mankoo, P.K.; Reiter, G.F. The vibrational proton potential in bulk liquid water and ice. J. Chem. Phys. 2008, 128, 154519. [Google Scholar] [CrossRef]
- Medders, G.R.; Babin, V.; Paesani, F. Development of a “First-Principles” Water Potential with Flexible Monomers. III. Liquid Phase Properties. J. Chem. Theory Comput. 2014, 10, 2906–2910. [Google Scholar] [CrossRef]
- Medders, G.R.; Paesani, F. Infrared and Raman Spectroscopy of Liquid Water through “First-Principles” Many-Body Molecular Dynamics. J. Chem. Theory Comput. 2015, 11, 1145–1154. [Google Scholar] [CrossRef]
- Tang, K.T.; Toennies, J.P. An improved simple model for the van der Waals potential based on universal damping functions for the dispersion coefficients. J. Chem. Phys. 1984, 80, 3726–3741. [Google Scholar] [CrossRef]
- Born, M.; Mayer, J.E. Zur Gittertheorie der Ionenkristalle. Z. Phys. 1932, 75, 1–18. [Google Scholar] [CrossRef]
- Otero-de-la Roza, A.; Johnson, E.R. Non-covalent interactions and thermochemistry using XDM-corrected hybrid and range-separated hybrid density functionals. J. Chem. Phys. 2013, 138, 204109. [Google Scholar] [CrossRef]
- Boys, S.; Bernardi, F. The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol. Phys. 1970, 19, 553–566. [Google Scholar] [CrossRef]
- Iwamatsu, M. Applying evolutionary programming to structural optimization of atomic clusters. Comput. Phys. Commun. 2001, 142, 214–218. [Google Scholar] [CrossRef]
- Fogel, D. Evolutionary Computation: Toward a New Philosophy of Machine Intelligence; IEEE Press-Wiley: Hoboken, NJ, USA, 2006. [Google Scholar]
- Pérez de Tudela, R.; Barragán, P.; Valdés, A.; Prosmiti, R. Energetics and Solvation Structure of a Dihalogen Dopant (I2) in 4He Clusters. J. Phys. Chem. A 2014, 118, 6492–6500. [Google Scholar] [CrossRef] [Green Version]
- Alharzali, N.; Rodríguez-Segundo, R.; Prosmiti, R. Modelling interactions of cationic dimers in He droplets: Microsolvation trends in HenK2+ clusters. Phys. Chem. Chem. Phys. 2021, 23, 7849–7859. [Google Scholar] [CrossRef]
- The Cambridge Cluster Database. 2021. Available online: http://www-wales.ch.cam.ac.uk/CCD.html (accessed on 30 December 2021).
- DENEB 1.30 Beta: The Nanotechnology Software by Atelgraphics. 2020. Available online: https://www.atelgraphics.com (accessed on 28 January 2022).
- Bajaj, P.; Riera, M.; Lin, J.K.; Mendoza Montijo, Y.E.; Gazca, J.; Paesani, F. Halide Ion Microhydration: Structure, Energetics, and Spectroscopy of Small Halide—Water Clusters. J. Phys. Chem. A 2019, 123, 2843–2852. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Segundo, R.; Arismendi-Arrieta, D.; Prosmiti, R. A bottom-up approach for ion-water interactions: From clusters to bulk. In State-of-the-Art Reviews on Energetic Ion-Atom and Ion-Molecule Collisions; World Scientific: Singapore, 2019; Chapter 8; pp. 205–225. [Google Scholar]
- Werner, H.J.; Knowles, P.J.; Knizia, G.; Manby, F.R.; Schütz, M. Molpro: A general-purpose quantum chemistry program package. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 242–253. [Google Scholar] [CrossRef]
- Dunning, T.H. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Woon, D.E.; Dunning, T.H. Gaussian basis sets for use in correlated molecular calculations. IV. Calculation of static electrical response properties. J. Chem. Phys. 1994, 100, 2975–2988. [Google Scholar] [CrossRef] [Green Version]
- Peterson, K.A.; Figgen, D.; Goll, E.; Stoll, H.; Dolg, M. Systematically convergent basis sets with relativistic pseudopotentials. II. Small-core pseudopotentials and correlation consistent basis sets for the post-d group 16–18 elements. J. Chem. Phys. 2003, 119, 11113–11123. [Google Scholar] [CrossRef] [Green Version]
- Peterson, K.A.; Shepler, B.C.; Figgen, D.; Stoll, H. On the Spectroscopic and Thermochemical Properties of ClO, BrO, IO, and Their Anions. J. Phys. Chem. A 2006, 110, 13877–13883. [Google Scholar] [CrossRef]
- Hill, J.G.; Peterson, K.A. Correlation consistent basis sets for explicitly correlated wavefunctions: Pseudopotential-based basis sets for the post-d main group elements Ga-Rn. J. Chem. Phys. 2014, 141, 094106. [Google Scholar] [CrossRef] [PubMed]
- Universität-Bonn. A Dispersion Correction for Density Functionals, Hartree-Fock and Semi-Empirical Quantum Chemical Methods. Available online: https://www.chemie.uni-bonn.de/pctc/mulliken-center/software/dft-d3/dft-d3 (accessed on 28 January 2022).
- Universität-Bonn. D4—A Generally Applicable Atomic-Charge Dependent London Dispersion Correction. Available online: https://www.chemie.uni-bonn.de/pctc/mulliken-center/software/dftd4 (accessed on 28 January 2022).
- Tao, J.; Perdew, J.P.; Staroverov, V.N.; Scuseria, G.E. Climbing the Density Functional Ladder: Non empirical Meta-Generalized Gradient Approximation Designed for Molecules and Solids. Phys. Rev. Lett. 2003, 91, 146401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y. andTruhlar, D.G. A new local density functional for main-group thermochemistry, transition metal bonding, thermochemicalkinetics, and noncovalent interactions. J. Chem. Phys. 2006, 125, 194101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, non-covalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215. [Google Scholar]
- Yanai, T.; Tew, D.P.; Handy, N.C. A new hybrid exchange-correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51. [Google Scholar] [CrossRef] [Green Version]
- Vydrov, O.A.; Scuseria, G.E. Assessment of a long-range corrected hybrid functional. J. Chem. Phys. 2006, 125, 234109. [Google Scholar] [CrossRef]
- Chai, J.; Head-Gordon, M. Systematic optimization of long-range corrected hybrid density functionals. J. Chem. Phys. 2008, 128, 084106. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [Green Version]
- Smith, D.; Burns, L.; Patkowski, K.; Sherrill, C. Revised Damping Parameters for the D3 Dispersion Correction to Density Functional Theory. J. Phys. Chem. Lett. 2016, 7, 2197–2203. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodríguez-Segundo, R.; Arismendi-Arrieta, D.J.; Prosmiti, R. A Benchmark Protocol for DFT Approaches and Data-Driven Models for Halide-Water Clusters. Molecules 2022, 27, 1654. https://doi.org/10.3390/molecules27051654
Rodríguez-Segundo R, Arismendi-Arrieta DJ, Prosmiti R. A Benchmark Protocol for DFT Approaches and Data-Driven Models for Halide-Water Clusters. Molecules. 2022; 27(5):1654. https://doi.org/10.3390/molecules27051654
Chicago/Turabian StyleRodríguez-Segundo, Raúl, Daniel J. Arismendi-Arrieta, and Rita Prosmiti. 2022. "A Benchmark Protocol for DFT Approaches and Data-Driven Models for Halide-Water Clusters" Molecules 27, no. 5: 1654. https://doi.org/10.3390/molecules27051654
APA StyleRodríguez-Segundo, R., Arismendi-Arrieta, D. J., & Prosmiti, R. (2022). A Benchmark Protocol for DFT Approaches and Data-Driven Models for Halide-Water Clusters. Molecules, 27(5), 1654. https://doi.org/10.3390/molecules27051654