
Aziridine Ring Opening as Regio- and Stereoselective Access to C-Glycosyl-Aminoethyl Sulfide Derivatives


Abstract
:1. Introduction
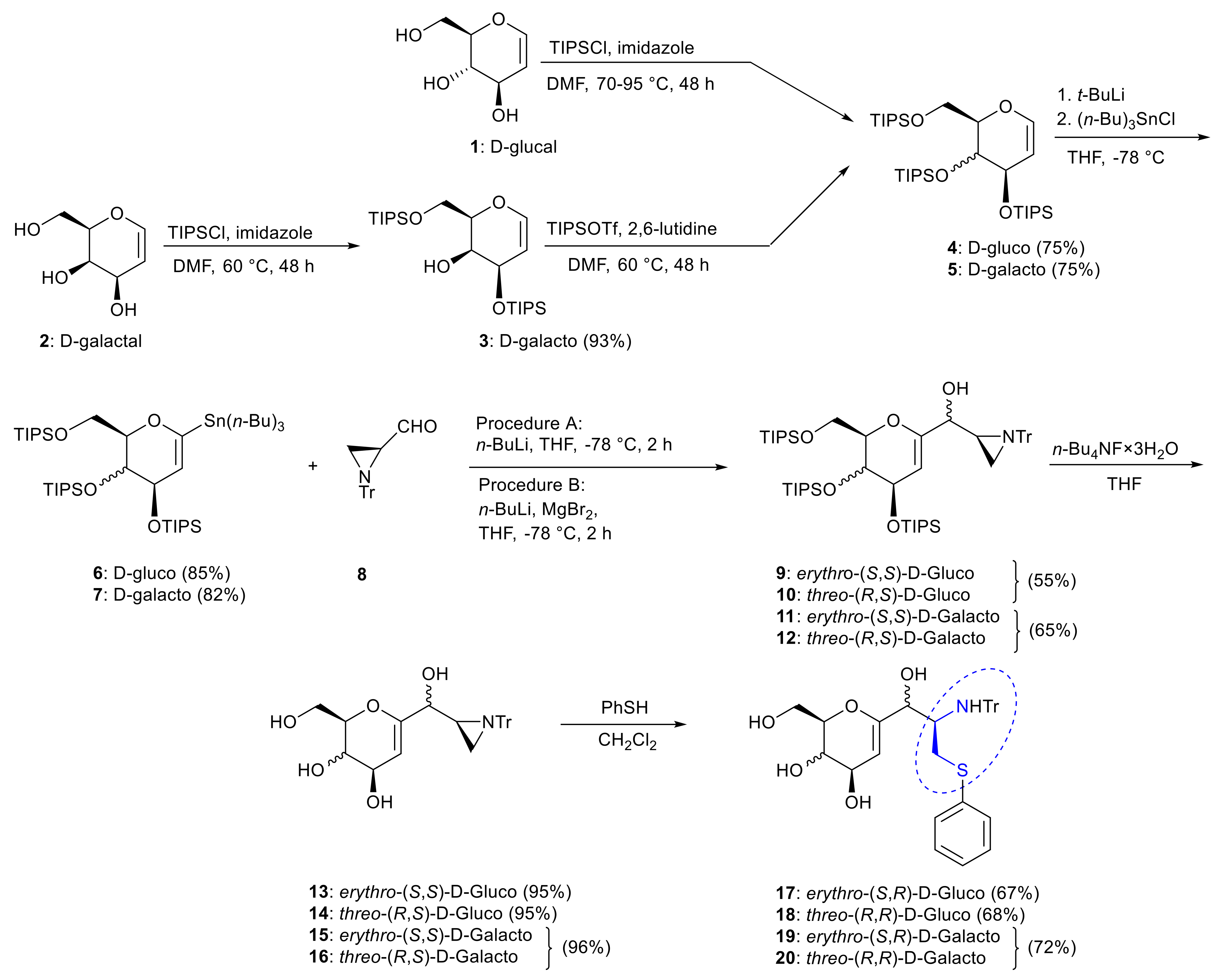
2. Results and Discussion
3. Materials and Methods
3.1. General Procedure for the Synthesis of Glycals
3.1.1. D-Glucal (1)
3.1.2. D-Galactal (2)
3.1.3. 3,6-Di-O-(triisopropylsilyl)-D-galactal (3)
3.1.4. 3,4,6-Tris-O-(triisopropylsilyl)-D-glucal (4)
3.1.5. 3,4,6-Tri-O-(triisopropylsilyl)-D-galactal (5)
3.2. General Procedure for the Synthesis of Tributhyltin Derivatives of Glycals
3.2.1. 1-(Tributylstannyl)-3,4,6-tris-O-(triisopropylsilyl)-D-glucal (6)
3.2.2. 1-(Tributylstannyl)-3,4,6-tris-O-(triisopropylsilyl)-D-galactal (7)
3.3. General Procedure for the Reaction of Derivatives of Glycals with Aziridine Aldehyde 8
3.3.1. erythro-(S)-[3,4,6-Tris-O-(triisopropylsilyl)-D-glucal-1-yl][(S)-1-triphenylmethylaziridin-2-yl]methanol (9)
3.3.2. threo-(R)-[3,4,6-Tris-O-(triisopropylsilyl)-D-glucal-1-yl][(S)-1-triphenylmethylaziridin-2-yl]methanol (10)
3.3.3. erythro-(S)-[3,4,6-Tris-O-(triisopropylsilyl)-D-galactal-1-yl][(S)-1-triphenylmethylaziridin-2-yl]methanol (11) and threo-(R)-[3,4,6-Tris-O-(triisopropylsilyl)-D-galactal-1-yl][(S)1-triphenylmethylaziridin-2-yl]methanol (12)
3.4. General Procedure for Deprotection of Hydroxyl Groups
3.4.1. erythro-(S)-[D-Glucal-1-yl][(S)-1-triphenylmethylaziridin-2-yl]methanol (13)
3.4.2. threo-(R)-[D-Glucal-1-yl][(S)-1-triphenylmethylaziridin-2-yl]methanol (14)
3.4.3. erythro-(S)-[D-Glalactal-1-yl][(S)-1-triphenylmethylaziridin-2-yl]methanol (15) and threo-(R)-[D-Galactal-1-yl][(S)-1-triphenylmethylaziridin-2-yl]methanol (16)
3.5. General Procedure for Aziridine Ring Opening Reaction
3.5.1. erytro-(1S,2R)-1-[(1-Hydroxy-3-(phenylthio)-2-(triphenylmethylamino)propyl)]-D-glucal (17)
3.5.2. threo-(1R,2R)-1-[(1-Hydroxy-3-(phenylthio)-2-(triphenylmethylamino)propyl)]-D-glucal (18)
3.5.3. erytro-(1S,2R)-1-[(1-Hydroxy-3-(phenylthio)-2-(triphenylmethylamino)propyl)]-D-galactal (19) and threo-((1R,2R)-1-[(1-hydroxy-3-(phenylthio)-2-(triphenylmethylamino)propyl)]-D-galactal (20)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Farina, V.; Reeves, J.T.; Senanayake, C.H.; Song, J.J. Asymmetric synthesis of active pharmaceutical ingredients. Chem. Rev. 2006, 106, 2734–2793. [Google Scholar] [CrossRef] [PubMed]
- Caner, H.; Groner, E.; Levy, L.; Agranat, I. Trends in the development of chiral drugs. Drug Discov. Today 2004, 9, 105–110. [Google Scholar] [CrossRef]
- Nag, A. Asymmetric Synthesis of Drugs and Natural Products; CRC Press: Boca Raton, FL, USA, 2018. [Google Scholar]
- Varki, A.; Cummings, R.; Esko, J.; Freeze, H.; Hart, G.; Marth, J. Essentials of Glycobiology; Cold Spring Harbor Laboratory Press: New York, NY, USA, 1999. [Google Scholar]
- Wong, C.-H. Carbohydrate-Based Drug Discovery; Wiley-VCH: Weinheim, Germany, 2003; Volumes 1 and 5. [Google Scholar]
- Nuzzi, A.; Massi, A.; Dondoni, A. General Synthesis of C-Glycosyl Amino Acids via Proline-Catalyzed Direct Electrophilic α-Amination of C-Glycosylalkyl Aldehyde. Org. Lett. 2008, 10, 4485–4488. [Google Scholar] [CrossRef] [PubMed]
- Schäfer, A.; Henkensmeier, D.; Kröger, L.; Thiem, J. Aziridine ring opening as regio- and stereoselective access to O-glycosyl amino acids and their transformation into O-glycopeptide mimetics. Tetrahedron Asymmetry 2009, 20, 902–909. [Google Scholar] [CrossRef]
- Seitz, O. Glycopeptide Synthesis and the Effects of Glycosylation on Protein Structure and Activity. ChemBioChem 2000, 1, 214–246. [Google Scholar] [CrossRef]
- Sears, P.; Wong, C.-H. Carbohydrate Mimetics: A New Strategy for Tackling the Problem of Carbohydrate-Mediated Biological Recognition. Angew. Chem. Int. Ed. 1999, 38, 2301–2324. [Google Scholar] [CrossRef]
- Dondoni, A.; Marra, A.; Massi, A. Stereoselective synthesis of the C-linked analogue of β-D-galactopyranosyl-L-serine. Tetrahedron 1998, 54, 2827–2832. [Google Scholar] [CrossRef]
- Lay, L.; Meldal, M.; Nicotra, F.; Panza, L.; Russo, G. Stereoselective synthesis of the C-analogue of β-D-glucopyranosyl serin. J. Chem. Soc. Chem. Commun. 1997, 1469–1470. [Google Scholar] [CrossRef]
- Debenham, S.D.; Debenham, J.S.; Burk, M.J.; Toone, E.J. Synthesis of Carbon-Linked Glycopeptides through Catalytic Asymmetric Hydrogenation. J. Am. Chem. Soc. 1997, 119, 9897–9898. [Google Scholar] [CrossRef]
- Fisher, J.F.; Harrison, A.W.; Bundy, G.L.; Wilkinson, K.F.; Bush, B.D.; Ruwart, M.J. Peptide to glycopeptide: Glycosylated oligopeptide renin inhibitors with attenuated in vivo clearance properties. J. Med. Chem. 1991, 34, 3140–3143. [Google Scholar] [CrossRef]
- Kröger, L.; Henkensmeier, D.; Schäfe, A.; Thiem, J. Novel O-glycosyl amino acid mimetics as building blocks for O-glycopeptides act as inhibitors of galactosidases. Bioorgan. Med. Chem. Lett. 2004, 14, 73–75. [Google Scholar] [CrossRef] [PubMed]
- Ernst, B.; Magnani, J. From carbohydrate leads to glycomimetic drugs. Nat. Rev. Drug. Disc. 2009, 8, 661–677. [Google Scholar] [CrossRef] [PubMed]
- Levy, D.E.; Tang, C. The Chemistry of C-Glycosides; Pergamon: Oxford, UK, 1995. [Google Scholar]
- Dondoni, A.; Mariotti, D.; Marra, A. Synthesis of α- and β-Glycosyl Asparagine Ethylene Isosteres (C-Glycosyl Asparagines) via Sugar Acetylenes and Garner Aldehyde Coupling. J. Org. Chem. 2002, 67, 4475–4486. [Google Scholar] [CrossRef] [PubMed]
- Marcaurelle, L.A.; Bertozzi, C.R. New Directions in the Synthesis of Glycopeptide Mimetics. Chem. Eur. J. 1999, 5, 1384–1390. [Google Scholar] [CrossRef]
- Schäfer, A.; Thiem, J. Synthesis of Novel Donor Mimetics of UDP-Gal, UDP-GlcNAc, and UDP-GalNAc as Potential Transferase Inhibitors. J. Org. Chem. 2000, 65, 24–29. [Google Scholar] [CrossRef]
- Wittmann, V.; Kessler, H. Stereoselective Synthesis of C-Glycoside with a Glycosyl Dianio. Angew. Chem. Int. Ed. Engl. 1993, 105, 1091–1093. [Google Scholar] [CrossRef] [Green Version]
- Westermann, B.; Walter, A.; Florke, U.; Altenbach, H.-J. Chiral Auxiliary Based Approach Toward the Synthesis of C-Glycosylated Amino Acids. Org. Lett. 2001, 3, 1375–1378. [Google Scholar] [CrossRef]
- Togo, H.; He, W.; Waki, Y.; Yokoyama, M. C-Glycosidation Technology with Free Radical Reactions. Synlett 1998, 7, 700–717. [Google Scholar] [CrossRef]
- Paterson, D.E.; Griffin, F.K.; Alcaraz, M.-L.; Taylor, R.J.K. A Ramberg−Bäcklund Approach to the Synthesis of C-Glycosides, C-Linked Disaccharides, and C-Glycosyl Amino Acids. Eur. J. Org. Chem. 2002, 7, 1323–1336. [Google Scholar] [CrossRef]
- Dondoni, A.; Marra, A. Methods for anomeric carbon-linked and fused sugar amino acid synthesis: The gateway to artificial glycopeptides. Chem. Rev. 2000, 100, 4395–4422. [Google Scholar] [CrossRef]
- Xu, X.; Fakha, G.; Sinou, D. Stereoselective synthesis of C-glycosyl analoguesphenylalanine. Tetrahedron 2002, 58, 7539–7544. [Google Scholar] [CrossRef]
- Li, X.; Takahashi, H.; Ohtake, H.; Ikegami, S. Synthesis of ketosyl spiro-isoxazolidine by 1,3-dipolar cycloaddition of 1-methylenesugars with nitrones—A new access to C-glycosyl amino acids. Heterocycles 2003, 59, 547–571. [Google Scholar] [CrossRef]
- Gustafsson, T.; Saxin, M.; Kihlberg, J. Synthesis of a C-Glycoside Analogue of β-D-Galactosylthreonine. J. Org. Chem. 2003, 68, 2506–2509. [Google Scholar] [CrossRef] [PubMed]
- Dondoni, A.; Giovannini, P.P.; Massi, A. Assembling heterocycle-tethered C-glycosyl and alpha-amino acid residues via 1,3-dipolar cycloaddition reactions. Org. Lett. 2004, 6, 2929–2932. [Google Scholar] [CrossRef] [PubMed]
- Postema, M.H.D.; Piper, J.L. Synthesis of Some Biologically Relevant β-C-Glycoconjugates. Org. Lett. 2003, 5, 1721–1723. [Google Scholar] [CrossRef] [PubMed]
- Chambers, D.J.; Evans, G.R.; Fairbanks, A.J. Synthesis of C-glycosyl amino acids: Scope and limitations of the tandem Tebbe/Claisen approach. Tetrahedron Asymmetry 2005, 16, 45–55. [Google Scholar] [CrossRef]
- Dondoni, A.; Massi, A.; Aldhoun, M. Hantzsch-Type Three-Component Approach to a New Family of Carbon-Linked Glycosyl Amino Acids. Synthesis of C-Glycosylmethyl Pyridylalanines. J. Org. Chem. 2007, 72, 7677–7687. [Google Scholar] [CrossRef]
- McGarvey, G.; Benedum, T.; Schmidtmann, F. Development of Co- and Post-Translational Synthetic Strategies to C-Neoglycopeptides. Org. Lett. 2002, 4, 3591–3594. [Google Scholar] [CrossRef]
- Müller, P.; Nury, P. Copper-Catalyzed Desymmetrization of N-Sulfonylaziridines with Methylmagnesium Halides. Org. Lett. 1999, 1, 439–442. [Google Scholar] [CrossRef]
- Hu, X.E.; Kim, N.K.; Ledoussal, B.; Colson, A.-O. Regio- and stereo-controlled copper organometallic addition to a piperidinyl aziridine: Synthesis of trans 3-amino-4-alkyl-piperidines. Tetrahedron Lett. 2002, 43, 4289–4293. [Google Scholar] [CrossRef]
- Crousse, B.; Narizuka, S.; Bonnet-Delpon, D.; Begue, J.-P. First Stereoselective Synthesis of cis 3-CF3-Aziridine-2-carboxylates. A Route to New (Trifluoromethyl) α-Functionalised β-Amino Acids. Synlett 2001, 5, 679–681. [Google Scholar] [CrossRef]
- Lugiņina, J.; Uzuleņ, J.; Posevins, D.; Turks, M. Ring-Opening of Carbamate-Protected Aziridines and Azetidines in Liquid Sulfur Dioxide. Eur. J. Org. Chem. 2016, 9, 1760–1771. [Google Scholar] [CrossRef]
- Chakraborty, T.K.; Ghosh, A.; Raju, T.V. Efficient Ring Opening Reactions of N-Tosyl Aziridines with Amines and Water in Presence of Catalytic Amount of Cerium(IV) Ammonium Nitrate. Chem. Lett. 2003, 32, 82–83. [Google Scholar] [CrossRef] [Green Version]
- O’Neil, I.A.; Woolley, J.C.; Southern, J.M.; Hobbs, H. The synthesis of β-N-tosylamino hydroxylamines via the ring opening of N-tosylaziridines and their use in reverse Cope cyclisation. Tetrahedron Lett. 2001, 42, 8243–8245. [Google Scholar] [CrossRef]
- Nishikawa, T.; Ishikawa, M.; Wade, K.; Isobe, M. Total Synthesis of α-C-Mannosyltryptophan, a Naturally Occurring C-Glycosyl Amino Acid. Synlett 2001, 945–947. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.W.; Hou, X.-L.; Dai, L.-X. Effective Ring-Opening Reaction of Aziridines with Trimethylsilyl Compounds: A Facile Access to β-Amino Acids and 1,2-Diamine Derivatives. J. Org. Chem. 2000, 65, 1344–1348. [Google Scholar] [CrossRef]
- Matsubara, S.; Kodama, T.; Utimoto, K. Yb(CN)3-catalyzed reaction of aziridines with cyanotrimethylsilane. A facile synthesis of optically pure β-amino nitriles. Tetrahedron Lett. 1990, 31, 6379–6380. [Google Scholar] [CrossRef]
- Wu, B.; Gallucci, J.C.; Parquett, J.R.; RajanBabu, T.V. Regiodivergent Ring Opening of Chiral Aziridine. Angew. Chem. Int. Ed. 2009, 48, 1126–1129. [Google Scholar] [CrossRef]
- Sugihara, Y.; Limura, S.; Nakayama, J. Aza-pinacol rearrangement: Acid-catalyzed rearrangement of aziridines to imines. Chem. Commun. 2002, 134–135. [Google Scholar] [CrossRef]
- Prasad, B.A.B.; Sekar, G.; Singh, V.K. An efficient method for the cleavage of aziridines using hydroxyl compound. Tetrahedron Lett. 2000, 41, 4677–4679. [Google Scholar] [CrossRef]
- Mao, H.; Joly, G.J.; Peeters, K.; Hoornaert, G.J.; Compernolle, F. Synthesis of 1-deoxymannojirimycin analogues using N-tosyl and N-nosyl activated aziridines derived from 1-amino-1-deoxyglucitol. Tetrahedron 2001, 57, 6955–6967. [Google Scholar] [CrossRef]
- Satoh, T.; Matsue, R.; Fujii, T.; Morikawa, S. Cross-coupling of nonstabilized aziridinylmagnesiums with alkylhalides catalyzed by Cu(I) iodide: A new synthesis of amines bearing a quaternary chiral center and an asymmetric synthesis of both enantiomers of the amines from one chiral starting material. Tetrahedron 2001, 57, 3891–3898. [Google Scholar] [CrossRef]
- Davis, F.A.; Liu, H.; Reddy, G.V. 2-Methyl N-(p-toluenesulfinyl)aziridine-2-carboxylic acid: Asymmetric synthesis of α-methylphenylalanine and α-methyl-β-phenylserine. Tetrahedron Lett. 1996, 37, 5473–5476. [Google Scholar] [CrossRef]
- Jarzyński, S.; Leśniak, S.; Rachwalski, M. Synthesis of enantiomerically pure 2-(N-aryl, N-alkyl-aminomethyl)aziridines: A new class of ligands for highly enantioselective asymmetric synthesis. Tetrahedron Asymmetry 2017, 28, 1808–1816. [Google Scholar] [CrossRef]
- Olofsson, B.; Somfai, P. Divergent Synthesis of D-erythro-Sphingosine, L-threo-Sphingosine, and Their Regioisomers. J. Org. Chem. 2003, 68, 2514–2517. [Google Scholar] [CrossRef]
- Fukuta, Y.; Mita, T.; Fukuda, N.; Kanai, M.; Shibasaki, M. De Novo Synthesis of Tamiflu via a Catalytic Asymmetric Ring-Opening of meso-Aziridines with TMSN3. J. Am. Chem. Soc. 2006, 128, 6312–6313. [Google Scholar] [CrossRef]
- Frappa, I.; Kryczka, B.; Lhoste, P.; Porwański, S.; Sinou, D.; Zawisza, A. Palladium(0)-Mediated Synthesis of Acetylated Unsaturated 1,4-Disaccharides. J. Carbohydr. Chem. 1998, 17, 1117–1130. [Google Scholar] [CrossRef]
- Zawisza, A.; Kryczka, B.; Lhoste, P.; Porwański, S.; Sinou, D. Efficient Palladium(0)-Catalyzed Synthesis of Alkenyl 1-Thioglycosides and Thiodisaccharides. J. Carbohydr. Chem. 2000, 19, 795–804. [Google Scholar] [CrossRef]
- Jarosz, S.; Szewczyk, K.; Zawisza, A. Synthesis and thermal stability of secondary sugar allyltin derivatives. Tetrahedron: Asymmetry 2003, 14, 1715–1723. [Google Scholar] [CrossRef]
- Szulc, I.; Kołodziuk, R.; Kryczka, B.; Zawisza, A. New phosphine–imine ligands derived from D-gluco- and D-galactosamine in Pd-catalysed asymmetric allylic alkylation. Tetrahedron Lett. 2015, 56, 4740–4743. [Google Scholar] [CrossRef]
- Kubiak, A.; Kołodziuk, R.; Porwański, S.; Zawisza, A. Palladium(0)-catalysed synthesis of 2,3- and 3,4-unsaturated aryl β-O-glycosides. Carbohydr. Res. 2015, 417, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Janasik, B.; Zawisza, A.; Malachowska, B.; Fendler, W.; Stanislawska, M.; Kuras, R.; Wąsowicz, W. Relationship between arsenic and selenium in workers occupationally exposed to inorganic arsenic. J. Trace Elem. Med. Biol. 2017, 42, 76–80. [Google Scholar] [CrossRef] [PubMed]
- Szulc, I.; Kołodziuk, R.; Zawisza, A. New phosphine-imine and phosphine-amine ligands derived from D-gluco-, D-galacto- and D-allosamine in Pd-catalysed asymmetric allylic alkylation. Tetrahedron 2018, 74, 1476–1485. [Google Scholar] [CrossRef]
- Robak, J.; Koselak, K.; Zawisza, A.; Porwański, S. Studies on the influence of saccharide fragment of urea organocatalysts on the yield and enantioselectivity of aza-Henry reaction. Arkivoc 2020, 8, 150–160. [Google Scholar] [CrossRef]
- Leśniak, S.; Rachwalski, M.; Sznajder, E.; Kiełbasiński, P. New Highly Efficient Aziridine-functionalized Tridentate Sulfinyl Catalysts for Enantioselective Diethylzinc Addition to Carbonyl Compounds. Tetrahedron Asymmetry 2009, 20, 2311–2314. [Google Scholar] [CrossRef]
- Rachwalski, M.; Jarzyński, S.; Leśniak, S. Aziridine Ring-containing Chiral Ligands as Highly Efficient Catalysts in Asymmetric Synthesis. Tetrahedron Asymmetry 2013, 24, 421–425. [Google Scholar] [CrossRef]
- Rachwalski, M.; Jarzyński, S.; Jasiński, M.; Leśniak, S. Mandelic Acid Derived α-Aziridinyl Alcohols as Highly Efficient Ligands for Asymmetric Additions of Zinc Organyls to Aldehydes. Tetrahedron Asymmetry 2013, 24, 689–693. [Google Scholar] [CrossRef]
- Buchcic, A.; Zawisza, A.; Leśniak, S.; Adamczyk, J.; Pieczonka, A.M.; Rachwalski, M. Enantioselective Mannich Reaction Promoted by Chiral Phos-phinoyl-Aziridines. Catalysts 2019, 9, 837. [Google Scholar] [CrossRef] [Green Version]
- Wujkowska, Z.; Zawisza, A.; Leśniak, S.; Rachwalski, M. Phosphinoyl-aziridines as a New Class of Chiral Catalysts for Enantioselective Michael Addition. Tetrahedron 2019, 75, 230–235. [Google Scholar] [CrossRef]
- Buchcic, A.; Zawisza, A.; Leśniak, S.; Rachwalski, M. Asymmetric Friedel–Crafts Alkylation of Indoles Catalyzed by Chiral Aziridine-Phosphines. Catalysts 2020, 10, 971–980. [Google Scholar] [CrossRef]
- Buchcic-Szychowska, A.; Adamczyk, J.; Marciniak, L.; Pieczonka, A.M.; Zawisza, A.; Leśniak, S.; Rachwalski, M. Efficient Asymmetric Simmons-Smith Cyclopropanation and Diethylzinc Addition to Aldehydes Promoted by Enantiomeric Aziridine-Phosphines. Catalysts 2021, 11, 968–978. [Google Scholar] [CrossRef]
- May, S.W.; Phillips, R.S. Asymmetric sulfoxidation by dopamine beta-hydroxylase, an oxygenase heretofore considered specific for methylene hydroxylation. J. Am. Chem. Soc. 1980, 102, 5981–5983. [Google Scholar] [CrossRef]
- May, S.W.; Phillips, R.S.; Mueller, P.W.; Herman, H.H. Dopamine beta-hydroxylase. Comperative specificities and mechanisms of the oxygenation reaction. J. Biol. Chem. 1981, 256, 8470–8475. [Google Scholar] [CrossRef]
- Padgette, S.R.; Herman, H.H.; Han, J.H.; Pollock, S.H.; May, S.W. Antihypertensive Activities of Phenyl Aminoethyl Sulfides, a Class of Synthetic Substrates for Dopamine β-Hydroxylase. J. Med. Chem. 1984, 27, 1354–1357. [Google Scholar] [CrossRef] [PubMed]
- Herman, H.H.; Husain, P.A.; Colbert, J.E.; Schweri, M.M.; Pollock, S.H.; Fowler, L.C.; May, S.W. The Enantiomeric Specificity of the Antihypertensive Activity of l-(Phenylthio)-2-aminopropane, a Synthetic Substrate Analogue for Dopamine β-Monooxygenase. J. Med. Chem. 1991, 34, 1082–1085. [Google Scholar] [CrossRef]
- Kandalkar, S.R.; Ramaiah, P.A.; Joshi, M.; Wavhal, A.; Waman, Y.; Raje, A.A.; Tambe, A.; Ansari, S.; De, S.; Palle, V.P.; et al. Modifications of flexible nonyl chain and nucleobase head group of (+)-erythro-9-(2′s-hydroxy-3′s-nonyl)adenine [(+)-EHNA] as adenosine deaminase inhibitors. Bioorgan. Med. Chem. 2017, 25, 5799–5819. [Google Scholar] [CrossRef] [PubMed]
- Tucker, H.; Coope, J.F. β-Adrenergic blocking agents. 18. 1-(Aryloxy)-3-(arylthioalkylamino)propan-2-ols and 1-substituted alkylthioamino-3-(aryloxy)propan-2-ols. J. Med. Chem. 1978, 21, 769–773. [Google Scholar] [CrossRef]
- Erdmann, A.; Menon, Y.; Gros, C.; Molinier, N.; Novosad, N.; Samson, A.; Gregoire, J.-M.; Long, C.; Ausseil, F.; Halby, L.; et al. Design and synthesis of new non nucleoside inhibitors of DNMT3A. Bioorgan. Med. Chem. 2015, 23, 5946–5953. [Google Scholar] [CrossRef]
- Bartlett, M.J.; Turner, C.A.; Harvey, J.E. Pd-Catalyzed Allylic Alkylation Cascade with Dihydropyrans: Regioselective Synthesis of Furo[3,2-c]pyrans. Org. Lett. 2013, 15, 2430–2433. [Google Scholar] [CrossRef]
- Bearder, J.R.; Dewis, M.L.; Whiting, D.A. Short synthetic route to congeners of the undecose antibiotic herbicidin. J. Chem. Soc. Perkin Trans. 1995, 227–233. [Google Scholar] [CrossRef]
- Zhang, S.; Niu, Y.-H.; Ye, X.-S. General Approach to Five-Membered Nitrogen Heteroaryl C-Glycosides Using a Palladium/Copper Cocatalyzed C–H Functionalization Strategy. Org. Lett. 2017, 19, 3608–3611. [Google Scholar] [CrossRef] [PubMed]
- Hnnesian, S.; Martin, M.; Desai, R. Formation of C-Glycosides by Polarity Inversion at the Anomeric Centre. J. Chem. Soc. Chem. Commun. 1986, 926–927. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Hwang, C.-K.; Duggan, M.E. Stereospecific Synthesis of 1,1-Dialkylglycosides. J. Chem. Soc. Chem. Commun. 1986, 12, 925–926. [Google Scholar] [CrossRef]
- Lesimple, P.; Beau, J.-M.; Jaurand, G.; Sinaÿ, P. Preparation and use of lithiated glycals: Vinylic deprotonation versus tin-lithium exchange from 1-tributylstannyl glycals. Tetrahedron Lett. 1986, 27, 6201–6204. [Google Scholar] [CrossRef]
- Crich, D.; Ritchie, T.J. Preparation and reactions of some cyclic orthoester derivatives. Tetrahedron 1988, 44, 2319–2328. [Google Scholar] [CrossRef]
- Friesen, R.W.; Sturino, C.F.; Daljeet, A.K.; Kolaczewska, A. Observation of .alpha.-silyl carbanions in the metalation of 3,4,6-tri-O-(tert-butyldimethylsilyl)-D-glucal. J. Org. Chem. 1991, 56, 1944–1947. [Google Scholar] [CrossRef]
- Utsunomiya, I.; Fuji, M.; Sato, T.; Natsume, M. Preparation of Alkyl-Substituted Indoles in the Benzene Portion. Part 9. Synthesis of (1aS,8bS)-1-tert-Butyloxycarbonyl-8-formyl-1,1a,2,8b-tetrahydroazirino[2’,3’:3,4]pyrrolo[1,2-a]indole. Model Study for the Enantiospecific Synthesis of Aziridinomitosenes. Chem. Pharm. Bull. 1993, 41, 854–860. [Google Scholar] [CrossRef] [Green Version]
- Kulshrestha, A.; Schomaker, J.M.; Holmes, D.; Staples, R.J.; Jackson, J.E.; Borhan, B. Selectivity in the Addition Reactions of Organometallic Reagents to Aziridine-2-carboxaldehydes: The Effects of Protecting Groups and Substitution Patterns. Chem. Eur. J. 2011, 17, 12326–12339. [Google Scholar] [CrossRef]
- Hwang, G.-I.; Chung, J.-H.; Lee, W.K. Efficient Synthesis of Ephedra Alkaloid Analogues Using an Enantiomerically Pure N-[(R)-(+)-α-Methylbenzyl]aziridine-2-carboxaldehyde. J. Org. Chem. 1996, 61, 6183–6188. [Google Scholar] [CrossRef]
- Goument, B.; Duhamel, L.; Maugé, R. Synthesis of (S)-fenfluramine from (R) or (S) 1-[3-(trifluoromethyl)phenyl]propan-2-ol. Bull. Soc. Chim. Fr. 1993, 130, 450–458. [Google Scholar]
- Van der Zeijden, A.A.H. A novel chiral cyclopentadienyl ligand based on ephedrine. Tetrahedron Asymmetry 1995, 6, 913–918. [Google Scholar] [CrossRef]
- Hyne, J.B. Preferred residence conformations of diastereoisomeric α-β amino alcohols: An, N.M.R. study of the ephedrines. Can. J. Chem. 1961, 39, 2536–2542. [Google Scholar] [CrossRef] [Green Version]
- Cheng, J.C.Y.; Hacksell, U.; Daves, G.D., Jr. Differentially Protected Ribofuranoid Glycals. J. Org. Chem. 1985, 50, 2778–2780. [Google Scholar] [CrossRef]
- Bae, J.H.; Shin, S.-H.; Park, C.S.; Lee, W.K. Preparation of cysteinol derivatives by highly regioselective ring opening of nonactivated chiral aziridines by thiols. Tetrahedron 1999, 55, 10041–10046. [Google Scholar] [CrossRef]
- Powers, J.C.; Asgian, J.L.; Ekici, Ö.D.; James, K.E. Irreversible inhibitors of serine, cysteine, and threonine proteases. Chem. Rev. 2002, 102, 4639–4750. [Google Scholar] [CrossRef] [PubMed]
- González, M.; Gándara, Z.; Pazos, G.; Gómez, G.; Fall, Y. Synthesis of (–)-Muricatacin from Tri-O-acetyl-D-glucal. Synthesis 2013, 45, 625–632. [Google Scholar] [CrossRef]
- Di Bussolo, V.; Caselli, M.; Pineschi, M.; Crotti, P. New Stereoselective β-Glycosylation via a Vinyl Oxirane Derived from D-Glucal. Org. Lett. 2002, 4, 3695–3698. [Google Scholar] [CrossRef]
- Kozikowski, A.P.; Lee, J. A synthetic approach to the cis-fused marine pyranopyrans,(3E)-and (3Z)-dactomelyne. X-ray structure of a rare organomercurial. J. Org. Chem. 1990, 55, 863–870. [Google Scholar] [CrossRef]
- Moore, P.W.; Schuster, J.K.; Stone, R.J.M.; Rhia, L.; Teesdale-Spittle, P.H.; Harvey, J.E. Divergent synthesis of 2-C-branched pyranosides and oxepines from 1, 2-gem-dibromocyclopropyl carbohydrates. Tetrahedron 2014, 70, 7032–7043. [Google Scholar] [CrossRef]
- Steunenberg, P.; Jeanneret, V.; Zhu, Y.-H.; Vogel, P. C (1→4)-linked disaccharides through carbonylative Stille cross-coupling. Tetrahedron Asymmetry 2005, 16, 337–346. [Google Scholar] [CrossRef]
- Linker, T.; Schanzenbach, D.; Elamparuthi, E. Remarkable Oxidation Stability of Glycals: Excellent Substrates for Cerium(IV)-Mediated Radical Reactions. J. Am. Chem. Soc. 2008, 130, 16003–16010. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
| Entry | Glycal | Procedure | Yield (%) 1 | erythro:threo2 |
|---|---|---|---|---|
| 1 | 6 | A | 40 | 1:3 |
| 2 | 6 | B | 55 | 4:5 |
| 3 | 7 | A | 45 | 1:9 |
| 4 | 7 | B | 65 | 1:9 |
| Compound | CHOH δ (ppm) | CHN-CHOH J (Hz) | Compound | CHOH δ (ppm) |
|---|---|---|---|---|
| erythro (S,S)-9 | 4.40 | 2.2 | (S,S)-13 | 4.30 |
| threo (R,S)-10 | 3.92 | 5.8 | (R,S)-14 | 3.94 |
| erythro (S,S)-11 | 4.33 | 2.4 | (S,S)-15 | 4.28 |
| threo (R,S)-12 | 3.91 | 5.8 | (R,S)-16 | 4.04 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tracz, A.; Malinowska, M.; Leśniak, S.; Zawisza, A. Aziridine Ring Opening as Regio- and Stereoselective Access to C-Glycosyl-Aminoethyl Sulfide Derivatives. Molecules 2022, 27, 1764. https://doi.org/10.3390/molecules27061764
Tracz A, Malinowska M, Leśniak S, Zawisza A. Aziridine Ring Opening as Regio- and Stereoselective Access to C-Glycosyl-Aminoethyl Sulfide Derivatives. Molecules. 2022; 27(6):1764. https://doi.org/10.3390/molecules27061764
Chicago/Turabian StyleTracz, Aleksandra, Martyna Malinowska, Stanisław Leśniak, and Anna Zawisza. 2022. "Aziridine Ring Opening as Regio- and Stereoselective Access to C-Glycosyl-Aminoethyl Sulfide Derivatives" Molecules 27, no. 6: 1764. https://doi.org/10.3390/molecules27061764