
Synthesis and Reactivity of Martin’s Spirosilane-Derived Chloromethylsilicate


Abstract
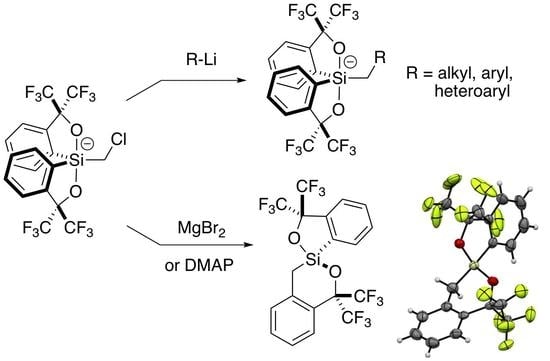
:
1. Introduction
2. Results and Discussion
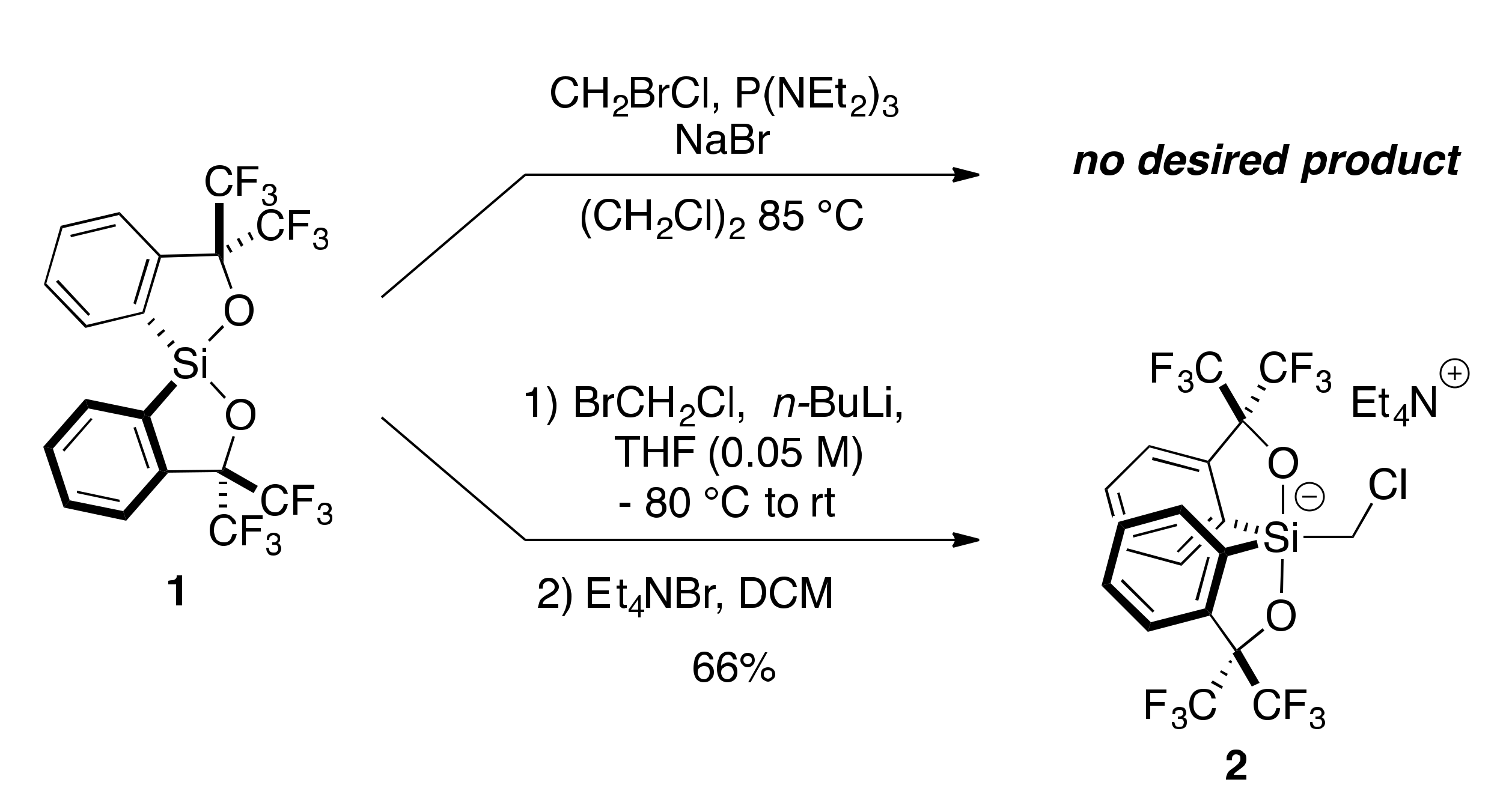
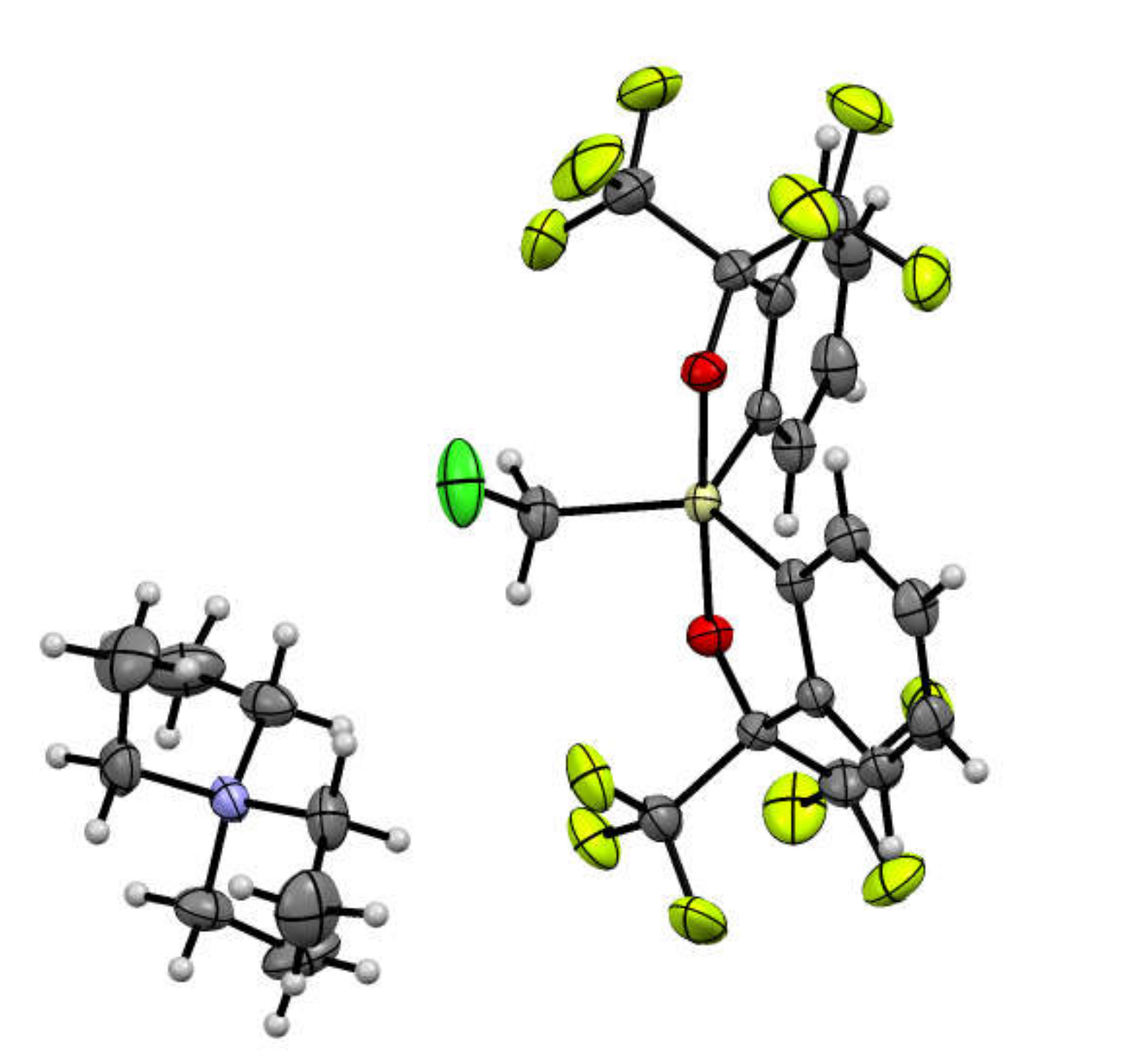
2.1. Synthesis of Chloromethylsilicate 2
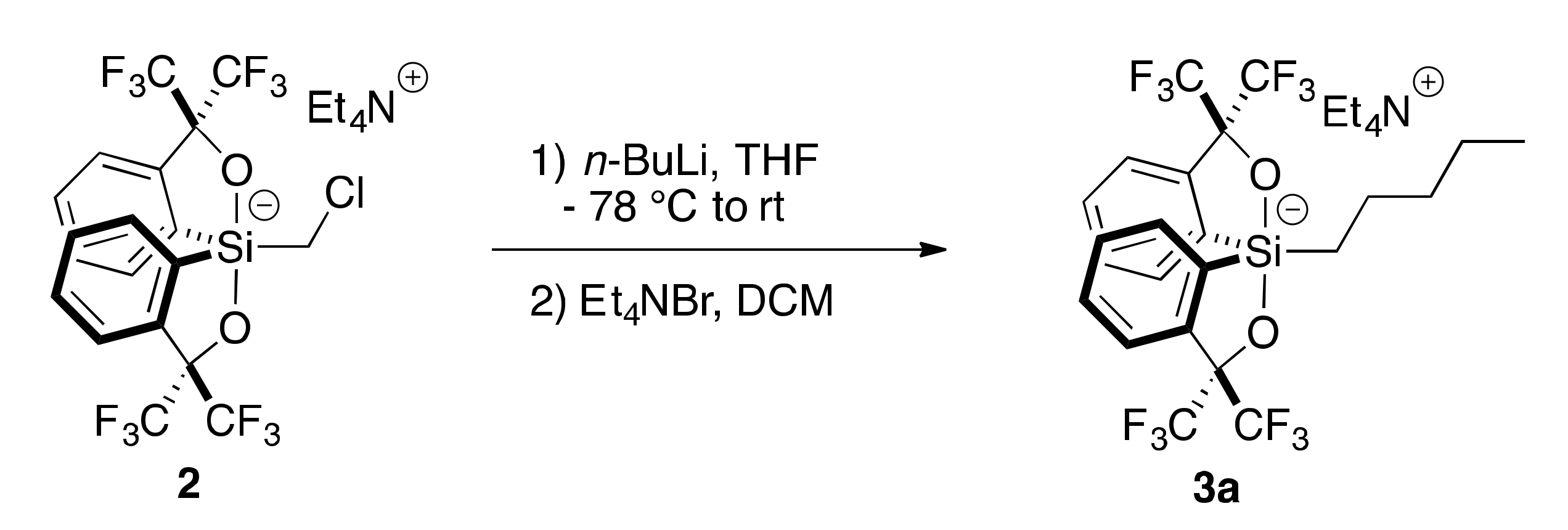
2.2. Addition of Organolithiums
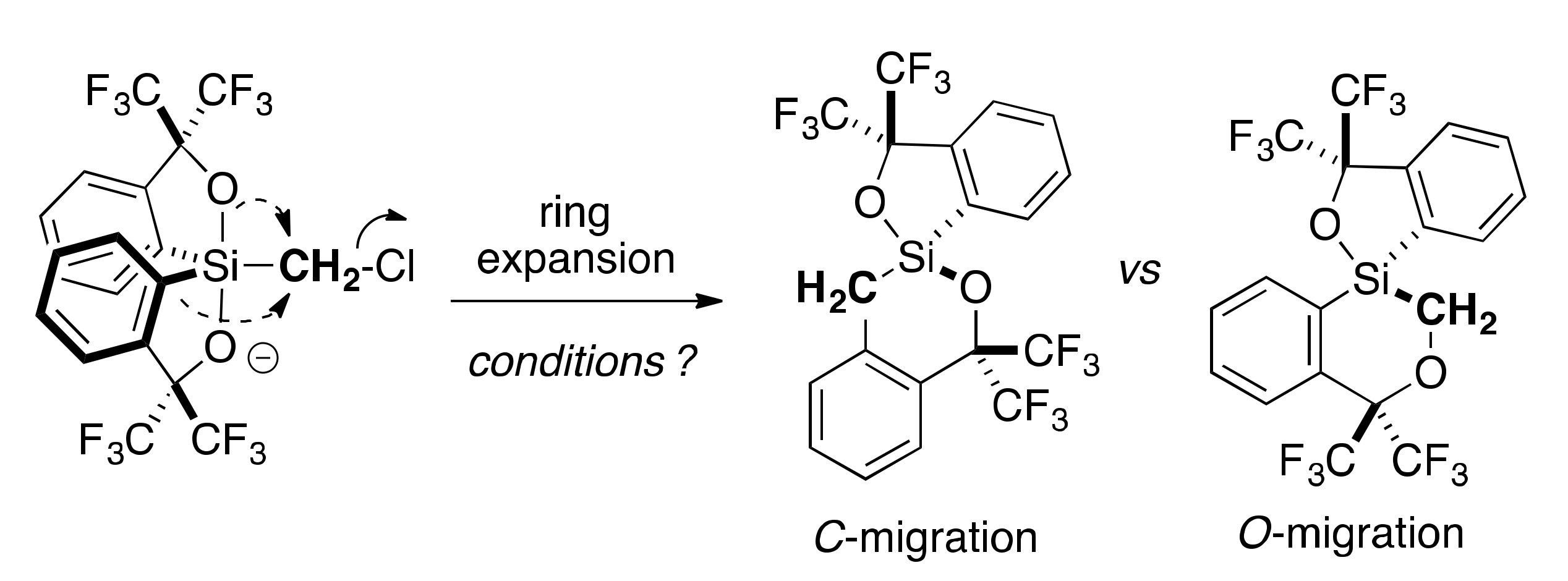
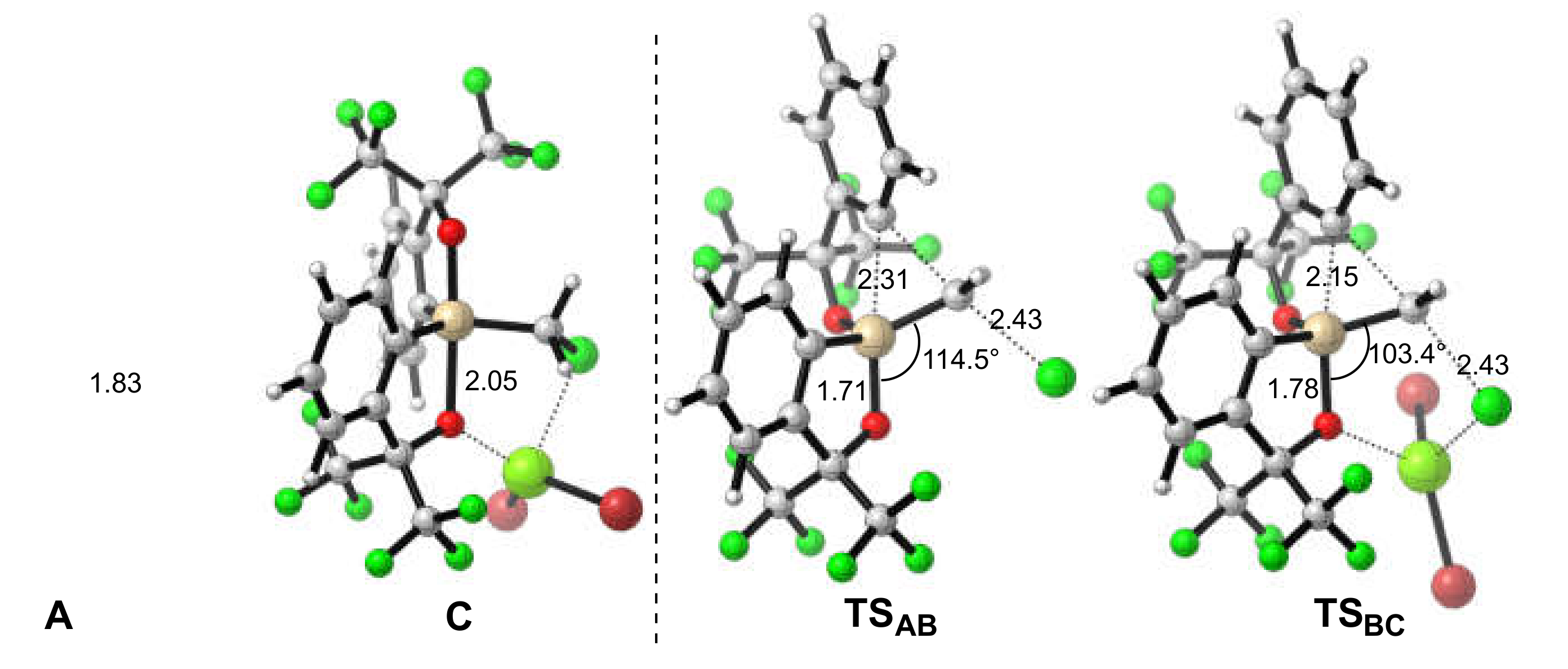
2.3. Lewis Acid-Assisted Ring Expansion
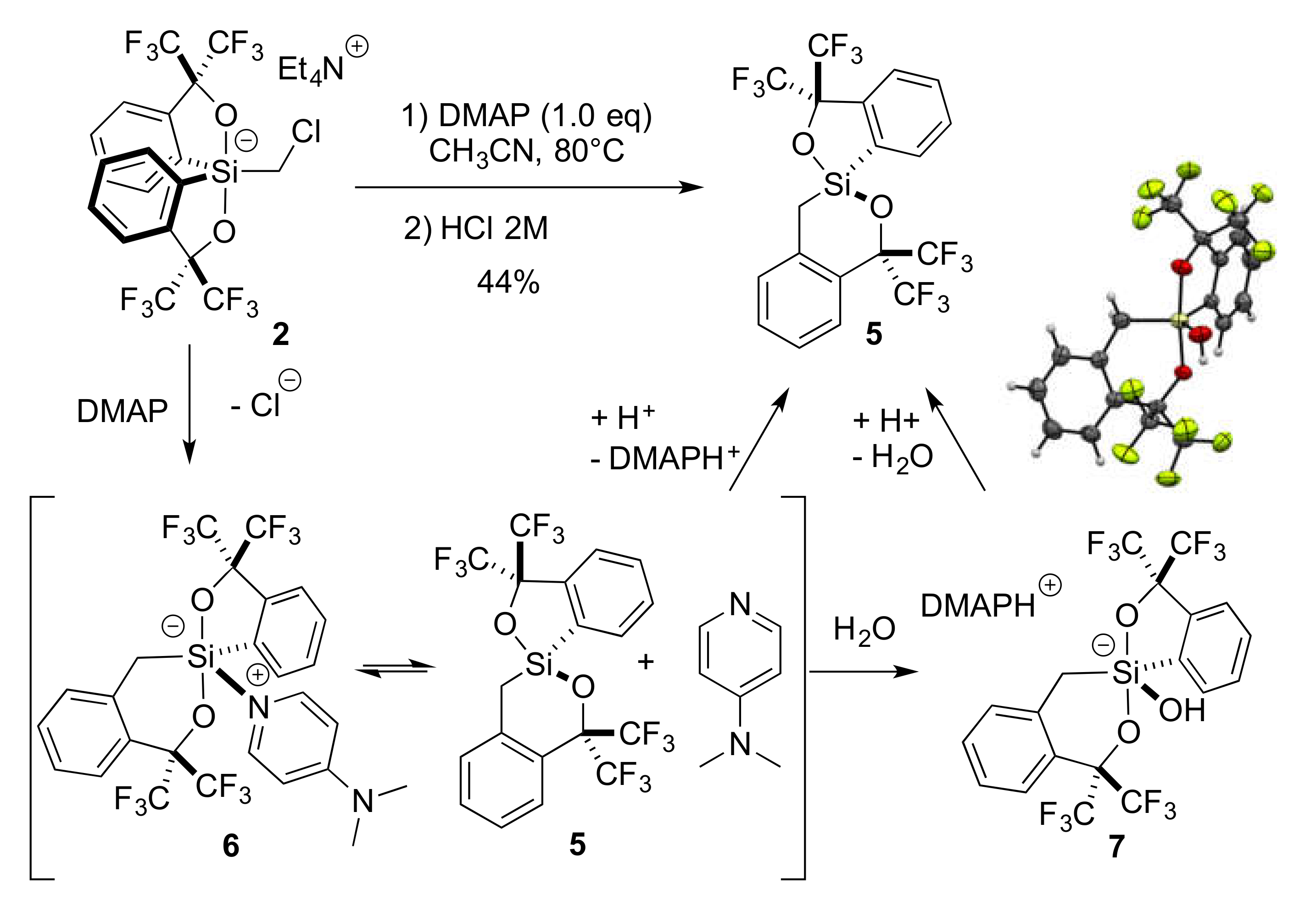
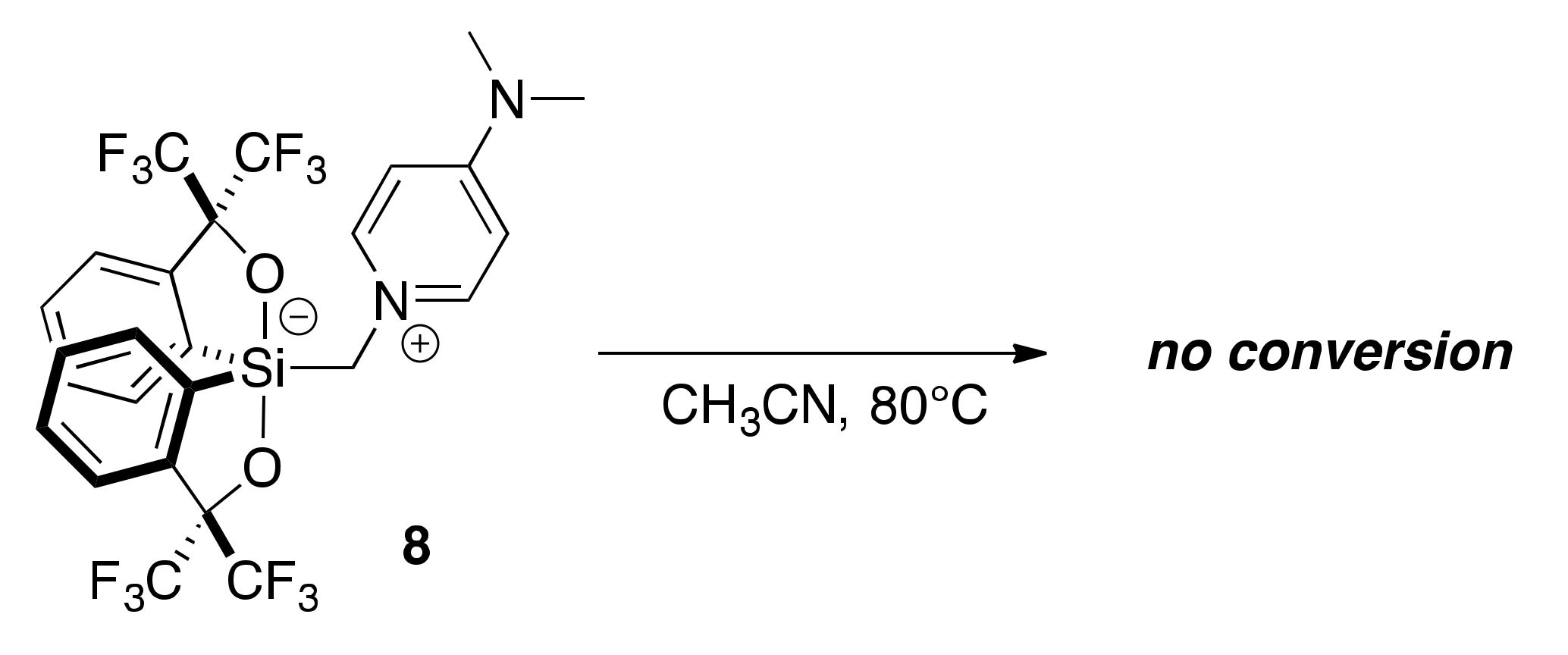
2.4. Lewis Base-Assisted Ring Expansion
3. Experimental
3.1. General Information
3.2. Synthesis and Characterization
3.2.1. Synthesis of Chloromethylsilicate 2
3.2.2. Addition of Organolithium
3.2.3. Synthesis of Spirosilane 5
3.2.4. Obtaining Crystals of Hydroxysilicate 7
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Brook, M.A. Silicon in Organic, Organometallic and Polymer Chemistry; Wiley: New York, NY, USA, 2000. [Google Scholar]
- Tandura, S.N.; Voronkov, M.G.; Alekseev, N.V. Molecular and electronic structure of penta- and hexacoordinate silicon compounds. Top. Curr. Chem. 1986, 131, 99–189. [Google Scholar]
- Lee, V.Y. (Ed.) Kano in Organosilicon Compounds Ch.11 Penta- and Hexacoordinated Silicon(IV) Compounds; Academic Press: Cambridge, MA, USA; Elsevier: London, UK, 2017. [Google Scholar]
- Corcé, V.; Chamoreau, L.; Derat, E.; Goddard, J.; Ollivier, C.; Fensterbank, L. Silicates as Latent Alkyl Radical Precursors: Visible-Light Photocatalytic Oxidation of Hypervalent Bis-Catecholato Silicon Compounds. Angew. Chem. Int. Ed. 2015, 54, 11414–11418. [Google Scholar] [CrossRef] [PubMed]
- Phelan, J.P.; Lang, S.B.; Compton, J.S.; Kelly, C.B.; Dykstra, R.; Gutierrez, O.; Molander, G.A. Redox-Neutral Photocatalytic Cyclopropanation via Radical/Pola Crossover. J. Am. Chem. Soc. 2018, 140, 8037–8047. [Google Scholar] [CrossRef] [PubMed]
- Guo, T.; Zhang, L.; Liu, X.; Fang, Y.; Jin, X.; Yang, Y.; Li, Y.; Chen, B.; Ouyang, M. Visible-Light-Promoted Redox-Neutral Cyclopropanation Reactions of a-Substituted Vinylphosphonates and Other Michael Acceptors with Chloromethyl Silicate as Methylene Transfer. Adv. Synth. Catal. 2018, 360, 4459–4463. [Google Scholar] [CrossRef]
- Luo, W.; Yang, Y.; Fang, Y.; Zhang, X.; Jin, X.; Zhao, G.; Zhang, L.; Li, Y.; Zhou, W.; Xia, T.; et al. Photoredox-Catalyzed Cyclopropanation of 1,1-Disubstituted Alkenes via Radical-Polar Crossover Process. Adv. Synth. Catal. 2019, 361, 4215–4221. [Google Scholar] [CrossRef]
- Pestunovich, V.A.; Kirpichenko, S.V.; Voronkov, M.G.; Rappoport, Z. Chemistry of Organic Silicon Compounds; Rappoport, Z., Apeloig, Y., Eds.; Wiley & Sons, Inc.: New York, NY, USA, 1998; p. 1447. [Google Scholar]
- Soldatenko, A.S.; Sterkhova, I.V.; Lazareva, N.F. Pentacoordinate silicon compounds based on 2,2′-dihydroxyazobenzene ligand. J. Organomet. Chem. 2019, 903, 120997. [Google Scholar] [CrossRef]
- Sivaramakrishna, A.; Kalikhman, I.; Kertsnus, E.; Korlyukov, A.A.; Kost, D. Donor-Stabilized Silyl Cations. 10. Pentacoordinate Siliconium-Ion Salts with a Triphenylphosphinimino-N Ligand Group: Two-Bond P-N-Si Coupling as a Measure for Coordination Strength. Organometallics 2006, 25, 3665–3669. [Google Scholar] [CrossRef]
- Lazareva, N.F.; Lazarev, I.M. A new method for the preparation of 1-(chloromethyl)- and 1-(dichloromethyl)silatranes. Russ. Chem. Bull. 2018, 67, 1742–1743. [Google Scholar] [CrossRef]
- Perozzi, E.F.; Martin, J.C. Facile syntheses of isolable organic derivatives of hypervalent sulfur, phosphorus, and silicon. Introduction of a stabilizing bidentate ligand via its dilithio derivative. J. Am. Chem. Soc. 1979, 101, 1591–1593. [Google Scholar] [CrossRef]
- Perozzi, E.F.; Michalak, R.S.; Figuly, G.D.; Stevenson, W.H.; Dess, D.; Ross, M.R.; Martin, J.C. Directed Dilithiation of Hexafluorocumyl Alcohol-Formation of a Reagent for the Facile Introduction of a Stabilizing Bidentate Ligand in Compounds of Hypervalent Sulfur (10-S-4), Phosphorus (10-P-5), Silicon (lO-Si-5), and Iodine (10-1-3). J. Org. Chem. 1981, 46, 1049–1053. [Google Scholar] [CrossRef]
- Lemière, G.; Millanvois, A.; Ollivier, C.; Fensterbank, L. A Parisian Vision of the Chemistry of Hypercoordianted Silicon Derivatives. Chem. Rec. 2021, 21, 1119–1129. [Google Scholar] [CrossRef] [PubMed]
- Medici, F.; Gontard, G.; Derat, E.; Lemière, G.; Fensterbank, L. Synthesis of Stable Pentacoordinate Silicon(IV)−NHC Adducts: An Entry to Anionic N-Heterocyclic Carbene Ligands. Organometallics 2018, 37, 517–520. [Google Scholar] [CrossRef]
- Medici, F.; Maury, J.; Lemière, G.; Fensterbank, L. Interaction between Spirosilanes and Lewis Bases: From Coordination to Frustration. Chem. Eur. J. 2019, 25, 9438–9442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deis, T.; Medici, F.; Poussard-Schulz, A.; Lemière, G.; Fensterbank, L. Synthesis and reactivity of an anionic NHC-borane featuring a weakly coordinating silicate anion. J. Organomet. Chem. 2019, 956, 122120. [Google Scholar] [CrossRef]
- Iii, W.H.S.; Wilson, S.; Martin, J.C.; Farnham, W.B. Pseudorotational mechanism for the inversion of 10-Si-5 siliconates: Ligand Structure and Reactivity. J. Am. Chem. Soc. 1985, 107, 6340–6352. [Google Scholar]
- Ikarashi, G.; Morofuji, T.; Kano, N. Terminal-oxidant-free photocatalytic C–Halkylations of heteroarenes with alkylsilicates asalkyl radical precursors. Chem. Commun. 2020, 56, 10006–10009. [Google Scholar] [CrossRef]
- Kobayashi, T.; Pannell, K.H. Synthesis of (Chloromethy1)silanes by the Low-Temperature Reaction of Chlorosilanes and in Situ Generated (Chloromethy1)lithium in Tetrahydrofuran. Organometallics 1991, 10, 1960–1964. [Google Scholar] [CrossRef]
- Shiragami, H.; Kawamoto, T.; Imi, K.; Matsubara, S.; Utimoto, K.; Nozaki, H. Lithium Carbenoids Induced Ring Enlargement of Silacyclobutane into 2-Halo-1-silacyclopentane and its Use in Organic Synthesis. Tetrahedron 1988, 44, 4009–4022. [Google Scholar] [CrossRef]
- François, C.; Boddaert, T.; Durandetti, M.; Querolle, O.; Van Hijfte, L.; Meerpoel, L.; Angibaud, P.; Maddaluno, J. Intramolecular Sila-Matteson Rearrangement: A General Access to Silylated Heterocycles. Org. Lett. 2012, 14, 2074–2077. [Google Scholar] [CrossRef]
- Boddaert, T.; François, C.; Mistico, L.; Querolle, O.; Meerpoel, L.; Angibaud, P.; Durandetti, M.; Maddaluno, J. Anionic Access to Silylated and Germylated Binuclear Heterocycles. Chem. Eur. J. 2014, 20, 10131–10139. [Google Scholar] [CrossRef]
- Karlov, S.S.; Selina, A.A.; Chernyshova, E.S.; Oprunenko, Y.F.; Merkulov, A.A.; Tafeenko, V.A.; Churakov, A.V.; Howard, J.A.; Zaitseva, G.S. Synthesis and characterization of metallatranes with phenyl substituents in atrane cage. Inorg. Chim. Acta 2007, 360, 563–578. [Google Scholar] [CrossRef]
- Kalikhman, I.; Girshberg, O.; Lameyer, L.; Stalke, A.D.; Kost, D. Irreversible Rearrangement in Hexacoordinate Silicon Complexes: From Neutral Bis(N→Si) Chelates to Mono(N→Si) Zwitterionic λ6-Silicates. Organometallics 2000, 19, 1927–1934. [Google Scholar] [CrossRef]
- Farnham, W.B.; Whitney, J.F. Stereomutation at hexacoordinate silicon by a ligand-dissociation process. J. Am. Chem. Soc. 1984, 106, 3992–3994. [Google Scholar] [CrossRef]
- Deis, T.; Maury, J.; Medici, F.; Jean, M.; Forte, J.; Vanthuyne, N.; Fensterbank, L.; Lemière, G. Synthesis and Optical Resolution of Configurationally Stable Zwitterionic Pentacoordinate Silicon Derivatives. Angew. Chem. Int. Ed. 2022, 134, e202113836. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
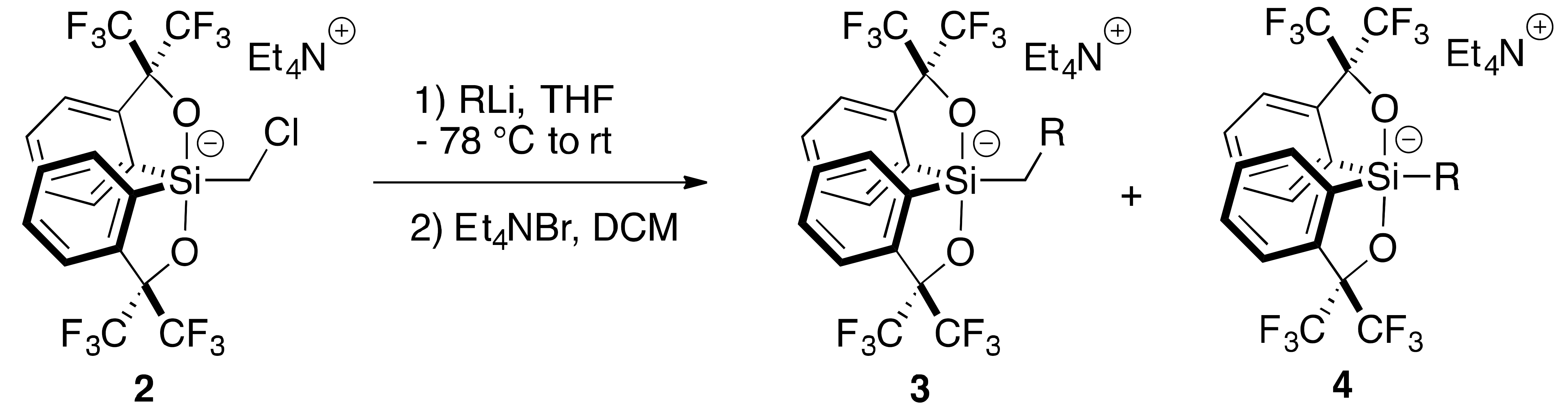 | |||
|---|---|---|---|
| Entry | RLi | Product (Ratio 3/4) | Overall Yield 3 + 4 |
| 1 |  | 3b | 74% |
| 2 | 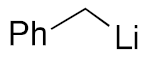 | 3c | 73% |
| 3 | 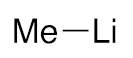 | 3d/4d (55/45) | 36% |
| 4 | 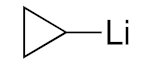 | 3e/4e (83/17) | 81% |
| 5 | 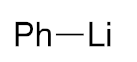 | 3f | 61% |
| 6 | 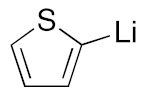 | 3g | 69% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Deis, T.; Forte, J.; Fensterbank, L.; Lemière, G. Synthesis and Reactivity of Martin’s Spirosilane-Derived Chloromethylsilicate. Molecules 2022, 27, 1767. https://doi.org/10.3390/molecules27061767
Deis T, Forte J, Fensterbank L, Lemière G. Synthesis and Reactivity of Martin’s Spirosilane-Derived Chloromethylsilicate. Molecules. 2022; 27(6):1767. https://doi.org/10.3390/molecules27061767
Chicago/Turabian StyleDeis, Thomas, Jérémy Forte, Louis Fensterbank, and Gilles Lemière. 2022. "Synthesis and Reactivity of Martin’s Spirosilane-Derived Chloromethylsilicate" Molecules 27, no. 6: 1767. https://doi.org/10.3390/molecules27061767