
Synthesis and Structural Characterization of Pyridine-2,6-dicarboxamide and Furan-2,5-dicarboxamide Derivatives


 and
and 
Abstract
:1. Introduction
2. Results and Discussion
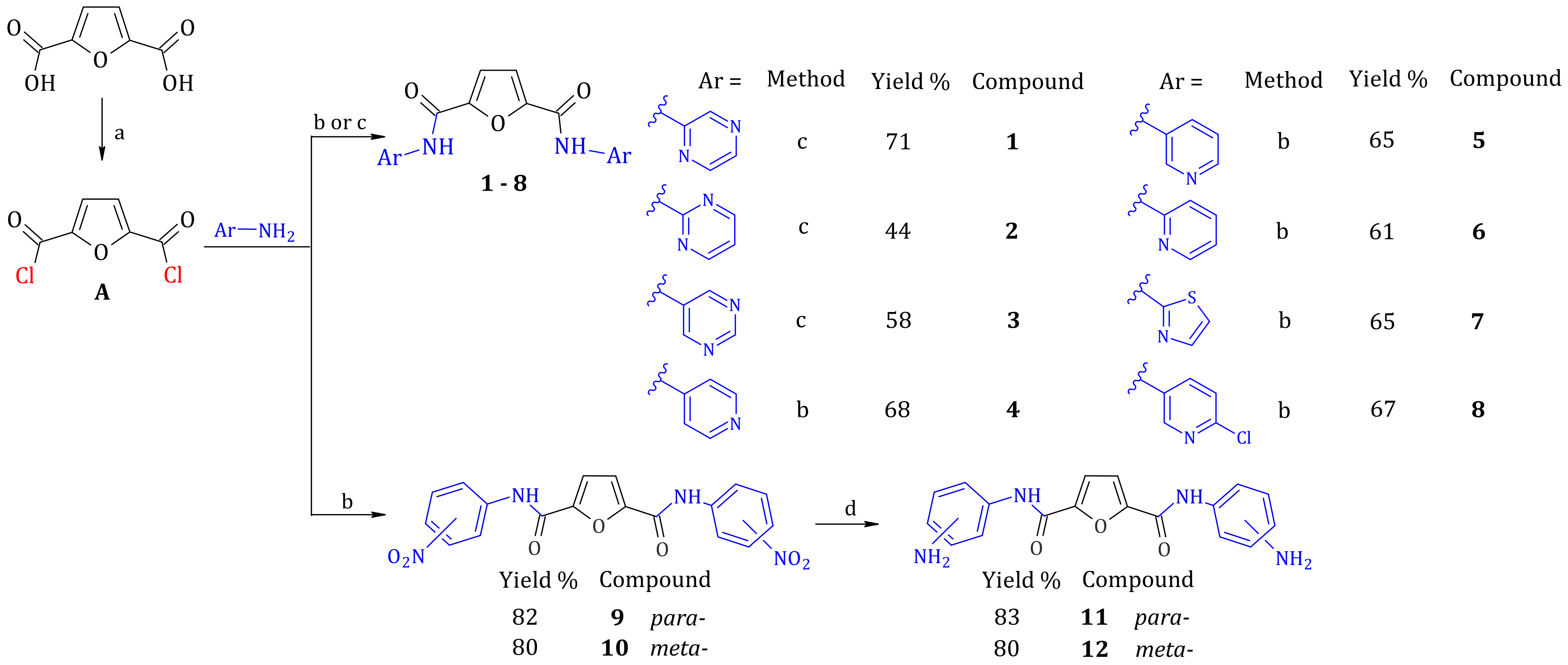
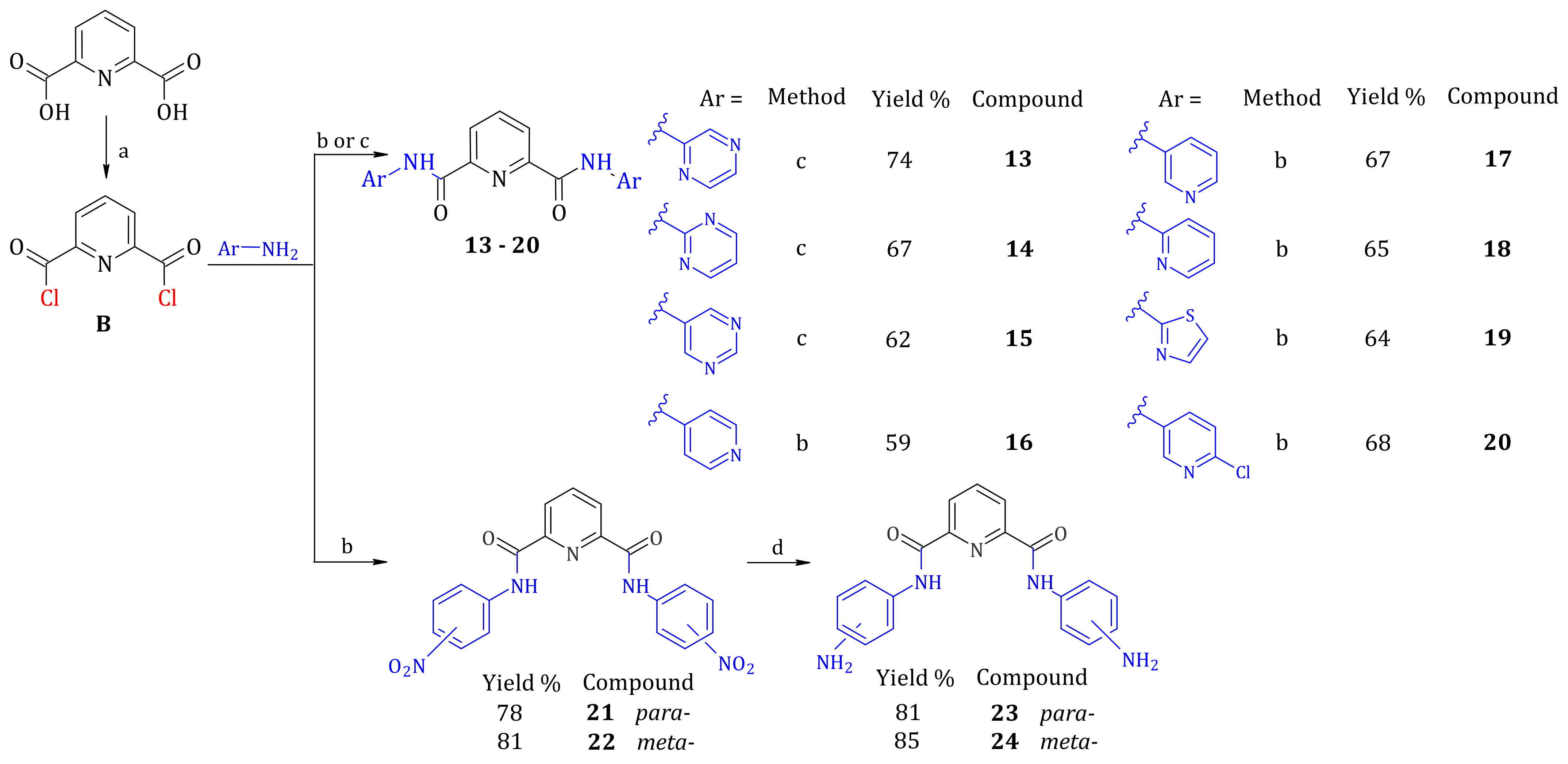
2.1. Synthesis of Furan-2,5-dicarboxamides and Pyridine-2,6-dicarboxamides
2.2. Crystallographic Studies of Furan-2,5-dicarboxamides and Pyridine-2,6-dicarboxamides
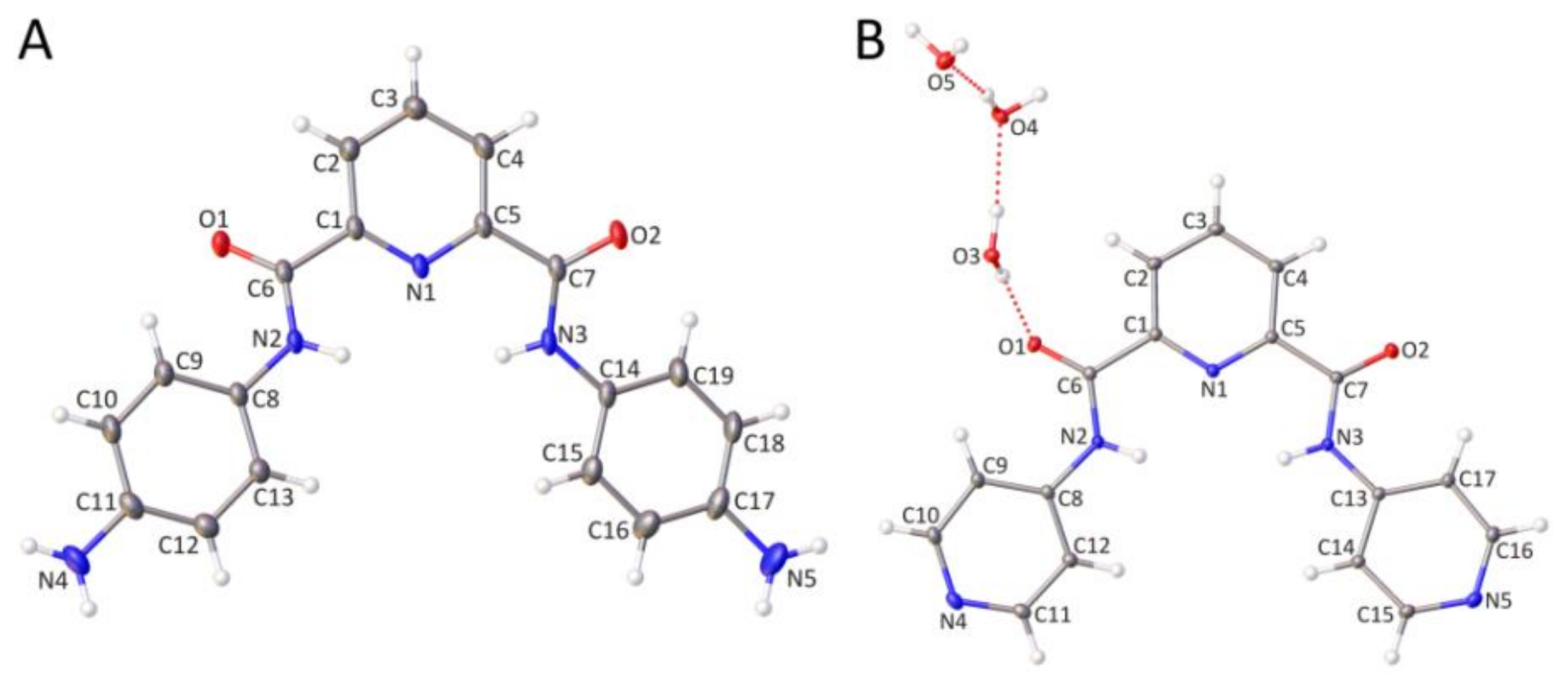
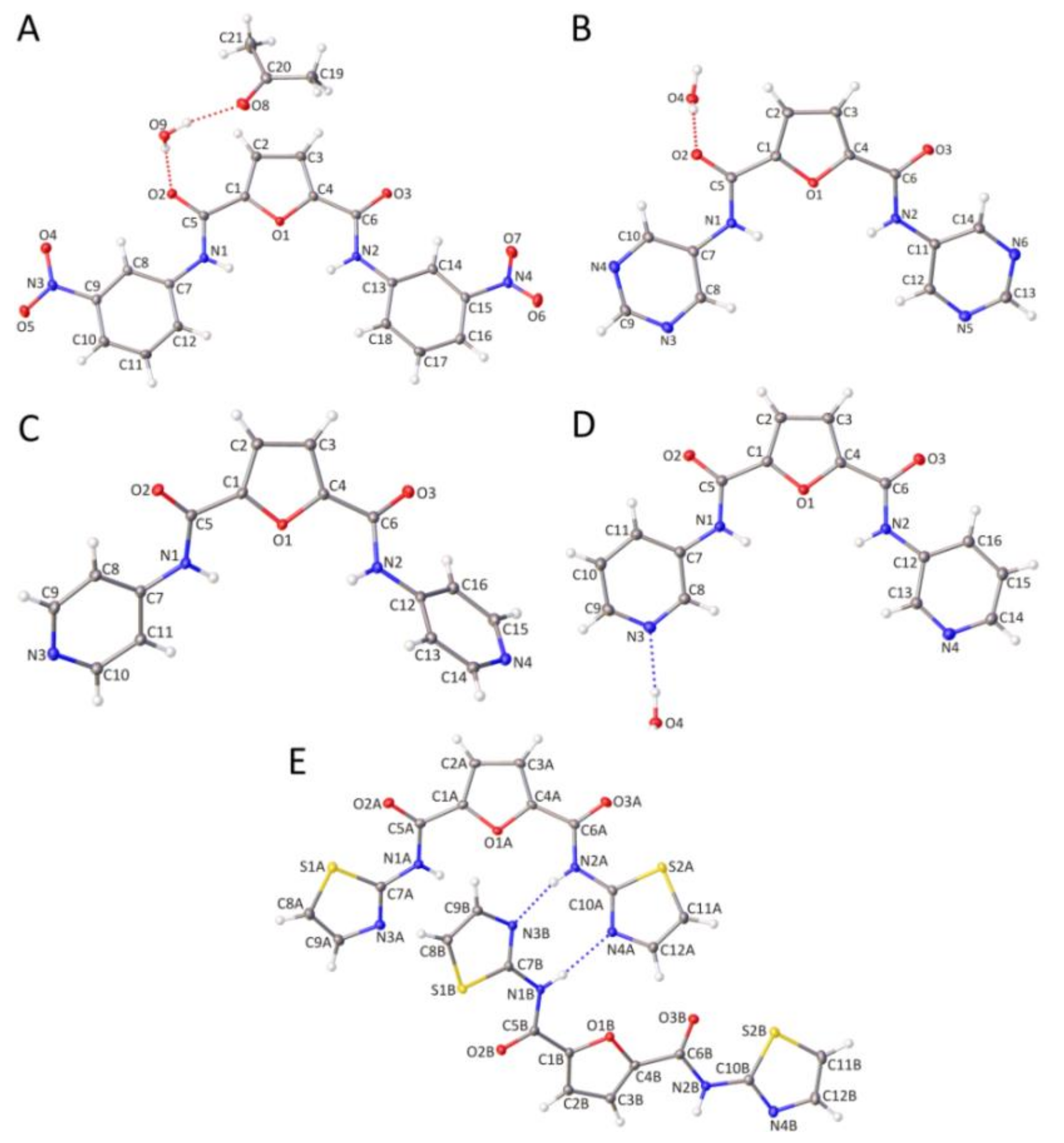
2.2.1. Molecular Structure of Furan-2,5-dicarboxamides and Pyridine-2,6-dicarboxamides
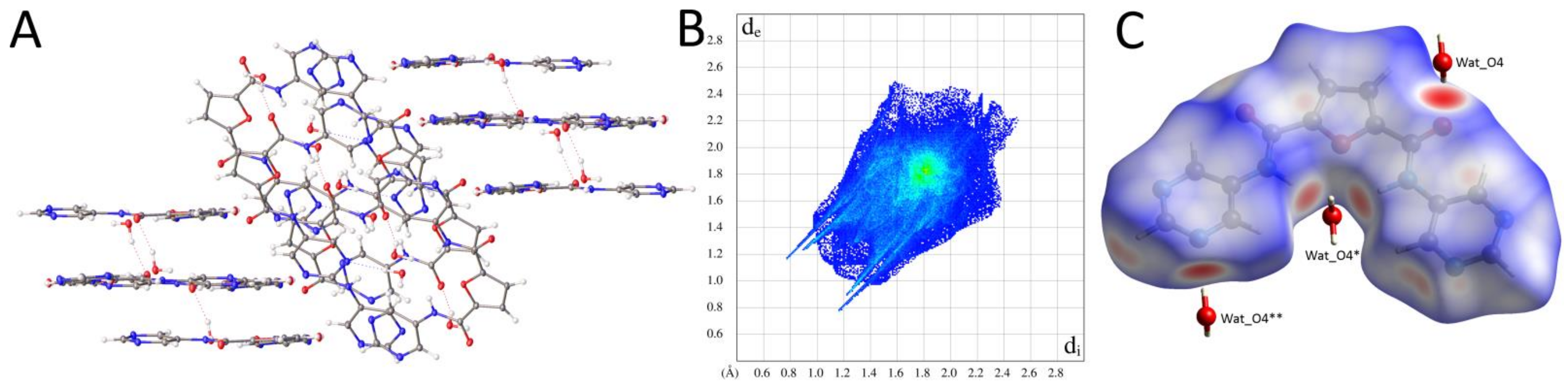
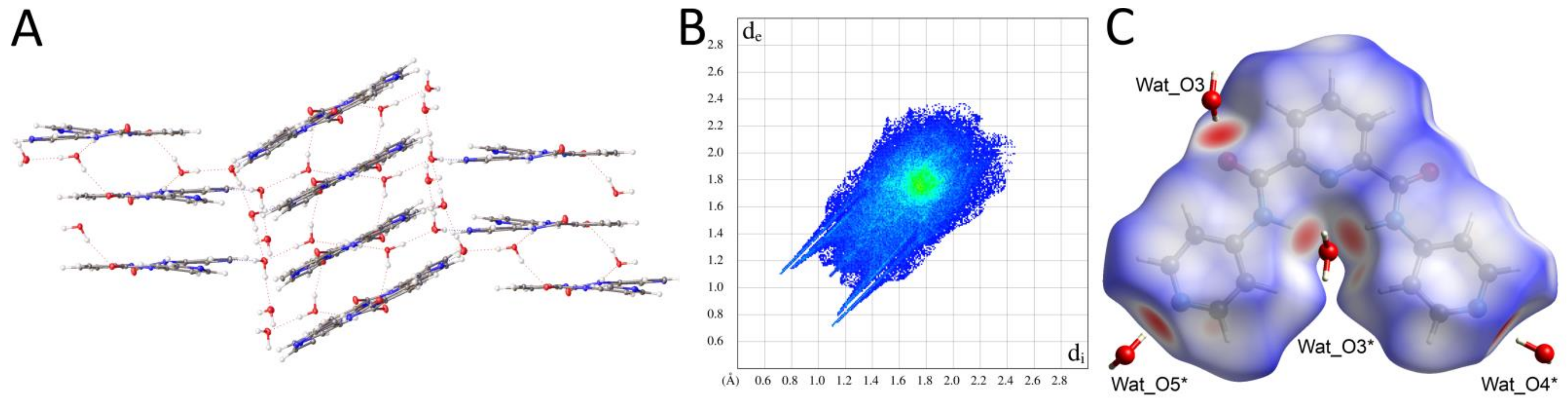
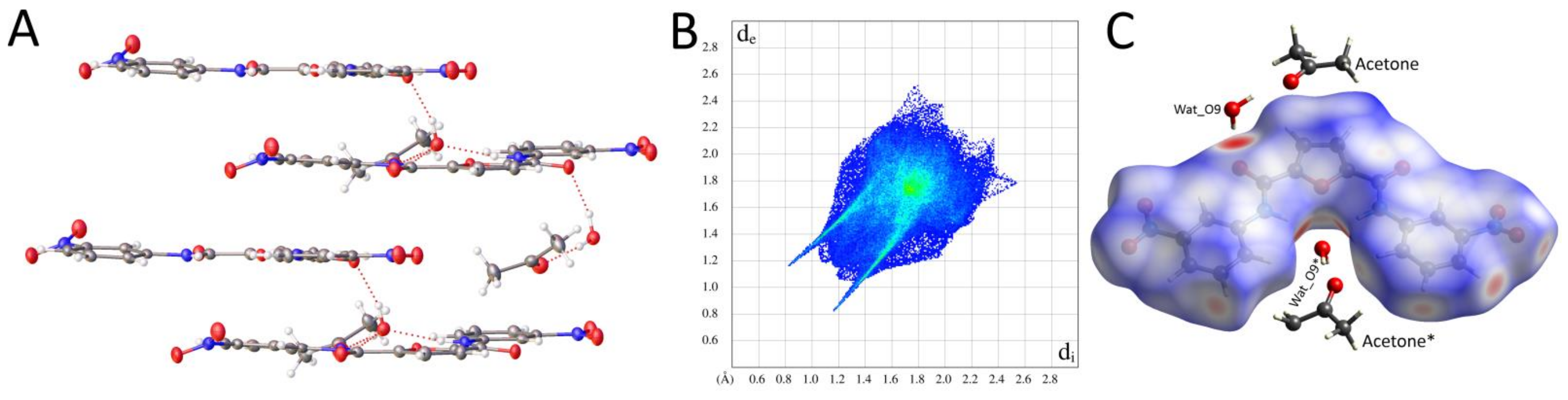
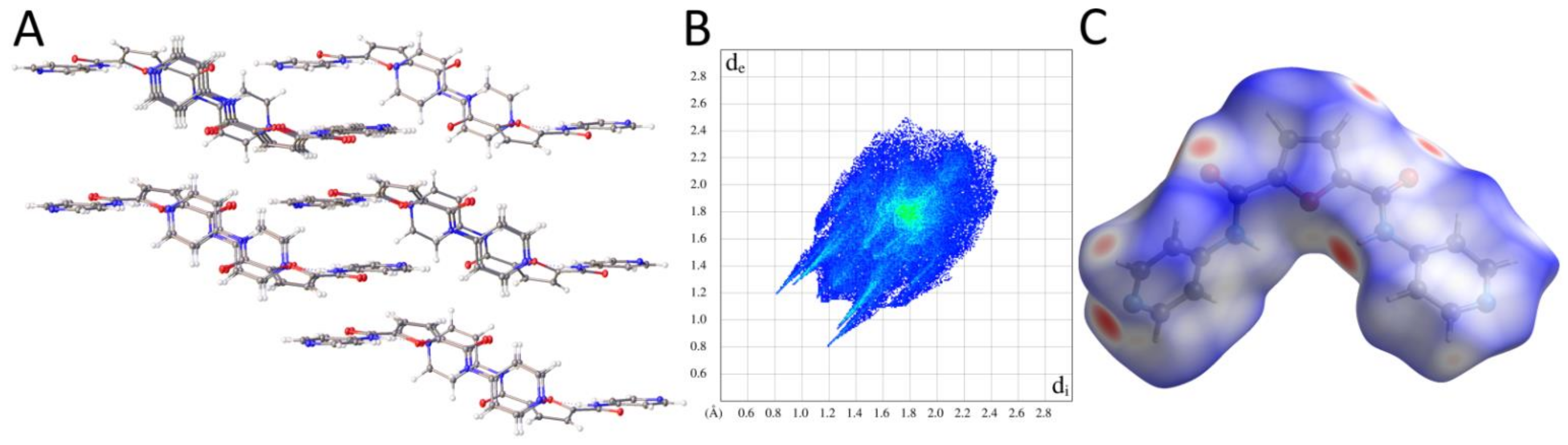
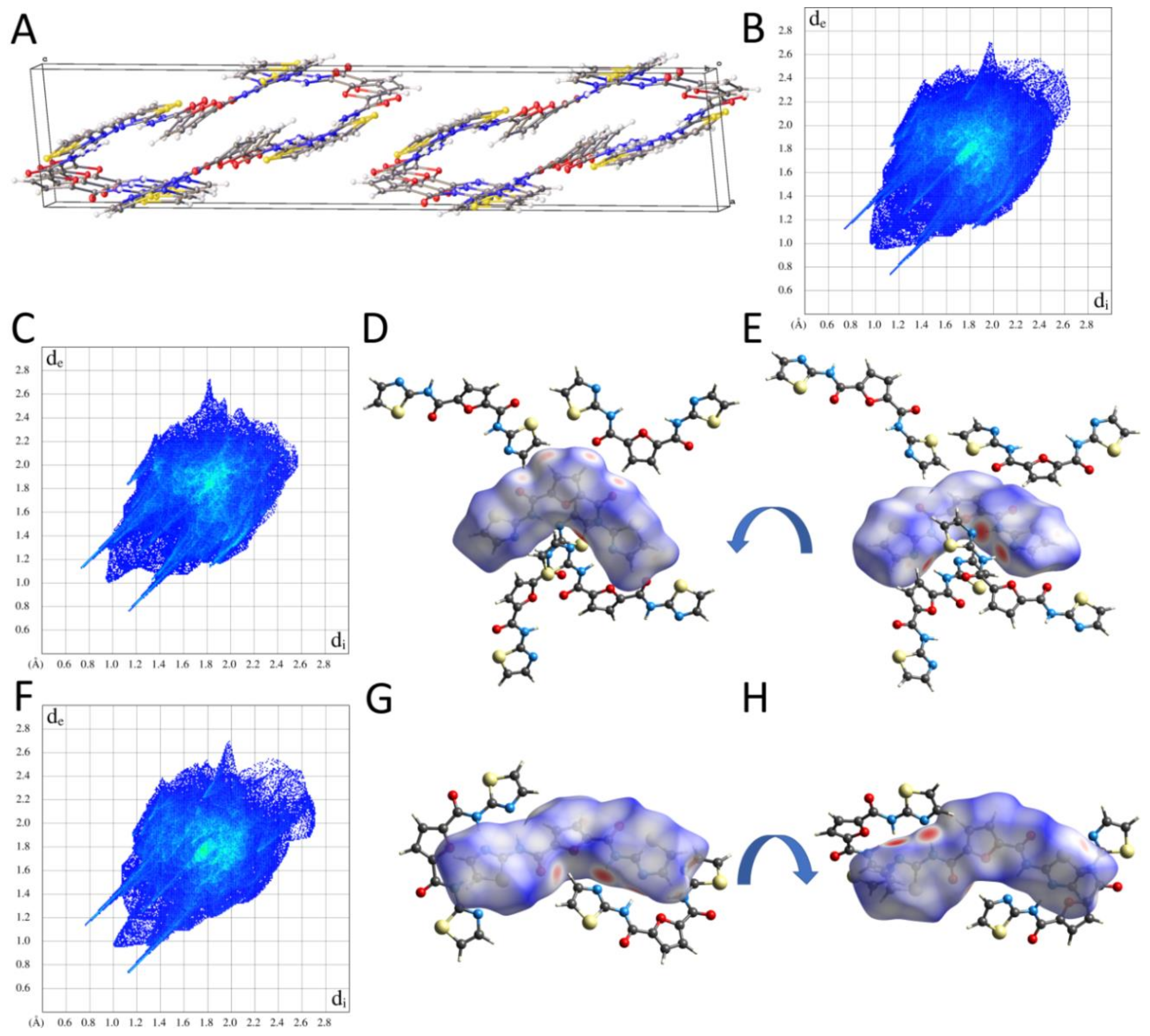
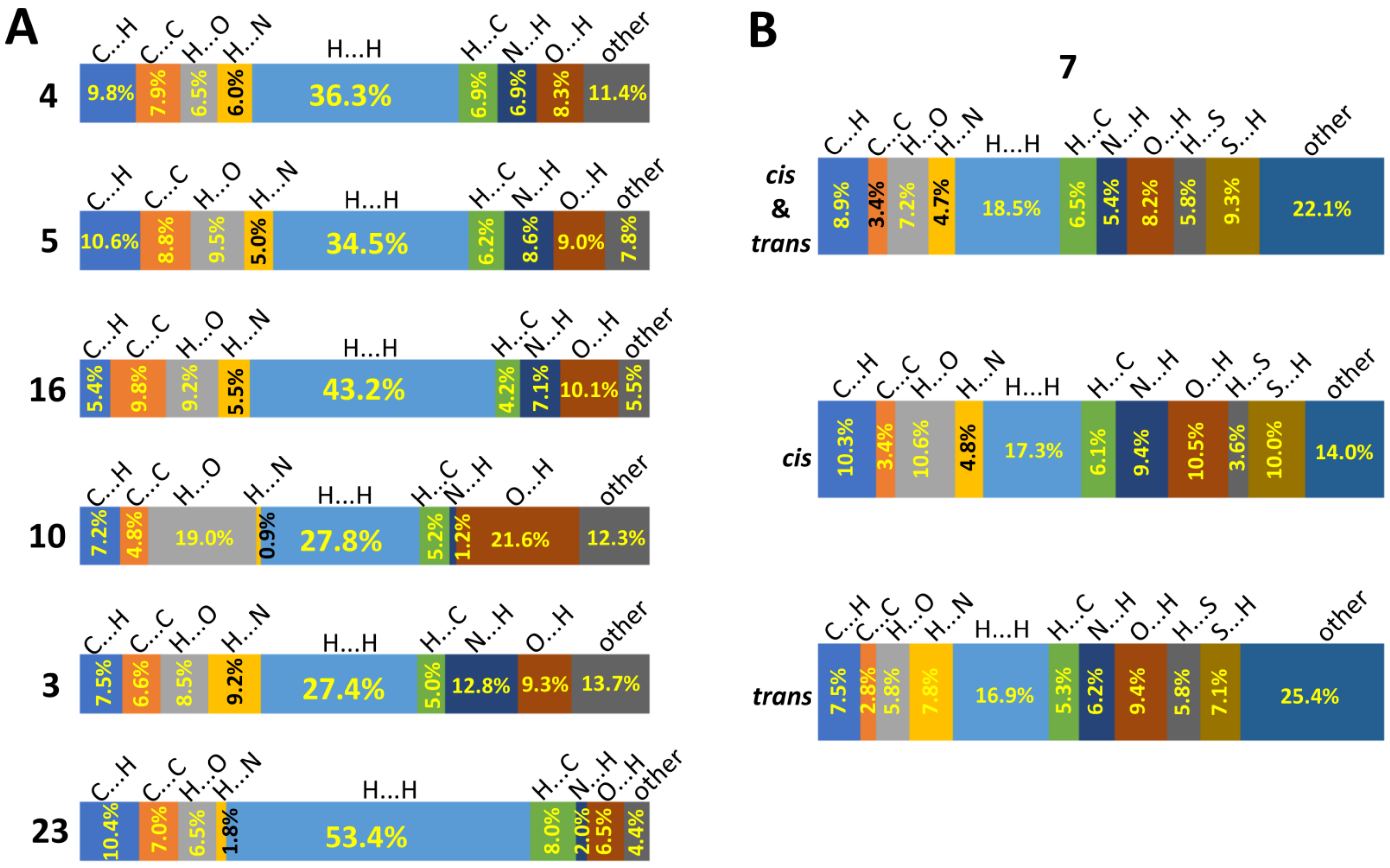
2.2.2. Supramolecular Features and Hirshfeld Surface Analysis
2.2.3. Comparison of Crystal Packing and Intermolecular Interactions
3. Materials and Methods
3.1. Materials
3.2. Structural Characterization
3.2.1. NMR Spectra
3.2.2. X-ray Crystallography and Theoretical Studies
3.3. Synthesis of Compounds
3.3.1. General Procedure for Synthesis of Acid Chlorides (A, B)
3.3.2. General Procedure for Synthesis of Diarylamides
General Procedure for Synthesis of Diarylamides 1–3 and 13–15
General Procedure for Synthesis of Diarylamides 4–10 and 16–22
3.3.3. General Procedure for Synthesis of Diamines from Dinitro Compounds 11, 12, 23 and 24
N2,N5-Di(pyrazin-2-yl)furan-2,5-dicarboxamide (Compound 1)
N2,N5-Di(pyrimidin-2-yl)furan-2,5-dicarboxamide (Compound 2)
N2,N5-Di(pyrimidin-5-yl)furan-2,5-dicarboxamide (Compound 3)
N2,N5-Di(pyridin-4-yl)furan-2,5-dicarboxamide (Compound 4)
N2,N5-Di(pyridin-3-yl)furan-2,5-dicarboxamide (Compound 5)
N2,N5-Di(pyridin-2-yl)furan-2,5-dicarboxamide (Compound 6)
N2,N5-Di(1,3-thiazol-2-yl)furan-2,5-dicarboxamide (Compound 7)
N2,N5-Bis(5-chloropyridin-2-yl)furan-2,5-dicarboxamide (Compound 8)
N2,N5-Di(4-nitrophenyl)furan-2,5-dicarboxamide (Compound 9)
N2,N5-Di(3-nitrophenyl)furan-2,5-dicarboxamide (Compound 10)
N2,N5-Di(4-aminophenyl)furan-2,5-dicarboxamide (Compound 11)
N2,N5-Di(3-aminophenyl)furan-2,5-dicarboxamide (Compound 12)
N2,N6-Di(pyrazin-2-yl)pyridine-2,6-dicarboxamide (Compound 13)
N2,N6-Di(pyrimidin-2-yl)pyridine-2,6-dicarboxamide (Compound 14)
N2,N6-Di(pyrimidin-5-yl)pyridine-2,6-dicarboxamide (Compound 15)
N2,N6-Di(pyridin-4-yl)pyridine-2,6-dicarboxamide (Compound 16)
N2,N6-Di(pyridin-3-yl)pyridine-2,6-dicarboxamide (Compound 17)
N2,N6-Di(pyridin-2-yl)pyridine-2,6-dicarboxamide (Compound 18)
N2,N6-Di(1,3-thiazol-2-yl)pyridine-2,6-dicarboxamide (Compound 19)
N2,N6-Bis(5-chloropyridin-2-yl)pyridine-2,6-dicarboxamide (Compound 20)
N2,N6-Di(4-nitrophenyl)pyridine-2,6-dicarboxamide (Compound 21)
N2,N6-Di(3-nitrophenyl)pyridine-2,6-dicarboxamide (Compound 22)
N2,N6-Di(4-aminophenyl)pyridine-2,6-dicarboxamide (Compound 23)
N2,N6-Di(3-aminophenyl)pyridine-2,6-dicarboxamide (Compound 24)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Kumar, P.; Gupta, R. The Wonderful World of Pyridine-2,6-Dicarboxamide Based Scaffolds. Dalton Trans. 2016, 45, 18769–18783. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.-H.; Tan, M.-Y. Synthesis of Novel Multifunctional Pyridine-2,6-Dicarboxylic Acid Derivatives. Synth. Commun. 2003, 33, 1113–1119. [Google Scholar] [CrossRef]
- Devi, P.; Barry, S.M.; Houlihan, K.M.; Murphy, M.J.; Turner, P.; Jensen, P.; Rutledge, P.J. Synthesis and Structural Characterisation of Amides from Picolinic Acid and Pyridine-2,6-Dicarboxylic Acid. Sci. Rep. 2015, 5, 9950. [Google Scholar] [CrossRef] [PubMed]
- Rahimi, H.; Hosseinzadeh, R.; Tajbakhsh, M. A New and Efficient Pyridine-2,6-Dicarboxamide-Based Fluorescent and Colorimetric Chemosensor for Sensitive and Selective Recognition of Pb2+ and Cu2+. J. Photochem. Photobiol. A Chem. 2021, 407, 113049. [Google Scholar] [CrossRef]
- Sudheer; Kumar, V.; Kumar, P.; Gupta, R. Detection of Al3+ and Fe3+ Ions by Nitrobenzoxadiazole Bearing Pyridine-2,6-Dicarboxamide Based Chemosensors: Effect of Solvents on Detection. New J. Chem. 2020, 44, 13285–13294. [Google Scholar] [CrossRef]
- Kang, S.-O.; Johnson, T.S.; Day, V.W.; Bowman-James, K. Pyridine-2,6-Dicarboxamide Pincer-Based Macrocycle: A Versatile Ligand for Oxoanions, Oxometallates, and Transition Metals. Supramol. Chem. 2018, 30, 305–314. [Google Scholar] [CrossRef]
- Dasgupta, M.; Nag, S.; Das, G.; Nethaji, M.; Bhattacharya, S. N,N′-Bis(Aryl)Pyridine-2,6-Dicarboxamide Complexes of Ruthenium: Synthesis, Structure and Redox Properties. Polyhedron 2008, 27, 139–150. [Google Scholar] [CrossRef]
- Que, L.; Ho, R.Y.N. Dioxygen Activation by Enzymes with Mononuclear Non-Heme Iron Active Sites. Chem. Rev. 1996, 96, 2607–2624. [Google Scholar] [CrossRef]
- Noveron, J.C.; Olmstead, M.M.; Mascharak, P.K. Co(III) Complexes with Carboxamido N and Thiolato S Donor Centers: Models for the Active Site of Co-Containing Nitrile Hydratases. J. Am. Chem. Soc. 1999, 121, 3553–3554. [Google Scholar] [CrossRef]
- Cadoni, E.; Magalhães, P.R.; Emídio, R.M.; Mendes, E.; Vítor, J.; Carvalho, J.; Cruz, C.; Victor, B.L.; Paulo, A. New (Iso)Quinolinyl-Pyridine-2,6-Dicarboxamide G-Quadruplex Stabilizers. A Structure-Activity Relationship Study. Pharmaceuticals 2021, 14, 669. [Google Scholar] [CrossRef]
- Ahamed, A.; Arif, I.A.; Mateen, M.; Surendra Kumar, R.; Idhayadhulla, A. Antimicrobial, Anticoagulant, and Cytotoxic Evaluation of Multidrug Resistance of New 1,4-Dihydropyridine Derivatives. Saudi J. Biol. Sci. 2018, 25, 1227–1235. [Google Scholar] [CrossRef] [PubMed]
- Pachisia, S.; Gupta, R. Architectural and Catalytic Aspects of Designer Materials Built Using Metalloligands of Pyridine-2,6-Dicarboxamide Based Ligands. Dalton Trans. 2020, 49, 14731–14748. [Google Scholar] [CrossRef] [PubMed]
- de Oliveira, M.I.; Chuy, G.P.; Vizzotto, B.S.; Burrow, R.A.; Lang, E.S.; dos Santos, S.S. Synthesis, Characterization and Biological Applications of Bismuth(III) Complexes with Aroylthiourea Ligands. Inorg. Chim. Acta 2020, 512, 119871. [Google Scholar] [CrossRef]
- Kumar, A.; Singh, N.P.; Agrawal, U.; Kumar, K. Synthesis, Characterization and Biological Activity of Ni(II), Cu(II) and Fe(III) ComplexesDerived from N2,N6-Bis(5-Mercapto-1,3,4-Thiadiazol-2-Yl)Pyridine-2,6-Dicarboxamide. Asian J. Chem. 2020, 32, 1691–1696. [Google Scholar] [CrossRef]
- Drozdowska, D.; Rusak, M.; Miltyk, W.; Midura-Nowaczek, K. Synthesis and Biological Evaluation of Distamycin Analogues—New Potential Anticancer Agents. Arch. Pharm. 2009, 342, 87–93. [Google Scholar] [CrossRef]
- Chen, L.; Li, H.; Liu, J.; Zhang, L.; Liu, H.; Jiang, H. Discovering Benzamide Derivatives as Glycogen Phosphorylase Inhibitors and Their Binding Site at the Enzyme. Bioorg. Med. Chem. 2007, 15, 6763–6774. [Google Scholar] [CrossRef]
- Al-Salahi, R.; Al-Omar, M.; Amr, A.E.-G. Synthesis of Chiral Macrocyclic or Linear Pyridine Carboxamides from Pyridine-2,6-Dicarbonyl Dichloride as Antimicrobial Agents. Molecules 2010, 15, 6588–6597. [Google Scholar] [CrossRef] [Green Version]
- Al-Omar, M.A.; Amr, A.E.-G.E. Synthesis of Some New Pyridine-2,6-Carboxamide-Derived Schiff Bases as Potential Antimicrobial Agents. Molecules 2010, 15, 4711–4721. [Google Scholar] [CrossRef]
- Schulze, B.; Schubert, U.S. Beyond Click Chemistry—Supramolecular Interactions of 1,2,3-Triazoles. Chem. Soc. Rev. 2014, 43, 2522. [Google Scholar] [CrossRef]
- Pavlidis, N.; Kofinas, A.; Papanikolaou, M.G.; Miras, H.N.; Drouza, C.; Kalampounias, A.G.; Kabanos, T.A.; Konstandi, M.; Leondaritis, G. Synthesis, Characterization and Pharmacological Evaluation of Quinoline Derivatives and Their Complexes with Copper(ΙΙ) in in Vitro Cell Models of Alzheimer’s Disease. J. Inorg. Biochem. 2021, 217, 111393. [Google Scholar] [CrossRef]
- Müller, S.; Sanders, D.A.; Di Antonio, M.; Matsis, S.; Riou, J.-F.; Rodriguez, R.; Balasubramanian, S. Pyridostatin Analogues Promote Telomere Dysfunction and Long-Term Growth Inhibition in Human Cancer Cells. Org. Biomol. Chem. 2012, 10, 6537. [Google Scholar] [CrossRef] [PubMed]
- Janczewski, Ł.; Zieliński, D.; Kolesińska, B. Synthesis of Amides and Esters Containing Furan Rings under Microwave-Assisted Conditions. Open Chem. 2021, 19, 265–280. [Google Scholar] [CrossRef]
- Ma, K.; Chen, G.; Zhang, Y. Thermal Cross-link between 2,5-furandicarboxylic Acid-based Polyimides and Bismaleimide via Diels–Alder Reaction. J. Polym. Sci. 2020, 58, 2951–2962. [Google Scholar] [CrossRef]
- Dyachenko, I.V.; Dyachenko, V.D.; Dorovatovskii, P.V.; Khrustalev, V.N.; Nenajdenko, V.G. Multicomponent Synthesis and Molecular Structure of 3-Amino-2-Aroyl(Alkoxycarbonyl, Arylcarbamoyl)-4-Aryl(Hetaryl)-5-Arylcarbamoyl-6-Methylthieno[2,3-b]Pyridines. Chem. Heterocycl. Comp. 2019, 55, 442–447. [Google Scholar] [CrossRef]
- Valente, S.; Mellini, P.; Spallotta, F.; Carafa, V.; Nebbioso, A.; Polletta, L.; Carnevale, I.; Saladini, S.; Trisciuoglio, D.; Gabellini, C.; et al. 1,4-Dihydropyridines Active on the SIRT1/AMPK Pathway Ameliorate Skin Repair and Mitochondrial Function and Exhibit Inhibition of Proliferation in Cancer Cells. J. Med. Chem. 2016, 59, 1471–1491. [Google Scholar] [CrossRef]
- Saudi, M.; Zmurko, J.; Kaptein, S.; Rozenski, J.; Neyts, J.; Van Aerschot, A. Synthesis and Evaluation of Imidazole-4,5- and Pyrazine-2,3-Dicarboxamides Targeting Dengue and Yellow Fever Virus. Eur. J. Med. Chem. 2014, 87, 529–539. [Google Scholar] [CrossRef] [Green Version]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Cryst. B Struct. Sci. Cryst. Eng. Mater. 2016, 72, 171–179. [Google Scholar] [CrossRef]
- Burley, S.K.; Bhikadiya, C.; Bi, C.; Bittrich, S.; Chen, L.; Crichlow, G.V.; Duarte, J.M.; Dutta, S.; Fayazi, M.; Feng, Z.; et al. RCSB Protein Data Bank: Celebrating 50 Years of the PDB with New Tools for Understanding and Visualizing Biological Macromolecules in 3D. Protein Sci. 2022, 31, 187–208. [Google Scholar] [CrossRef]
- Spackman, M.A.; Byrom, P.G. A Novel Definition of a Molecule in a Crystal. Chem. Phys. Lett. 1997, 267, 215–220. [Google Scholar] [CrossRef]
- Spackman, M.A.; Jayatilaka, D. Hirshfeld Surface Analysis. CrystEngComm 2009, 11, 19–32. [Google Scholar] [CrossRef]
- Kumara, K.; Dileep Kumar, A.; Naveen, S.; Ajay Kumar, K.; Lokanath, N.K. Synthesis, Spectral Characterization and X-Ray Crystal Structure Studies of 3-(Benzo[d][1,3]Dioxol-5-Yl)-5-(3-Methylthiophen-2-Yl)-4,5-Dihydro-1H-Pyrazole-1-Carboxamide: Hirshfeld Surface, DFT and Thermal Analysis. J. Mol. Struct. 2018, 1161, 285–298. [Google Scholar] [CrossRef]
- Kumara, K.; Shivalingegowda, N.; Mahadevaswamy, L.D.; Kariyappa, A.K.; Lokanath, N.K. Crystal Structure Studies and Hirshfeld Surface Analysis of 5-(4-Methoxyphenyl)-3-(Thiophen-2-Yl)-4,5-Dihydro-1 H -Pyrazole-1-Carbothioamide. Chem. Data Collect. 2017, 9–10, 251–262. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A Complete Structure Solution, Refinement and Analysis Program. J Appl Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT—Integrated Space-Group and Crystal-Structure Determination. Acta Cryst. A Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheldrick, G.M. Crystal Structure Refinement with SHELXL. Acta Cryst. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Spek, A.L. Structure Validation in Chemical Crystallography. Acta Cryst. D Biol. Cryst. 2009, 65, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Spackman, P.R.; Turner, M.J.; McKinnon, J.J.; Wolff, S.K.; Grimwood, D.J.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer: A Program for Hirshfeld Surface Analysis, Visualization and Quantitative Analysis of Molecular Crystals. J. Appl. Cryst. 2021, 54, 1006–1011. [Google Scholar] [CrossRef]
- Panja, S.; Ghosh, S.; Ghosh, K. Pyridine/Pyridinium Symmetrical Bisamides as Functional Materials: Aggregation, Selective Sensing and Drug Release. New J. Chem. 2018, 42, 6488–6497. [Google Scholar] [CrossRef]
- Ma, K.; Chen, G.; Wang, W.; Zhang, A.; Zhong, Y.; Zhang, Y.; Fang, X. Partially Bio-Based Aromatic Polyimides Derived from 2,5-Furandicarboxylic Acid with High Thermal and Mechanical Properties. J. Polym. Sci. Part A Polym. Chem. 2018, 56, 1058–1066. [Google Scholar] [CrossRef]
- Abdolmaleki, S.; Ghadermazi, M.; Fattahi, A.; Sheshmani, S. Synthesis, Characterization, Spectral Studies and Cytotoxic Effects of Mixed-Ligand Mono and Binuclear Copper(II) Complexes and Their Amide Ligands. Inorg. Chim. Acta 2016, 443, 284–298. [Google Scholar] [CrossRef]
- Wicht, K.J.; Combrinck, J.M.; Smith, P.J.; Hunter, R.; Egan, T.J. Identification and SAR Evaluation of Hemozoin-Inhibiting Benzamides Active against Plasmodium falciparum. J. Med. Chem. 2016, 59, 6512–6530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, K.; Sarkar, A.R.; Ghorai, A.; Ghosh, U. Design and Synthesis of Anthracene-Based Bispyridinium Amides: Anion Binding, Cell Staining and DNA Interaction Studies. New J. Chem. 2012, 36, 1231. [Google Scholar] [CrossRef]
- Subramanian, P.; Indu, S.; Kaliappan, K.P. A One-Pot Copper Catalyzed Biomimetic Route to N -Heterocyclic Amides from Methyl Ketones via Oxidative C–C Bond Cleavage. Org. Lett. 2014, 16, 6212–6215. [Google Scholar] [CrossRef] [PubMed]
- Wagner-Wysiecka, E.; Chojnacki, J. Chromogenic Amides of Pyridine-2,6-Dicarboxylic Acid as Anion Receptors. Supramol. Chem. 2012, 24, 684–695. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pyridine Derivative | N2-C6-C1-N1 [°] | N3-C7-C5-N1 [°] | N2-H2 [Å] | N3-H3 [Å] | H2∙∙∙H3 [Å] | Conformation |
|---|---|---|---|---|---|---|
| 23 | 20.4(3) | 21.5(3) | 0.86(2) | 0.84(2) | 3.39(3) | skew |
| 16 * | −1.89(1) | −10.1(1) | 0.86(2) | 0.90(2) | 2.91(3) | semi-skew |
| Furan derivative | N1-C5-C1-O1 [°] | N2-C6-C4-O1 [°] | N1-H1 [Å] | N2-H2 [Å] | H1∙∙∙H2 [Å] | |
| 7 (cis) | −19.9(2) | 0.8(2) | 0.84(2) | 0.83(2) | 3.85(2) | semi-skew |
| 7 (trans) | 17.7(2) | 10.2(1) | 0.84(2) | 0.86(2) | - | skew |
| 10 * | −6.3(4) | 0.1(4) | 0.85(4) | 0.94(3) | 3.25(4) | planar |
| 3 * | 5.0(2) | 0.1(2) | 0.87(2) | 0.84(2) | 3.38(2) | planar |
| 4 | −18.3(2) | −19.2(2) | 0.94(1) | 0.89(2) | 3.82(2) | skew |
| 5 * | −0.9(2) | −2.5(2) | 0.89(2) | 0.89(2) | 3.39(2) | planar |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Puckowska, A.; Gawel, M.; Komorowska, M.; Drozdzal, P.; Arning, A.; Pawelski, D.; Brzezinski, K.; Plonska-Brzezinska, M.E. Synthesis and Structural Characterization of Pyridine-2,6-dicarboxamide and Furan-2,5-dicarboxamide Derivatives. Molecules 2022, 27, 1819. https://doi.org/10.3390/molecules27061819
Puckowska A, Gawel M, Komorowska M, Drozdzal P, Arning A, Pawelski D, Brzezinski K, Plonska-Brzezinska ME. Synthesis and Structural Characterization of Pyridine-2,6-dicarboxamide and Furan-2,5-dicarboxamide Derivatives. Molecules. 2022; 27(6):1819. https://doi.org/10.3390/molecules27061819
Chicago/Turabian StylePuckowska, Anna, Magdalena Gawel, Marlena Komorowska, Pawel Drozdzal, Aleksandra Arning, Damian Pawelski, Krzysztof Brzezinski, and Marta E. Plonska-Brzezinska. 2022. "Synthesis and Structural Characterization of Pyridine-2,6-dicarboxamide and Furan-2,5-dicarboxamide Derivatives" Molecules 27, no. 6: 1819. https://doi.org/10.3390/molecules27061819
APA StylePuckowska, A., Gawel, M., Komorowska, M., Drozdzal, P., Arning, A., Pawelski, D., Brzezinski, K., & Plonska-Brzezinska, M. E. (2022). Synthesis and Structural Characterization of Pyridine-2,6-dicarboxamide and Furan-2,5-dicarboxamide Derivatives. Molecules, 27(6), 1819. https://doi.org/10.3390/molecules27061819