
Discovery of a New Drug-like Series of OGT Inhibitors by Virtual Screening


 , ,
, ,  and
and
Abstract
:1. Introduction
2. Results and Discussion
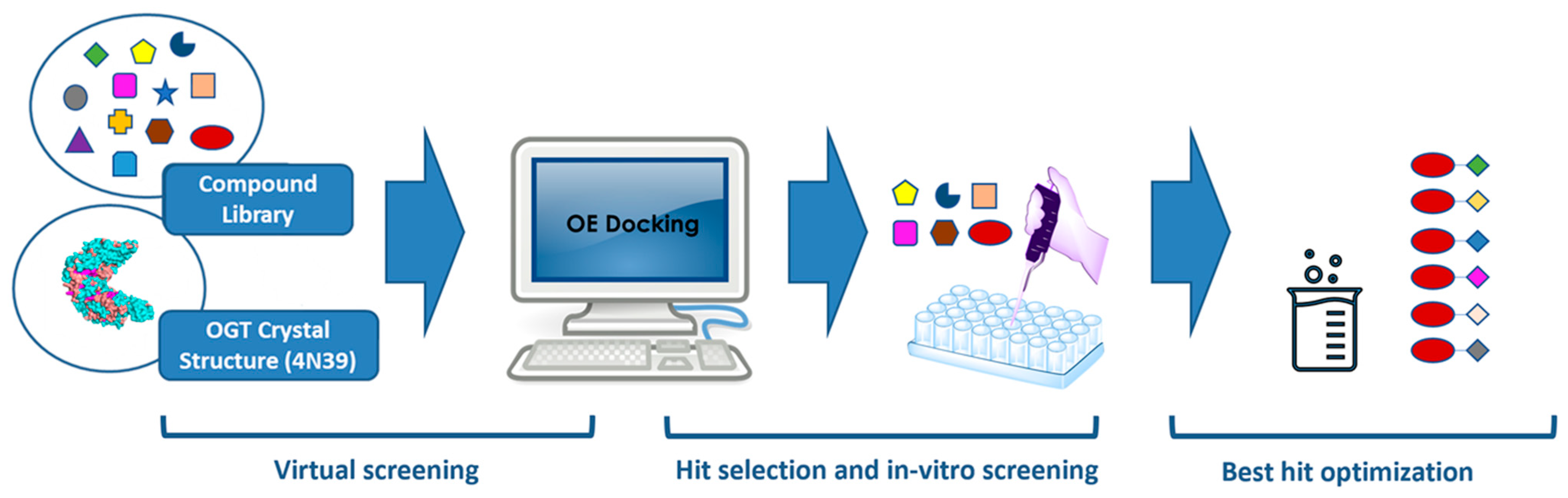
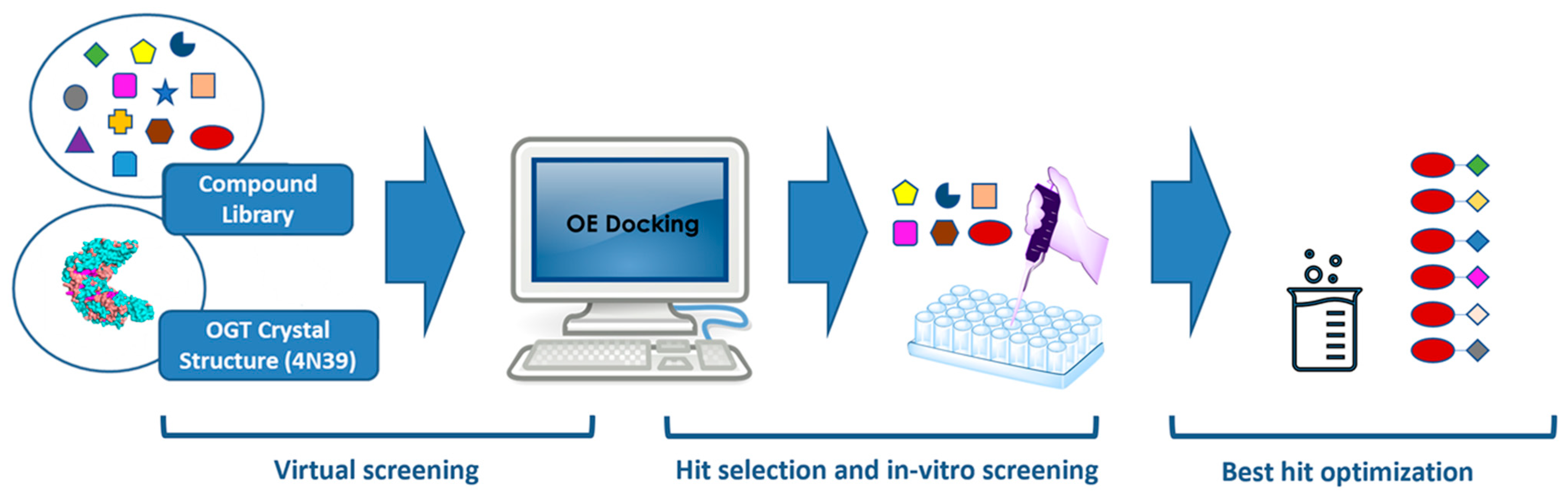
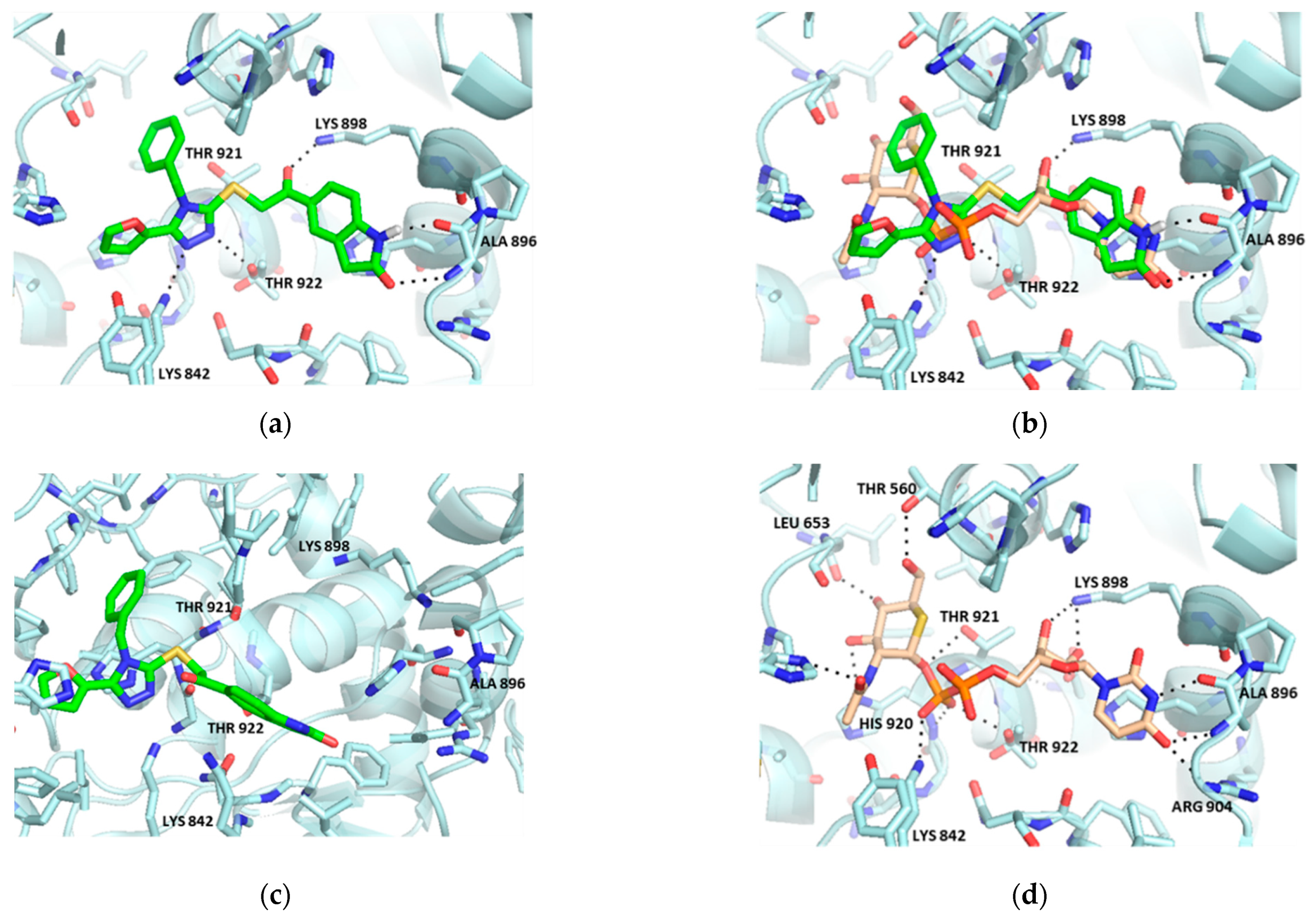
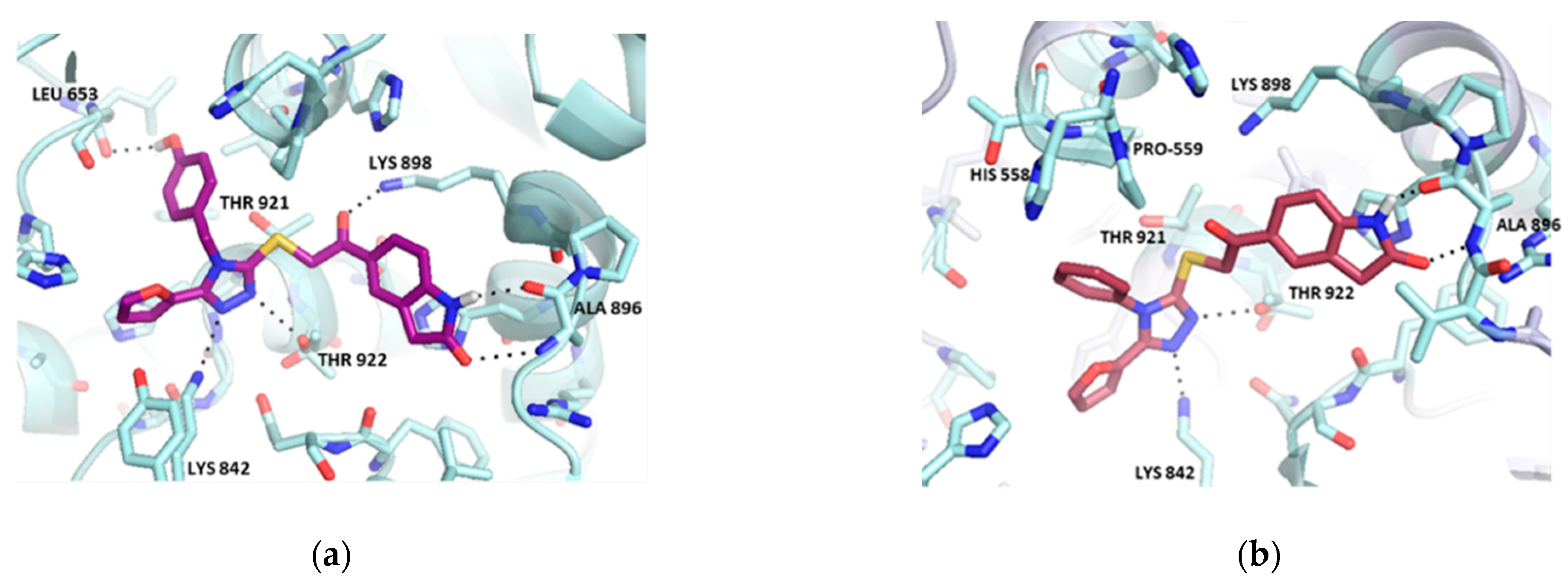
2.1. Virtual Screening
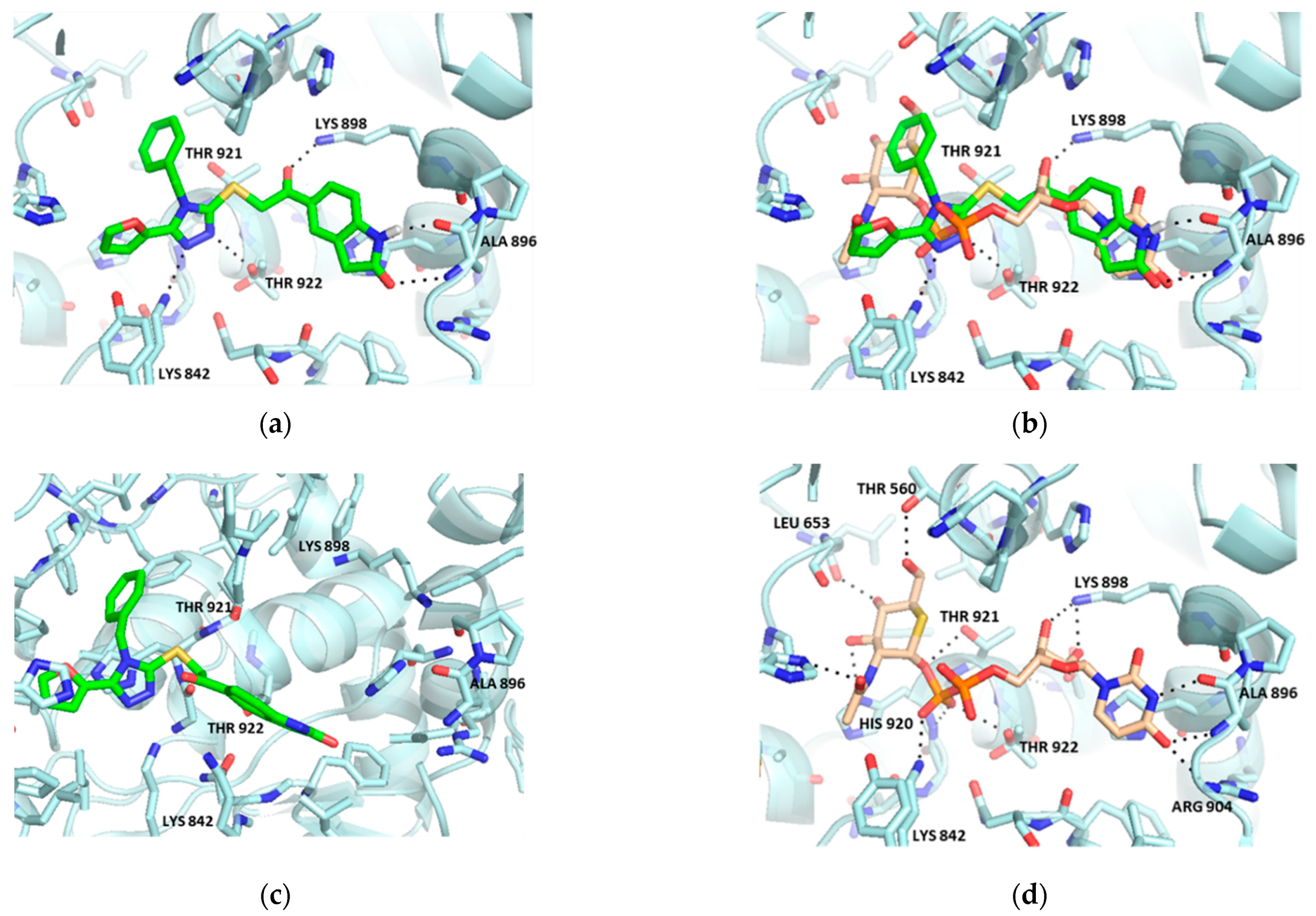
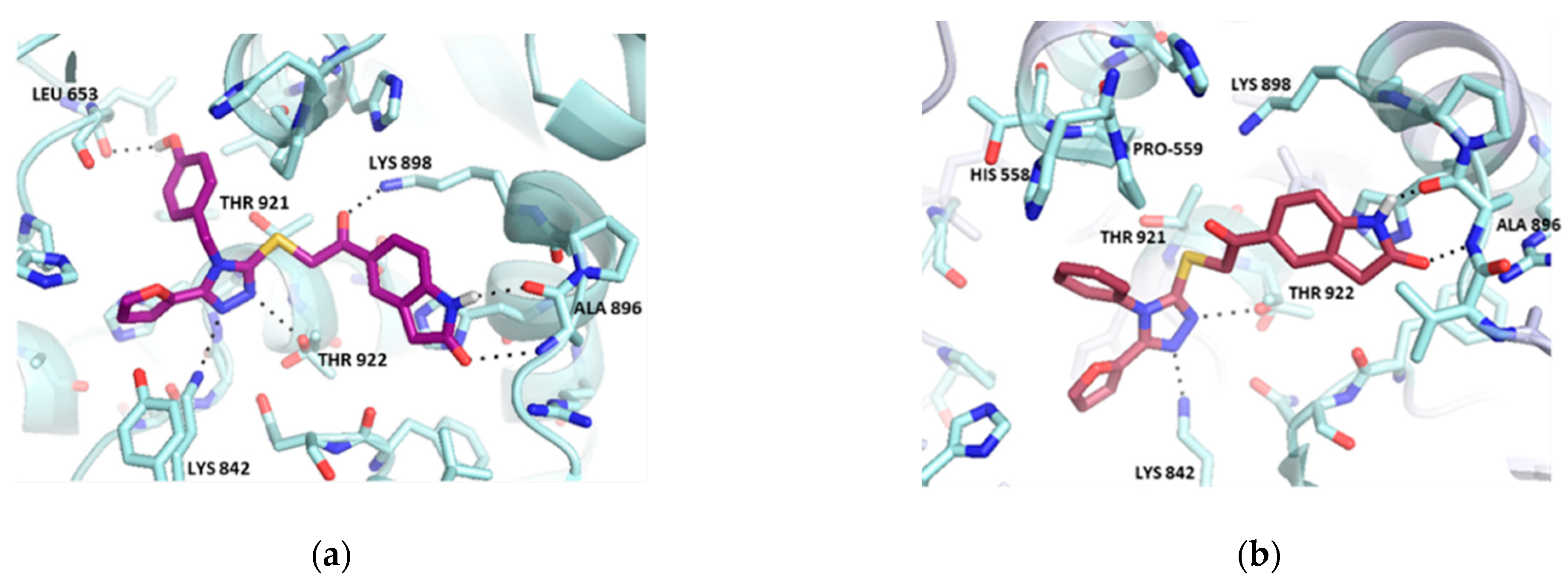
2.2. Hit Optimization
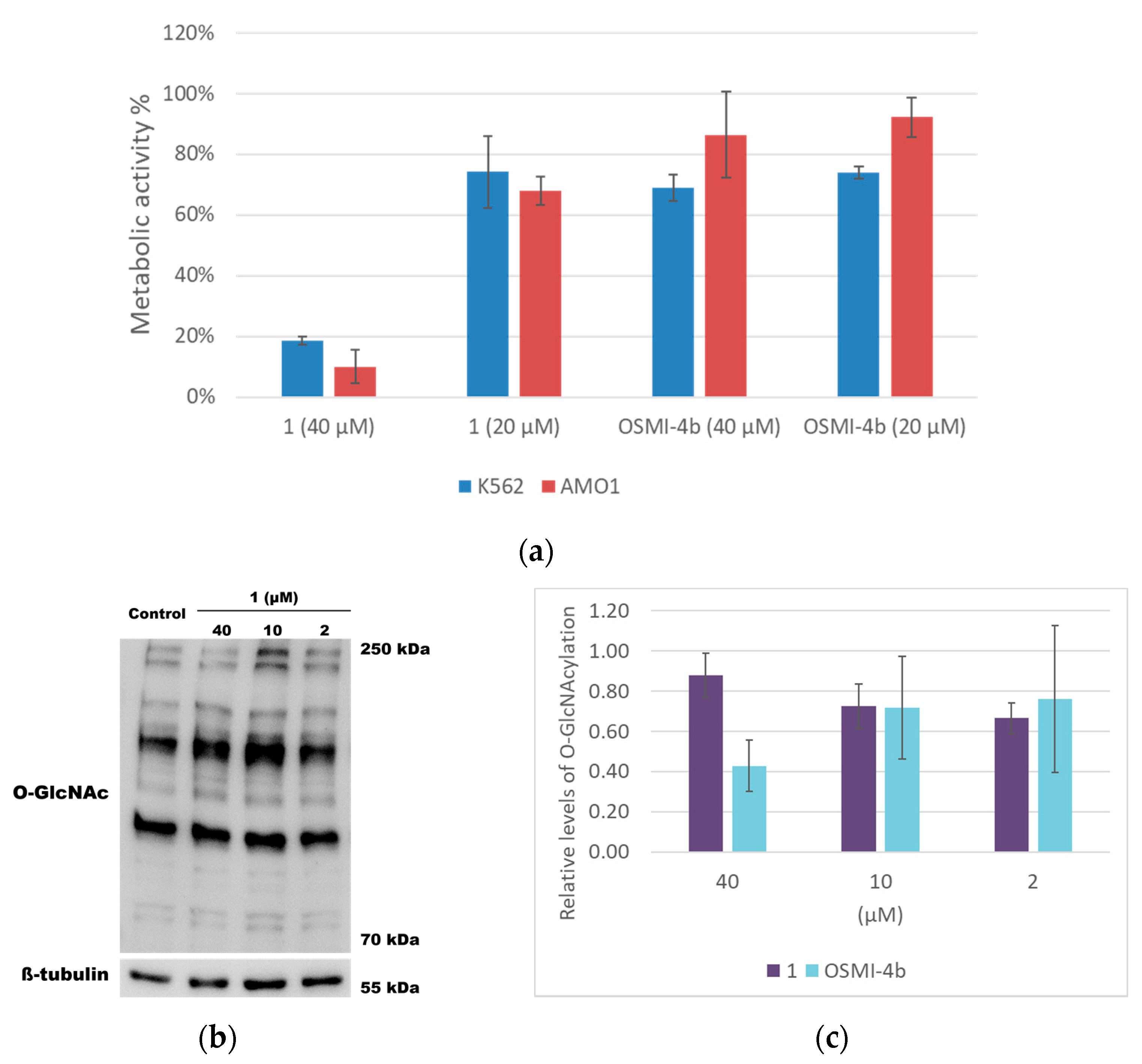
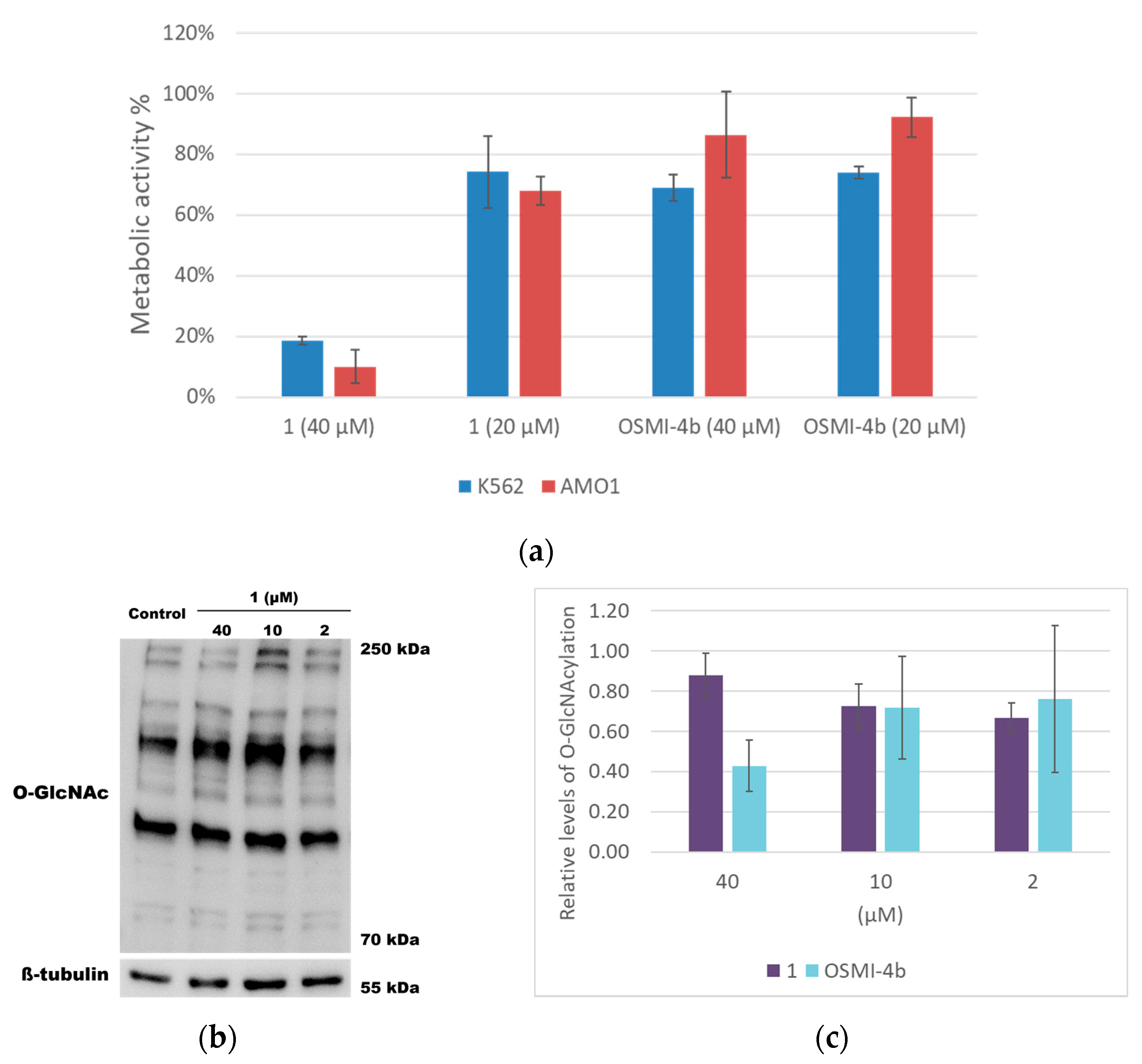
2.3. Cell-Based Assays
3. Materials and Methods
3.1. Chemistry-General
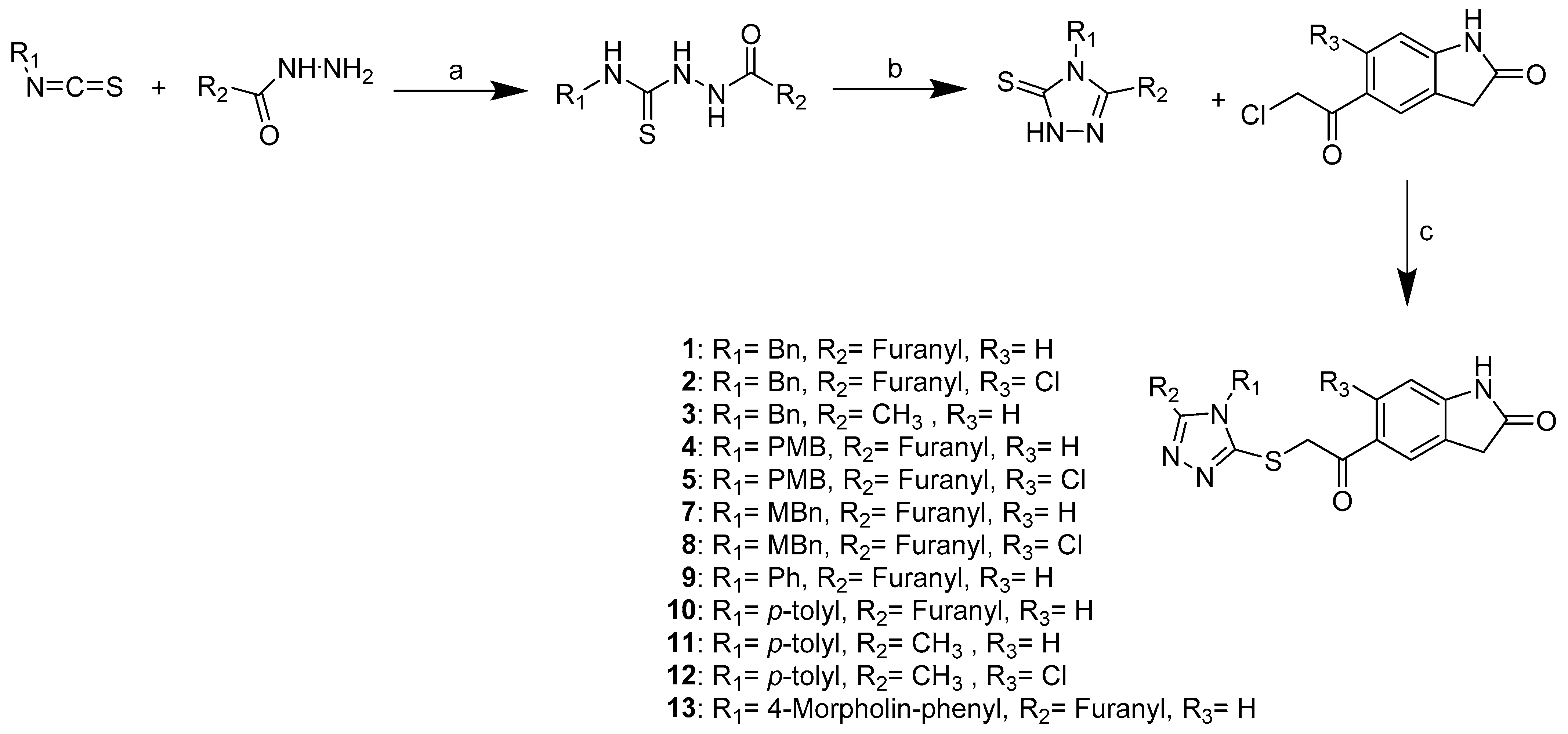
3.1.1. General Synthetic Procedures A, B and C
General Procedure A for the Synthesis of Compounds 14, 16, 19, 21, 23, 25, 27, 32
General Procedure B for the Synthesis of Compounds 15, 17, 20, 22, 24, 26, 28, 33
General Procedure C for the Synthesis of Compounds 1, 2, 3, 4, 5, 7, 8, 9, 10, 11, 12, 13
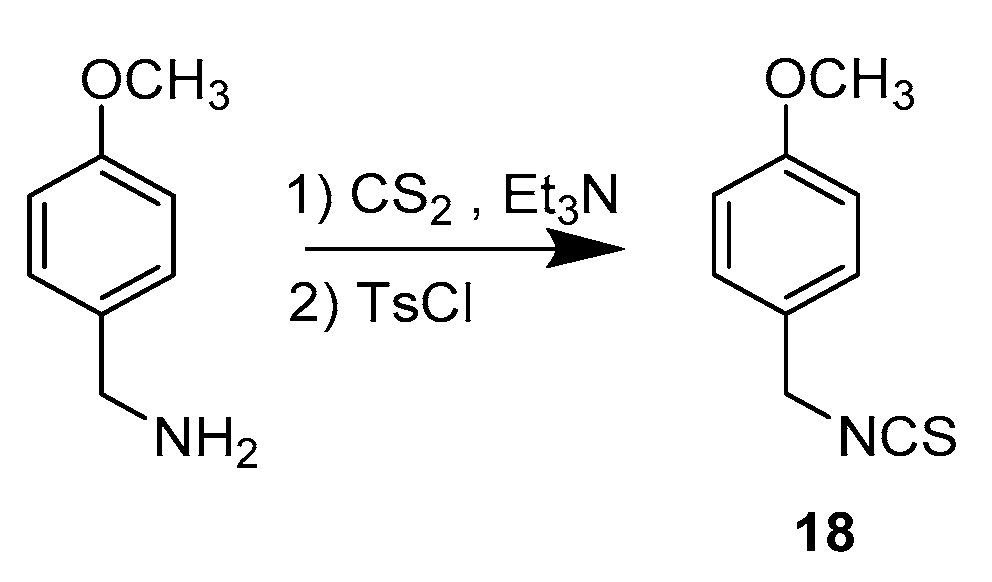
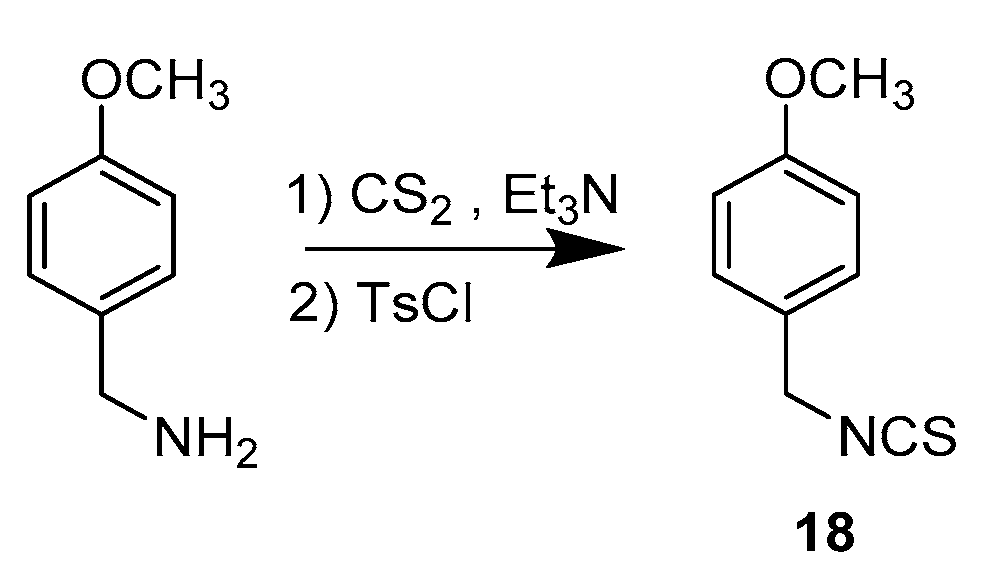
3.1.2. Synthetic Procedure for Compound 18
1-(Isothiocyanatomethyl)-4-methoxybenzene (18)
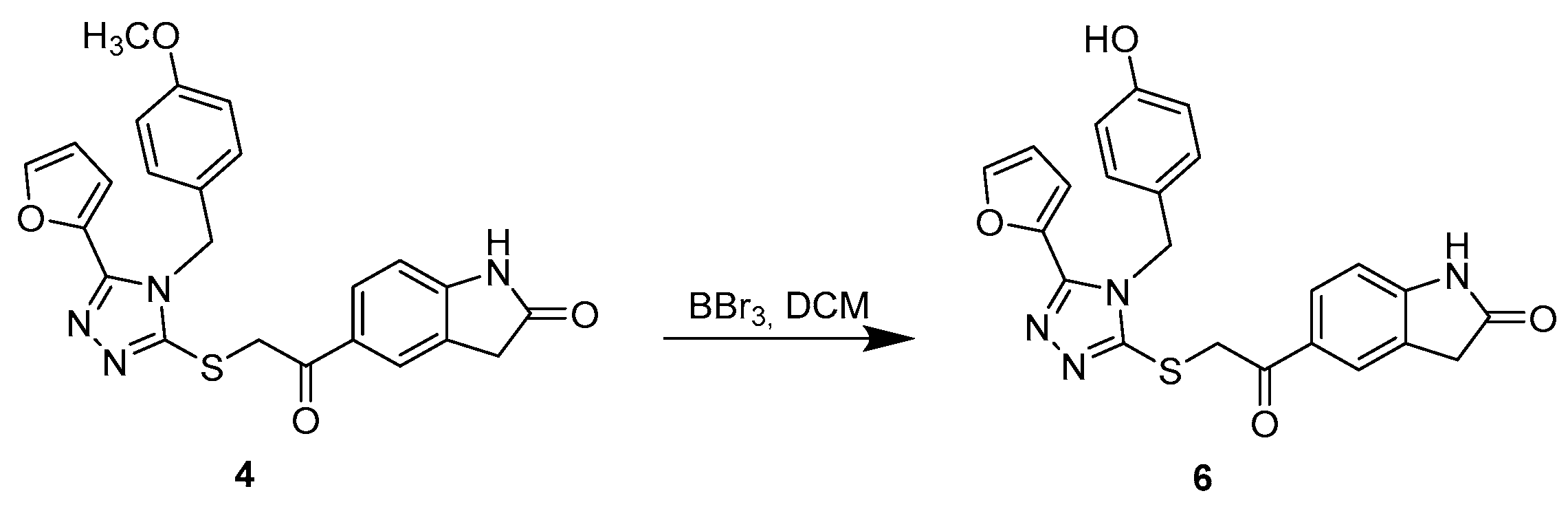
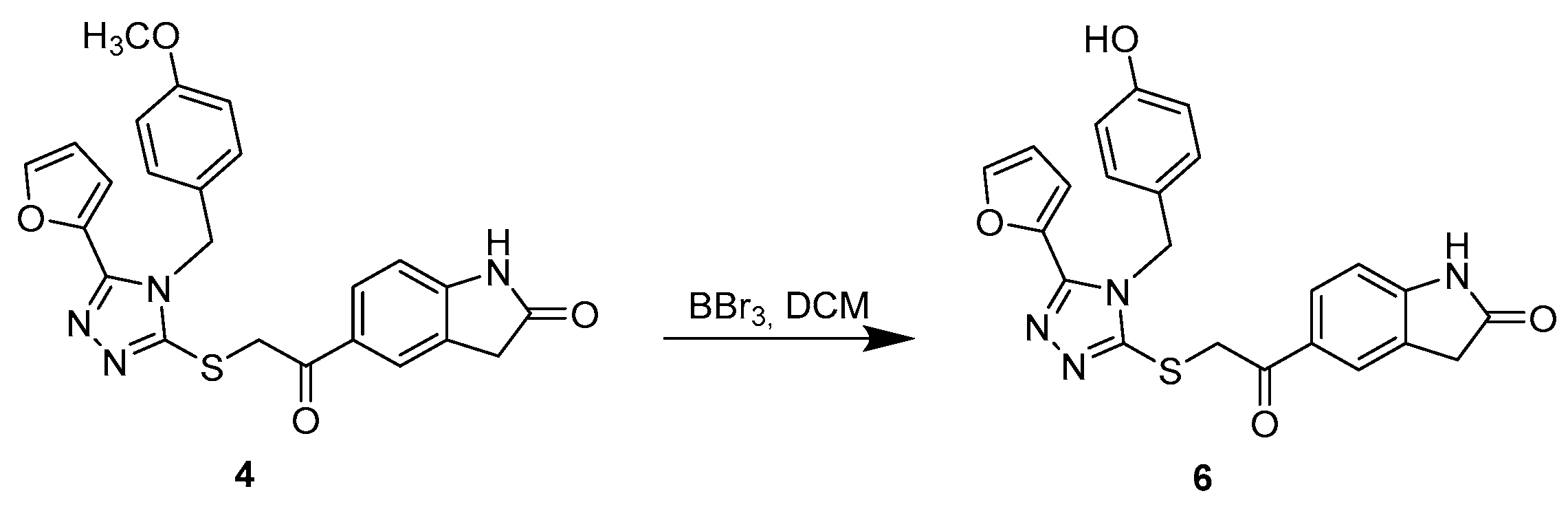
3.1.3. Synthetic Procedure for Compound 6
5-(2-((5-(Furan-2-yl)-4-(4-hydroxybenzyl)-4H-1,2,4-triazol-3-yl)thio)acetyl)indolin-2-one (6)
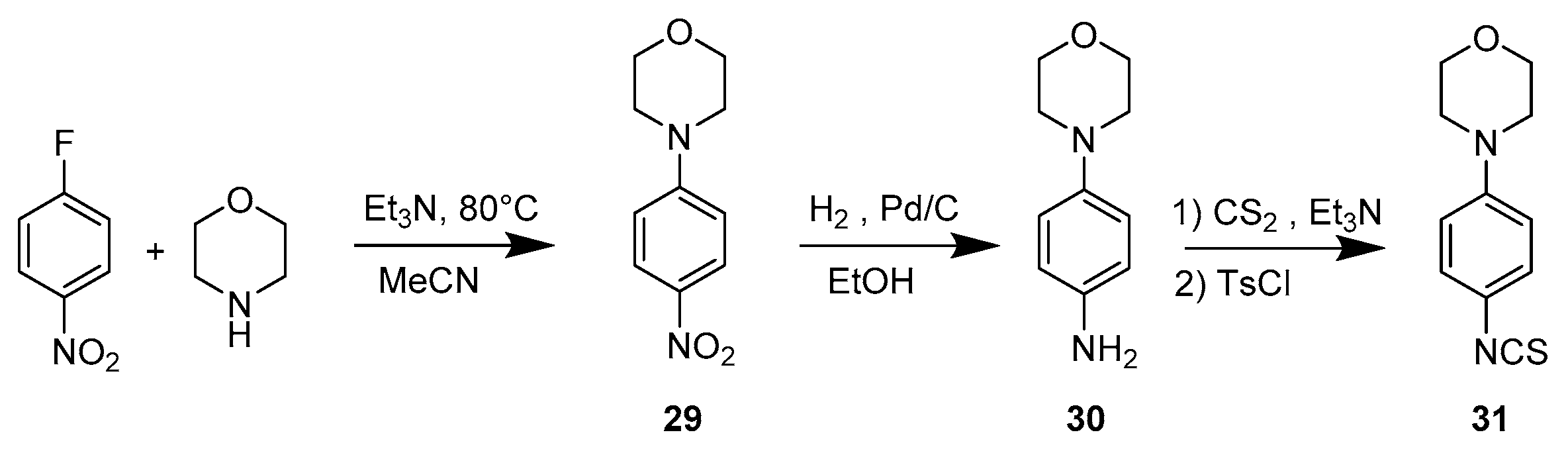
3.1.4. Synthetic Procedure for Compounds 29, 30, 31
4-(4-Nitrophenyl)morpholine (29)
4-Morpholinoaniline (30)
4-(4-Isothiocyanatophenyl)morpholine (31)
3.2. Molecular Modeling
3.2.1. Compound Library
3.2.2. Structure-Based Virtual Screening
3.2.3. Docking
3.2.4. Molecular Dynamics Simulation
3.2.5. Structure-Based Pharmacophore Modeling
3.3. cLogP Prediction
3.4. Fluorescent Activity Assay
3.5. Cell-Based Assays
3.5.1. Cell Cultures
3.5.2. Metabolic Activity Assay
3.5.3. Statistical Analysis
3.5.4. SDS-PAGE
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
Abbreviations
References
- Kreppel, L.K.; Blomberg, M.A.; Hart, G.W. Dynamic Glycosylation of Nuclear and Cytosolic Proteins: Cloning and Characterization of a Unique O-GlcNAc Transferase with Multiple Tetratricopeptide Repeats. J. Biol. Chem. 1997, 272, 9308–9315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, J.; Hart, G.W. O-GlcNAc Profiling: From Proteins to Proteomes. Clin. Proteom. 2014, 11, 8. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Chen, Q.; Zhang, N.; Zhang, K.; Dou, T.; Cao, Y.; Liu, Y.; Li, K.; Hao, X.; Xie, X.; et al. Proteomic Profiling and Genome-Wide Mapping of O-GlcNAc Chromatin-Associated Proteins Reveal an O-GlcNAc-Regulated Genotoxic Stress Response. Nat. Commun. 2020, 11, 5898. [Google Scholar] [CrossRef] [PubMed]
- Hanover, J.A.; Krause, M.W.; Love, D.C. Bittersweet Memories: Linking Metabolism to Epigenetics through O-GlcNAcylation. Nat. Reviews. Mol. Cell Biol. 2012, 13, 312–321. [Google Scholar] [CrossRef] [PubMed]
- Groves, J.A.; Lee, A.; Yildirir, G.; Zachara, N.E. Dynamic O-GlcNAcylation and Its Roles in the Cellular Stress Response and Homeostasis. Cell Stress Chaperones 2013, 18, 535–558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ong, Q.; Han, W.; Yang, X. O-GlcNAc as an Integrator of Signaling Pathways. Front. Endocrinol. 2018, 9, 599. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Liu, T.-W.; Cecioni, S.; Eskandari, R.; Zandberg, W.F.; Vocadlo, D.J. O-GlcNAc Occurs Cotranslationally to Stabilize Nascent Polypeptide Chains. Nat. Chem. Biol. 2015, 11, 319–325. [Google Scholar] [CrossRef]
- Shi, J.; Tomašič, T.; Sharif, S.; Brouwer, A.J.; Anderluh, M.; Ruijtenbeek, R.; Pieters, R.J. Peptide Microarray Analysis of the Cross-Talk between O-GlcNAcylation and Tyrosine Phosphorylation. FEBS Lett. 2017, 591, 1872–1883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hart, G.W.; Slawson, C.; Ramirez-Correa, G.; Lagerlof, O. Cross Talk Between O-GlcNAcylation and Phosphorylation: Roles in Signaling, Transcription, and Chronic Disease. Annu. Rev. Biochem. 2011, 80, 825–858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, D.L.Y.; Hart, G.W. Purification and Characterization of an O-GlcNAc Selective N-Acetyl-Beta-D-Glucosaminidase from Rat Spleen Cytosol. J. Biol. Chem. 1994, 269, 19321–19330. [Google Scholar] [CrossRef]
- Chiaradonna, F.; Ricciardiello, F.; Palorini, R. The Nutrient-Sensing Hexosamine Biosynthetic Pathway as the Hub of Cancer Metabolic Rewiring. Cells 2018, 7, 53. [Google Scholar] [CrossRef] [Green Version]
- Hart, G.W.; Housley, M.P.; Slawson, C. Cycling of O-Linked β-N-Acetylglucosamine on Nucleocytoplasmic Proteins. Nature 2007, 446, 1017–1022. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Hart, G.W. Protein O-GlcNAcylation in Diabetes and Diabetic Complications. Expert Rev. Proteom. 2013, 10, 365–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caldwell, S.A.; Jackson, S.R.; Shahriari, K.S.; Lynch, T.P.; Sethi, G.; Walker, S.; Vosseller, K.; Reginato, M.J. Nutrient Sensor O-GlcNAc Transferase Regulates Breast Cancer Tumorigenesis through Targeting of the Oncogenic Transcription Factor FoxM1. Oncogene 2010, 29, 2831–2842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makwana, V.; Ryan, P.; Patel, B.; Dukie, S.A.; Rudrawar, S. Essential Role of O-GlcNAcylation in Stabilization of Oncogenic Factors. Biochim. Et Biophys. Acta Gen. Subj. 2019, 1863, 1302–1317. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Pang, Y.; Chang, T.; Bounelis, P.; Chatham, J.C.; Marchase, R.B. Increased Hexosamine Biosynthesis and Protein O-GlcNAc Levels Associated with Myocardial Protection against Calcium Paradox and Ischemia. J. Mol. Cell. Cardiol. 2006, 40, 303–312. [Google Scholar] [CrossRef] [PubMed]
- Umapathi, P.; Mesubi, O.O.; Banerjee, P.S.; Abrol, N.; Wang, Q.; Luczak, E.D.; Wu, Y.; Granger, J.M.; Wei, A.-C.; Gaido, O.E.R.; et al. Excessive O-GlcNAcylation Causes Heart Failure and Sudden Death. Circulation 2021, 143, 1687–1703. [Google Scholar] [CrossRef]
- Xu, W.; Zhang, X.; Wu, J.; Fu, L.; Liu, K.; Liu, D.; Chen, G.G.; Lai, P.B.; Wong, N.; Yu, J. O-GlcNAc Transferase Promotes Fatty Liver-Associated Liver Cancer through Inducing Palmitic Acid and Activating Endoplasmic Reticulum Stress. J. Hepatol. 2017, 67, 310–320. [Google Scholar] [CrossRef] [Green Version]
- Akella, N.M.; le Minh, G.; Ciraku, L.; Mukherjee, A.; Bacigalupa, Z.A.; Mukhopadhyay, D.; Sodi, V.L.; Reginato, M.J. O-GlcNAc Transferase Regulates Cancer Stem-like Potential of Breast Cancer Cells. Mol. Cancer Res. 2020, 18, 585–598. [Google Scholar] [CrossRef]
- Gloster, T.M.; Zandberg, W.F.; Heinonen, J.E.; Shen, D.L.; Deng, L.; Vocadlo, D.J. Hijacking a Biosynthetic Pathway Yields a Glycosyltransferase Inhibitor within Cells. Nat. Chem. Biol. 2011, 7, 174–181. [Google Scholar] [CrossRef] [Green Version]
- Ortiz-Meoz, R.F.; Jiang, J.; Lazarus, M.B.; Orman, M.; Janetzko, J.; Fan, C.; Duveau, D.Y.; Tan, Z.W.; Thomas, C.J.; Walker, S. A Small Molecule That Inhibits OGT Activity in Cells. ACS Chem. Biol. 2015, 10, 1392–1397. [Google Scholar] [CrossRef] [PubMed]
- Martin, S.E.S.; Tan, Z.W.; Itkonen, H.M.; Duveau, D.Y.; Paulo, J.A.; Janetzko, J.; Boutz, P.L.; Törk, L.; Moss, F.A.; Thomas, C.J.; et al. Structure-Based Evolution of Low Nanomolar O-GlcNAc Transferase Inhibitors. J. Am. Chem. Soc. 2018, 140, 13542–13545. [Google Scholar] [CrossRef] [PubMed]
- Loi, E.M.; Weiss, M.; Pajk, S.; Gobec, M.; Tomašič, T.; Pieters, R.J.; Anderluh, M. Intracellular Hydrolysis of Small-Molecule O-Linked N-Acetylglucosamine Transferase Inhibitors Differs among Cells and Is Not Required for Its Inhibition. Molecules 2020, 25, 3381. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.-W.; Zandberg, W.F.; Gloster, T.M.; Deng, L.; Murray, K.D.; Shan, X.; Vocadlo, D.J. Metabolic inhibitors of o-glcnac transferase that act in vivo implicate decreased o-glcnac levels in leptin-mediated nutrient sensing. Angew. Chem. Int. Ed. 2018, 57, 7644–7648. [Google Scholar] [CrossRef]
- Gross, B.J.; Kraybill, B.C.; Walker, S. Discovery of O-GlcNAc Transferase Inhibitors. J. Am. Chem. Soc. 2005, 127, 14588–14589. [Google Scholar] [CrossRef] [PubMed]
- Borodkin, V.S.; Schimpl, M.; Gundogdu, M.; Rafie, K.; Dorfmueller, H.C.; Robinson, D.A.; van Aalten, D.M.F. Bisubstrate UDP-Peptide Conjugates as Human O-GlcNAc Transferase Inhibitors. Biochem. J. 2014, 457, 497–502. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Tomašič, T.; Shi, J.; Weiss, M.; Ruijtenbeek, R.; Anderluh, M.; Pieters, R.J. Inhibition of: O-GlcNAc Transferase (OGT) by Peptidic Hybrids. MedChemComm 2018, 9, 883–887. [Google Scholar] [CrossRef]
- Weiss, M.; Loi, E.M.; Sterle, M.; Balsollier, C.; Tomašič, T.; Pieters, R.J.; Gobec, M.; Anderluh, M. New Quinolinone O-GlcNAc Transferase Inhibitors Based on Fragment Growth. Front. Chem. 2021, 9, 205. [Google Scholar] [CrossRef]
- OpenEye. OEDocking 3.3.1.2: OpenEye Scientific Software. 2020. Available online: https://docs.eyesopen.com/applications/oedocking/releasenotes/version3_3_1.html (accessed on 17 March 2022).
- Jug, G.; Anderluh, M.; Tomašič, T. Comparative Evaluation of Several Docking Tools for Docking Small Molecule Ligands to DC-SIGN. J. Mol. Modeling 2015, 21, 164. [Google Scholar] [CrossRef]
- Lazarus, M.B.; Jiang, J.; Gloster, T.M.; Zandberg, W.F.; Whitworth, G.E.; Vocadlo, D.J.; Walker, S. Structural Snapshots of the Reaction Coordinate for O-GlcNAc Transferase. Nat. Chem. Biol. 2012, 8, 966–968. [Google Scholar] [CrossRef] [Green Version]
- Vocadlo, D.J.; Alteen, M.G.; Gros, C.; Meek, R.; Cardoso, D.A.; Busmann, J.; Sangouard, G.; Deen, M.C.; Tan, H.-Y.; Shen, D.L.; et al. A Direct Fluorescent Activity Assay for Glycosyltransferases Enables Convenient High-throughput Screening: Application to O-GlcNAc Transferase. Angew. Chem. 2020, 59, 9601–9609. [Google Scholar] [CrossRef]
- Balsollier, C.; Pieters, R.J.; Anderluh, M. Overview of the Assays to Probe O-Linked β-N-Acetylglucosamine Transferase Binding and Activity. Molecules 2021, 26, 1037. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A Free Web Tool to Evaluate Pharmacokinetics, Drug-Likeness and Medicinal Chemistry Friendliness of Small Molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- OMEGA 2.5.1: OpenEye Scientific Software. Available online: Http://Www.Molcalx.Com.Cn/Oe-Docs/Omega/Releasenotes/Version2_5_1.Html (accessed on 13 December 2021).
- OpenEye. OEDocking 3.2.0: OpenEye Scientific Software. 2020. Available online: https://docs.eyesopen.com/applications/oedocking/releasenotes/OEDocking_version3.2.0.html (accessed on 17 March 2022).
- Lazarus, M.B.; Jiang, J.; Kapuria, V.; Bhuiyan, T.; Janetzko, J.; Zandberg, W.F.; Vocadlo, D.J.; Herr, W.; Walker, S. HCF-1 Is Cleaved in the Active Site of O-GlcNAc Transferase. Science 2013, 342, 1235–1239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kalé, L.; Schulten, K. Scalable Molecular Dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacKerell, A.D.; Bashford, D.; Bellott, M.; Dunbrack, R.L.; Evanseck, J.D.; Field, M.J.; Fischer, S.; Gao, J.; Guo, H.; Ha, S.; et al. All-Atom Empirical Potential for Molecular Modeling and Dynamics Studies of Proteins. J. Phys. Chem. B 1998, 102, 3586–3616. [Google Scholar] [CrossRef] [PubMed]
- Mackerell, A.D.; Feig, M.; Brooks, C.L. Extending the Treatment of Backbone Energetics in Protein Force Fields: Limitations of Gas-Phase Quantum Mechanics in Reproducing Protein Conformational Distributions in Molecular Dynamics Simulations. J. Comput. Chem. 2004, 25, 1400–1415. [Google Scholar] [CrossRef]
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I.; et al. CHARMM General Force Field: A Force Field for Drug-like Molecules Compatible with the CHARMM All-Atom Additive Biological Force Fields. J. Comput. Chem. 2010, 31, 671–690. [Google Scholar] [CrossRef] [Green Version]
- Vanommeslaeghe, K.; MacKerell, A.D. Automation of the CHARMM General Force Field (CGenFF) I: Bond Perception and Atom Typing. J. Chem. Inf. Modeling 2012, 52, 3144–3154. [Google Scholar] [CrossRef]
- Vanommeslaeghe, K.; Raman, E.P.; MacKerell, A.D. Automation of the CHARMM General Force Field (CGenFF) II: Assignment of Bonded Parameters and Partial Atomic Charges. J. Chem. Inf. Modeling 2012, 52, 3155–3168. [Google Scholar] [CrossRef] [Green Version]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A Smooth Particle Mesh Ewald Method. J. Chem. Phys. 1995, 103, 8577. [Google Scholar] [CrossRef] [Green Version]
- Ryckaert, J.P.; Ciccotti, G.; Berendsen, H.J.C. Numerical Integration of the Cartesian Equations of Motion of a System with Constraints: Molecular Dynamics of n-Alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef] [Green Version]
- Wolber, G.; Langer, T. LigandScout: 3-D pharmacophores derived from protein-bound ligands and their use as virtual screening filters. J. Chem. Inf. Modeling 2004, 45, 160–169. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Structure | Inhibition% | Name | Structure | Inhibition% | Name | Structure | Inhibition% |
|---|---|---|---|---|---|---|---|---|
| Vs-1 |  | 41% | Vs-5 |  | 54% IC50 75 ± 14 µM | Vs-10 |  | 19% |
| Vs-4 |  | 18% | Vs-50 |  | 23% | Vs-11 | 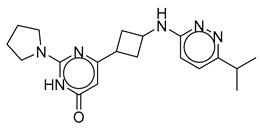 | 22% |
| Vs-3 | 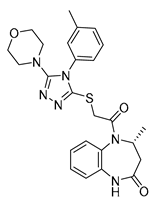 | 32% | Vs-51 | 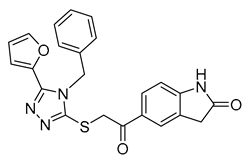 | 58% IC50 68 ± 15 µM | Vs-37 | 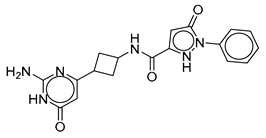 | 43% |
| Vs-13 | 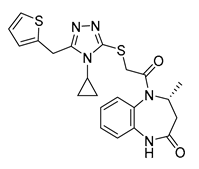 | 11% | Vs-9 | 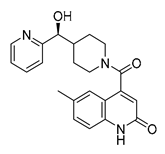 | 38% | Vs-19 | 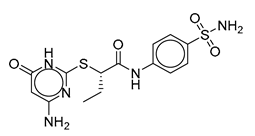 | 15% |
| Vs-24 | 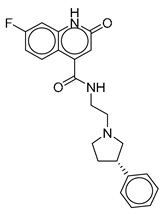 | 26% | Vs-56 | 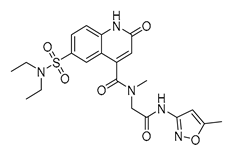 | 15% | Vs-68 | 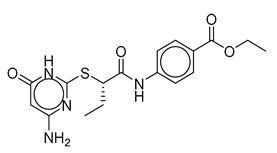 | 24% |
| Vs-30 | 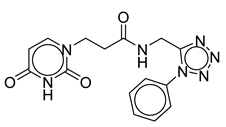 | 33% | Vs-32 | 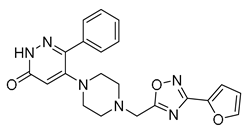 | 26% | Vs-83 | 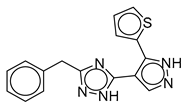 | 56% IC50 88 ± 16 µM |
| OSMI-4a | 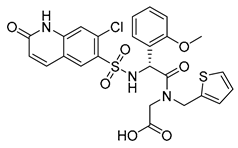 | 100% IC50 0.3 ± 0.1 µM | OSMI-4b | 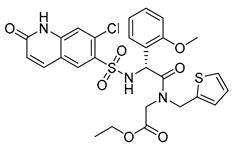 | 100% IC50 0.06 ± 0.02 µM |
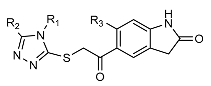
| Name | R1 | R2 | R3 | IC50 (µM) ± SD |
|---|---|---|---|---|
| 1 (Vs-51) | 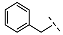 | 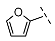 |  | 7 ± 5 |
| 2 |  | 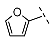 | 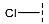 | 92 ± 25 |
| 3 | 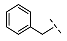 | 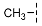 | 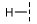 | 29 ± 4 |
| 4 | 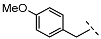 | 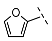 | 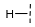 | 33 ± 16 |
| 5 | 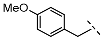 | 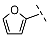 | 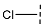 | 39 ± 6 |
| 6 | 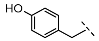 | 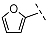 | 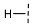 | 21 ± 16 |
| 7 | 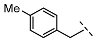 | 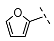 | 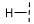 | 103 ± 9 |
| 8 | 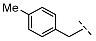 | 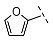 | 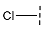 | 103 ± 18 |
| 9 | 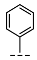 | 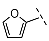 | 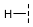 | 16 ± 1 |
| 10 |  | 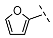 | 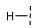 | 36 ± 3 |
| 11 | 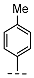 | 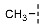 | 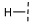 | 27 ± 13 |
| 12 |  |  |  | Not soluble |
| 13 | 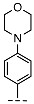 | 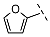 | 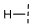 | No inhibition |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Loi, E.M.; Tomašič, T.; Balsollier, C.; van Eekelen, K.; Weiss, M.; Gobec, M.; Alteen, M.G.; Vocadlo, D.J.; Pieters, R.J.; Anderluh, M. Discovery of a New Drug-like Series of OGT Inhibitors by Virtual Screening. Molecules 2022, 27, 1996. https://doi.org/10.3390/molecules27061996
Loi EM, Tomašič T, Balsollier C, van Eekelen K, Weiss M, Gobec M, Alteen MG, Vocadlo DJ, Pieters RJ, Anderluh M. Discovery of a New Drug-like Series of OGT Inhibitors by Virtual Screening. Molecules. 2022; 27(6):1996. https://doi.org/10.3390/molecules27061996
Chicago/Turabian StyleLoi, Elena M., Tihomir Tomašič, Cyril Balsollier, Kevin van Eekelen, Matjaž Weiss, Martina Gobec, Matthew G. Alteen, David J. Vocadlo, Roland J. Pieters, and Marko Anderluh. 2022. "Discovery of a New Drug-like Series of OGT Inhibitors by Virtual Screening" Molecules 27, no. 6: 1996. https://doi.org/10.3390/molecules27061996
APA StyleLoi, E. M., Tomašič, T., Balsollier, C., van Eekelen, K., Weiss, M., Gobec, M., Alteen, M. G., Vocadlo, D. J., Pieters, R. J., & Anderluh, M. (2022). Discovery of a New Drug-like Series of OGT Inhibitors by Virtual Screening. Molecules, 27(6), 1996. https://doi.org/10.3390/molecules27061996