Abstract
Betanin and betanidin are compounds with extensive interest; they are effectively free radical scavengers. The present work aims to elucidate the differences between the mechanism of the antioxidant activity of betanin, betanidin, and their respective C15-epimers. Shape Theory establishes comparisons between the molecules’ geometries and determines parallelisms with the descriptors BDE, PA, ETE IP, PDE, and infrared spectra (IR) obtained from the molecule simulations. Furthermore, the molecules were optimized using the B3LYP/6-31+G(d,p) protocol. Finally, the molecular docking technique analyzes the antioxidant activity of the compounds in the complex with the therapeutic target xanthine oxidase (XO), based on a new proposal for the geometrical arrangement of the ligand atoms in the framework of Shape Theory. The results obtained indicate that the SPLET mechanism is the most favorable in all the molecules studied and that the first group that loses the hydrogen atom in the four molecules is the C17COOH, presenting less PA the isobetanidin. Furthermore, regarding the molecular docking, the interactions of these compounds with the target were favorable, standing out to a greater extent the interactions of isobetanidin with XO, which were analyzed after applying molecular dynamics.
1. Introduction
Antioxidants are chemicals that inhibit or delay unwanted oxidation reactions. They offer their electrons to free radicals and thus prevent cell damage. About the substrate subjected to oxidation, the antioxidant protects it from irreversible danger, preventing its oxidative conversion through reactive species. Thus, when antioxidant substances are present in food, they delay, control, or inhibit oxidation and deterioration of food quality [1,2,3]. Researches in recent years have focused on the search for species with high free radical-scavenging activity. In this line, there is evidence in the literature that some natural colorants, such as betalains, are compounds that act by inhibiting the action of free radicals that contribute to the appearance of several degenerative diseases [4]. As for pharmaceutical, cosmetic, and food applications, betalains are stable in a pH range of 3.5 to 7. This characteristic gives them the importance of being used in low acid and neutral products [5,6].
Betalains are derived from betalamic acid and are soluble in water. They consist of two groups of structures: betaxanthins, with yellow-orange coloration, and betacyanins, responsible for the red-purple color. Betaxanthins are immonium conjugate of betalamic acid with amino acids or amines, whereas betacyanins are immonium conjugate of betalamic acid with cyclo-DOPA, traditionally glycosylated. Although tyrosine is the precursor of this last element, in higher plants, the precursor of betalamic acid is 4,5-seco-DOPA, which constitutes the basic structure of the betalain skeleton [7]. Figure S1 of the Supplementary Material shows the essential structure representation of betacyanins.
The substitution via glycosylation or acylation of one or both hydroxyl groups located at positions 5 and 6 of this compound results in different betacyanin derivatives. Predominantly 5-O-glycosides and glycosylation of 5-O-glycoside and esterification with hydroxycinnamic acids are also common [7]. Thus, Betanin (betanidin-5-O--glucoside) is the most widespread betacyanin among plants; for example, red beet contains a concentration of betanin in the range of 300–600 mg/kg, and presents concentrations, in smaller amounts, of isobetanin (See Figure S2 in Supplemetary Material), betanidin, and betaxanthins [8,9].
Hydrolysis of Betacyanin produces Betanidin, and the epimer at C-15 produces Isobetanin or a mixture of the two aglycones. Moreover, their conjugated double bond is associated with their color, where the maximum light absorption at 540 nm is for the red Betacyanins [10].
When acting in biological systems, antioxidants produce changes in cellular elements’ chemical composition or structure. For instance, oxidation of xanthine takes place at the molybdopterin (Mo-pt) center, and the electrons thus introduced are rapidly distributed to the other centers by intramolecular electron transfer. The crystal structure of salicylate-XO was reported and provided for structure-based docking studies [11,12,13,14,15].
Several studies were developed and demonstrated the efficacy of betalains as free radical scavengers. For instance, it was demonstrated that the antioxidant capacity of betanin is pH-dependent; these results were analyzed from the calculation of phenolic OH homolytic bond dissociation energy (BDE) and ionization potential (IP). These parameters were obtained from DFT B3LYP/6-311+G** or B3LYP/6-31G** quantum-mechanical calculations level of theory [16].
The present work aims to extend the research in this line by approaching the modeling of betanin (Bn), isobetanin (IsoBn), betanidin (Bd), and isobetanidin (IsoBd) to point out how marked are the differences between the mechanism of the antioxidant activity of the molecules in question. The main reaction mechanisms HAT (hydrogen atom transfer), SPLET (sequential proton-loss electron transfer), and SET-PT (sequential electron-proton transfer) are analyzed in an aqueous medium to demonstrate which of them, from the thermodynamic point of view, is the most favorable mechanism for each molecule and to establish differences between the compounds studied concerning these mechanisms. For the achievement of this purpose, the named Shape Theory have used to establish similarities and dissimilarities between the molecules geometry and based on that to determine correspondences with the descriptors: BDE (bond dissociation enthalpy), related to the HAT mechanism, PA (proton affinity) and ETE (electron transfer enthalpy) associated with the SPLET mechanism, IP (ionization potential) and PDE (proton dissociation enthalpy) related to the SEP-PT mechanism. The theoretical study (DFT) achieves from the parent molecules’ structural geometries and their respective radical forms. Frontier orbitals (HOMO/LUMO) were also studied. Antioxidant activity of the betalains via implementing a formalism based on the distances between the molecules in the named Shape Theory, a subject included in mathematics and statistics, was analyzed.
Shape theory is a new area for comparing and estimating the average shape and variability between objects summarized by a finite number of reference points; a large list of applications can be found in various sciences [17]. The Gaussian statistics was used extensively [17,18]; now, robust models for shape theory are available for several transformations (affine, polar, SVD, QR) and for real normalized division algebras (real, complex, quaternion, and octonion) [19,20,21,22,23,24,25]. A crucial concept for our purposes, includes the Riemannian distance (RD) and was applied recently in the following fields: spectroscopic analysis [26] and rocking curves [27]; stochastic search of molecules [28]; and remote sensing of burning satellite products [29].
In the context of chemistry and the present paper, [28] sets the shape theory as a robust discrimination method for molecules, since it uses geometric invariances based on rigid Euclidean motions and measures the distances of molecules in equivalence class spaces. Instead of using the redundant methods based on random search and nongeometrical meaning changes of molecules for optimization, [28] proved the theoretical and computational efficiency of shape theory for finding non repeated equilibrium structures of nanoclusters, using their Riemannian geometry properties and considering the multiple local minima of the potential energy surface.
In the present work, we implement the shape theory in two new ways: firstly, as a discriminant method of optimized molecules and their antioxidant capacity, and secondly, as an alternative method for molecular docking based on the Riemannian geometry.
Another aspect considered is the set of IR spectra obtained from the simulations of the molecules under study, which allowed understanding the vibrational mechanisms of the molecules associated with their respective frequency and their correlation with the molecular descriptors associated with the antioxidant capacity.
IR spectra are a way to assess compounds’ chemical stability and physical capacity [30], with rapid quantification allowing the collection of specific molecular fingerprints from samples [31]. These capabilities and the shape theory allow a complete characterization of the compounds [26].
2. Methods
2.1. Dft Studies
The capacity to scavenge free radicals (R) through a hydrogen atom transfer (H) from phenolic hydroxyl groups to the free radical, either in one step or in two steps, was mainly approached through three mechanisms (i) HAT, a direct Hydrogen Atom Transfer between the antioxidant and the active radical (Equation (1)), (ii) SPLET (Sequential Proton Loss Electron Transfer): deprotonation of the antioxidant which resulting anion, followed by an electron transfer to the active radical, the final step is protonation of the anion of the active radical (Equation (2)) and (iii) SET-PT (Single Electron Transfer Proton Transfer): an electron is transferred from the antioxidant to the active radical, yielding a cation-radical and an anion, followed by proton transfer from the cation-radical to the anion (Equation (3)). These mechanisms may occur parallel but with different rates [11,29,32,33,34,35].
Descriptions that characterize each one of the mechanisms are: the bond dissociation enthalpy (BDE) characterizes the HAT mechanism, the SLEPT mechanism via the proton affinity (PA), and the electron transfer enthalpy (ETE) and SET-PT mechanism via the ionization potential (IP), and the proton dissociation enthalpy (PDE).
The following equations calculate the descriptors:
where Hx is the enthalpy of the system at 298.15 K and a pressure of 1 atm.
The package Gaussian 09 conducted the calculations [36]. In addition, PubChem’s repository facilitates the download files of the structures of Betanin and Betanidin used for the DFT studies, and from them, the stereoisomers were obtained [37]. The free radical-scavenging activity can be determined via studies with different levels of theory and have reported that the computational level DFT B3LYP/631+G(d,p) has allowed analyzing the system quite well at a low computational cost [11,16,32,38].
The parent molecules and their respective radicals, radical cations, and anions were optimized at the B3LYP level of theory and with the 6 − 31 + G(d,p) basis set without any geometry constraints [39]. The radical or ionic structures were optimization starting from the parent molecules’ optimized geometries, applying the available method (UB3LYP(6 − 31 + G(d,p). The systems involving open-shell species are sensitive to spin contamination. Spin contaminations of radicals showed a value between 0.76–0.78. After annihilating the first spin contaminant, they dropped to the correct value of 0.75. Therefore, spin contamination should not bias found reaction enthalpies.
The solvation via an implicit method with Integral-Equation-Formalism Polarizable Continuum Model (IEFPCM) as the model [40,41]. At the calculated vibrational frequencies, no imaginary frequency modes were obtained, confirming the stability of the optimized structures.
The numerical values-calculated to determine the most favorable antioxidant mechanism from the Equations (4)–(8) of the theoretical descriptors: BDE, PA, ETE, IP, PDE, and the energy values corresponding to the frontier molecular orbitals HOMO/LUMO. All enthalpies-calculated at 298 K and 1.0 atm. Previous reports provide the enthalpies in the water of the hydrogen atom, proton, and electron: the enthalpy of the proton H(H+) is −259.00 kcal/mol, the enthalpy of the electron H(e−) is −55.61 kcal/mol, and the enthalpy of the hydrogen atom H(H−) is −314.65 kcal/mol [32,42]. The IR-simulated spectra via the Gaussian 09 package for the parent molecules Bn, Bd, IsoBn, and IsoBd. The energetic study of the loses the hydrogen atom mechanisms was conducted based on IR spectra and comparing the group frequencies and the fingerprint frequencies [43].
2.2. Shape Spaces
In the context of chemistry, [28] define the shape of a molecule as all geometric information that remains after eliminating rotation, translation, scaling, or reflection of the cluster. For a mathematical treatment, molecules of k atoms in three dimensions are writing as kx3 matrices. After removing translation, rotation, scaling, or reflection, the shape of the molecule is just a point in a matrix space. The so-called Riemannian distance (RD) is one of the most important descriptors in shape theory. [28] and the present work uses the RD to find the similarity or dissimilarity of two molecules from a given optimization process. In that sense both molecules can be seen as two points in a hypersphere, understood as the space of equivalence classes of invariant matrices under translation and scaling [17,18]. Thus, the RD is an angle in hypersphere, ranged from 0 (equal molecules) to π/2 (extremely different molecule), and it serves as an excellent nonredundant descriptor (from the Riemannian geometrical meaning) that classifies molecules. The RD is also sensitive to small changes of the atom positions, which is important for measuring subtle discrepancies in the chemical processes of antioxidant activity.
The first application of shape theory in the present work involves the characterization of the Riemannian distances between deprotonated molecules and the respective parent molecules.This discrimination is studied from two perspectives: (1) we compare the complete deprotonated molecules to observe the global distance due to the effect of antioxidant activity; (2) we study the discrepancy in the subspaces defined by each chemical element, as an unusual geometric property implicit in the optimization process. Both geometric measures, global and partial, are related to other purely chemical descriptors.
We make the calculations with own algorithms implemented in R, a free software environment for statistical computing and graphics [44]; the routines collect the Shape Theory concepts provided by [17], and the chemical applications inspired by [28].
2.3. A Shape Theory Docking Method and Molecular Dynamics
The second method derived from shape theory, involves a new docking approach. It considers the intrinsic and the invariant Riemannian geometrical information of the ligands. The molecular docking analysis pursues a model for the protein-ligand complex, in order to elucidate the interactions with the amino acid residues in the active site that allows the inhibition of the XO enzyme. Then, the supporting description via RD can be used for as a suitable explanation of the antioxidant capacity of these compounds.
The new molecular docking method was also analyzed by proposing a preferential system for the ligand coordinates, which is invariant under similarity transformations (rotations, scaling and translations). It considers the maximal spatial variability of the ligand. Although, the spectral decomposition theorem can provide one preferential system based on variability, the strict order of the atoms can be perturbed by reflections involved in the rotation matrices given by the principal components method. This method can produce molecules without a chemistry framework. Our well-behaved invariant method generalizes Bookstein’s coordinates of shape theory [17]. In our case, the method proposes the two most distant ligand atoms as the baseline. Then, the new transform is rotated in such way that maximal variability lies of the x-axis. A second rotation is performed to the second perpendicular maximal variability is on the y-axis. In a similar manner a third rotation places the new ligand in such way that the third orthogonal variability remain in the z-axis. This invariant ligand is called the template. We complete the method by proposing nine ligands which are rotations (90, 180, and 270 degrees) of the template, originally placed in the xy, xz, and yz planes. Finally, we perform the molecular docking of the original template a the nine new ligands inside the given protein.
The new method avoids the usual random search which does not distinguish equivalent classes of ligands. The usual methods are redundant allowing repetitions with no meaningful new geometrical and invariant information.
We explore the probable interactions of the union of the compounds with XO via molecular docking with the AutoDock Vina program [45,46]. For docking studies, the X-ray crystal structure of bovine XO in complex with salicylate (PDB ID code 1FIQ, Protein Data Bank: http://www.rcsb.org/pdb accessed on 16 December 2021) was used [47] in 21 June 2021. For the development of this study, we selected the enzyme xanthine oxidase XO, which is present in some mammalian tissues, in various species, from bacteria to humans. Its importance lies in the fact that it produces reactive oxygen species either alone or in combination with other enzymes and compounds. XO belongs to the group of oxidoreductases that catalyzes the oxidation of hypoxanthine to xanthine and xanthine to uric acid in the last stage of the metabolic degradation of purines, with the production of reactive oxygen species, as mentioned. This phenomenon is related to various pathological states such as chronic heart failure, cardiomyopathy in diabetes, and gout [12,13,33].
Ligand preparation and receptor one include the remotion of the A- and B-chains of the protein and all small molecules. First, we added the polar hydrogen atoms to the macromolecule file, and then a grid box was defined to enclose the active site with dimensions of 20 Å × 20 Å × 20 Å, and a center set at the point x = 26, y = 10, z = 118. Finally, the final docked structure for each compound was analyzed using the VMD visualization program [48]. It compares the results with those obtained from docking of the Febuxostat inhibitor with the XO active site [15,33].
We carried out the molecular dynamics (MD) study to simulate the local conformational changes in the complex resulting from the union between the highest-ranked compound in molecular docking and the XO enzyme. The procedure allows both the ligand and all the protein residues to reach a steady-state conformation.
The NAMD (Nanoscale Molecular Dynamics) [49] and CHARMM-GUI (Chemistry at Harvard Macromolecular Mechanics- GUI) [50] were used regarding computer tools. The CharmM36 force field was used, integrated into the graphical user interface for CHARMM, which can calculate parameters of bound or unbound molecules. On the other hand, the TIP3P water model was chosen. Then an environment was created to put the molecule together with the water. For this, the protein was placed in an orthorhombic system with the number of water molecules needed to solvate the box. The simulation was carried out in 10 ns. For the molecular dynamics, we were using the best conformation obtained from the molecular docking stage. After doing the docking, we can start with the Dynamics Molecular. On the other hand, to better analyze the complexes formed in the computational study, the VMD graphic viewer was used to obtain the last trajectory (frame) corresponding to 10 ns and the most energetically stable of the molecular dynamics carried out. Analyzed the trajectories obtained, and the interactions with the enzyme were obtained.
Another novelty of the new shape theory method proposed here, connects the Riemannian distance with results of docking summarized by the protonic affinity. This important fact provides an inusited relation with the geometry properties emerging from the Riemannian invariance of the protein-ligand complex.
3. Results and Discussion
3.1. DFT Studies
The optimized Bn and C15 epimer (IsoBn), Bd and C15 epimer (IsoBd) structures feature three structural hydrogens that could be released from the molecule, one between the two hydroxyl groups attached to C5 and C6. The other three are formed: two by the oxygen atom of each carboxyl group at C2, C15, and C17 and the third with the hydrogen-bonded to N16. The carboxyl group of the Bd and Bn has an equatorial position, and the epimers in the carboxyl group have an axial position (Supplementary Information, Figure S1). After preparing the molecules, according to the required calculations, which includes O-H (or NH) group in the compound possesses the extractable or directed hydrogen for the radical attack, they were optimized to obtain the lowest energy structures for BnC2, BnC6, BnC15, BnC17, BdN16, BdC2, BdC5, BdC6, BdC15, BdC17, and BdN16, (see Figure 1C and Figure S1), in same way for IsoBn and IsoBd. The antioxidant capacity of these compounds is highly affected by the pH of the medium, but in aqueous solutions, they are stable in a pH range of 3.5 to 7 [16,38,51,52]. To explain the antioxidant activity, we calculated the descriptors BDE, IP, PDE, PA, and ETE on each compound; see Table 1.
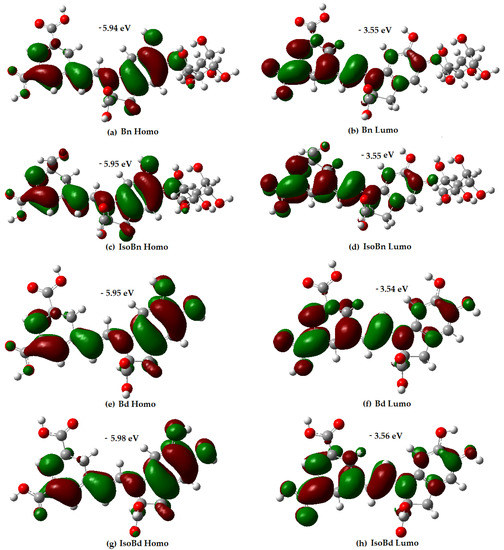
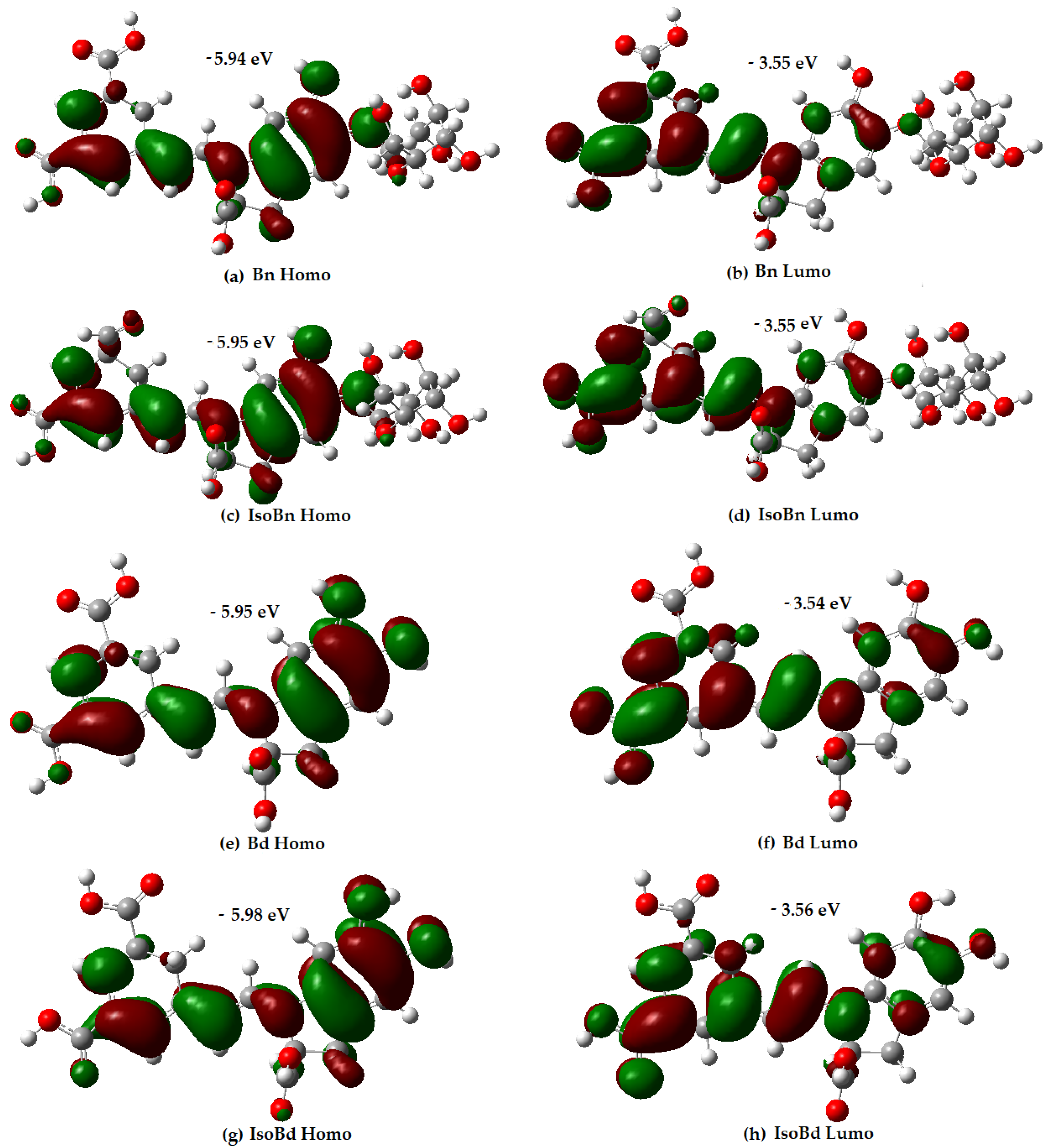
Figure 1.
Energy and distribution of HOMO and LUMO for Bn, IsoBn, Bd, IsoBd.

Table 1.
Calculated BDE, PA, ETE, IP, and PDE values for compounds studied. Data in kcal/mol, at B3LYP/6-31+G(d,p) level of theory in water.
The mechanism of scavenging free radicals can predict from the values of the descriptors of the O-H (or N-H) groups in the compounds. By comparing these descriptor values (the PA value is lower than for the rest of the descriptors), the most thermodynamically probable reaction in a polar medium such as water is sequential electron transfer with loss of protons (SPLET). For phenolic antioxidants, experimental and theoretical studies also confirmed the importance of the SPLET mechanism in polar solvents [53].
According to reported data, the carboxyl group is the first in loses the hydrogen, then the hydroxyl group, and finally the N16 group [16,52]. Other authors reported the computation of proton affinity in an aqueous medium. They determined that the ascending order of deprotonation of the Bn and Bd molecules is C17COOH (C17), COOHC15 (C15), and COOHC2 (C2) for Bn and Bd. The PA of the first group with respect to the second and third for Bn is 5.4 Kcal/mol and 9.6 Kcal/mol respectively; for Bd is 5.3 Kcal/mol and 6.8 Kcal/mol, respectively [54]. In a vacuum, the order of deprotonation is C17, C2, and C15 for Bn and Bd [55]. The C17COOH group loses the first hydrogen before all compounds, followed by the hydrogen belonging to the C15COOH (C15) group, and finally the hydrogen corresponding to the C2COOH (C2) group. This is true for most of the compounds except for IsoBn that the value of PA is slightly lower for the C2 group than groups C15, according to the results cited. Comparing the PA values of the C17 group with C15 for all molecules, the range is 2.1 to 3.6 Kcal/mol, and C2 is 2.1 to 3.8 Kcal/mol. The differences between the results obtained and those previously reported are of the same order [54,55]. The lowest PA value corresponds to the hydrogen of the COOH group (C17) attached to the non-chiral carbon of the Bd isomer, followed by the nonchiral group of the Bn isomer. The PA value in kcal/mol of the compounds increases in the following order: IsoBd < IsoBn < Bn = Bd, this indicates that the antioxidant capacity seems to be better at C17 of the Bd epimer. The antioxidant capacity of these compounds includes the next step of the mechanism, related to the descriptor ETE (Equation (2)). The compounds’ ETE value in kcal/mol increases in the following order: Bn < IsoBn < Bd < IsoBd. The fact that IsoBd loses hydrogen easier than the rest and at the same time corresponds to the highest value of the ETE descriptor could imply that the anion form is long-living, which would allow various transformations, thus being able to lose antioxidant capacity [32]. However, in general terms, the difference between the ETE values and all the descriptors is not very wide, and as discussed above, the most favorable antioxidant mechanism in water is the SPLET mechanism for all the compounds studied, IsoBd is the first compound to loss of hydrogen.
The compounds’ IP descriptor in kcal/mol increases as follows: Bn < IsoBn < IsoBd < Bd, implying that Bn possesses the best electron-donating ability [34].
The antioxidant capacity can be evaluated by different in vitro assays, either HAT, SEP assays, or a mixture of both. It depends on the chemical reaction considered and the structure under study [56,57].
Structurally, betalains are immonium derivatives of betalamic acid that contain an aromatic amino compound which can stabilize radicals. This stabilization is tightly linked to betalain’s electron donation ability.
As evidenced by Gandía–Herrero, et al. [51], the antioxidant activity of betalain molecules depends on their chemical structure and can be determined via Mixed-Mode Assays Trolox equivalent antiradical capacity (TEAC) value of betalains without aromatic resonance, charge, or hydroxy groups is 2.4 ± 0.1 units. This value is about 1.8 ± 0.1 units for betalains with charge and no aromatic resonance and increases to 2.8 ± 0.4 units for compounds carrying an aromatic ring. The TEAC value reaches 4.1 ± 0.3 units for betalains having a six-membered benzene ring fused to a five-membered nitrogen containing ring, forming an indoline group. The presence of a charged amino group ion seems to decrease the antiradical activity. Interestingly, no effect related to the carboxylation of these pigments was noted [51]. Specifically the Bn has reached a TEAC of 4.7 ± 0.3 [58].
More recent information presents the assessment of the antioxidant capacity of Bn through four assays, the HAT type: ORAC (10.52), the SET type: FRAP (11.82) and Mixed-Mode Assays are: TEAC (15.35) and DPPH (25.42) [59].
3.2. Frontiers Molecular Orbitals (HOMO/LUMO)
HOMO represents the ability to donate electrons, and LUMO the ability to accept electrons. The distribution of the Frontiers molecular orbitals (HOMO/LUMO) in a molecule is related to its antioxidant capacity, as it allows prediction of the most likely sites at which a compound can be affected by free radicals [34].
Analysis of the orbital distribution in Figure 1 shows that the HOMO and LUMO compositions of the compounds exhibit not very marked differences, and they are localized over almost the entire molecule, without any contribution of the glycoside motifs.
The distribution electron density of the LUMO for Bn and its C15-epimer is in higher proportion in the C17COOH group and the dihydropyridine ring concerning the aromatic ring. Regarding the HOMO compositions, the C17COOH group presents delocalization; the aromatic ring shows a slightly different delocalization than the dihydropyridine ring. The results express that the C17COOH group and the dihydropyridine ring are the most likely sites for radical attack for all molecules. The most probable sites for deprotonation are C17, C15, and C2 in all compounds [60]. Additionally, molecules with higher energy HOMO orbital have a stronger electron-donating ability [34]. In Figure 1, Bn has the highest HOMO energy (−5.94 eV), followed by IsoBn and Bd (−5.95 eV), lastly, IsoBd (−5.98 eV). It shows that Bn has the strongest electron-donating ability among the compounds studied, corresponding to the obtained IP value.
3.3. IR Analysis
Figure S4 of the Supplementary Material shows the evolution of parent molecules and C15-epimers, corresponding to a rotation. The magnitude of the interactions present is explained exclusively by kinetic effects. That is why the Bn and its Iso have glucose. It is a more significant vibrational load. There is a region of vibration of the glucose in the range of 905.10 cm to 849 cm. The first hydrogen that is lost in the reaction is affected by the number of additional bonds that the molecule has; that is to say, deprotonation is facilitated to the extent that the molecule has a greater number of atoms, which is correlated in lower energy in IR’s intensities with the following order Bn < IsoBn < Bd < IsoBd.
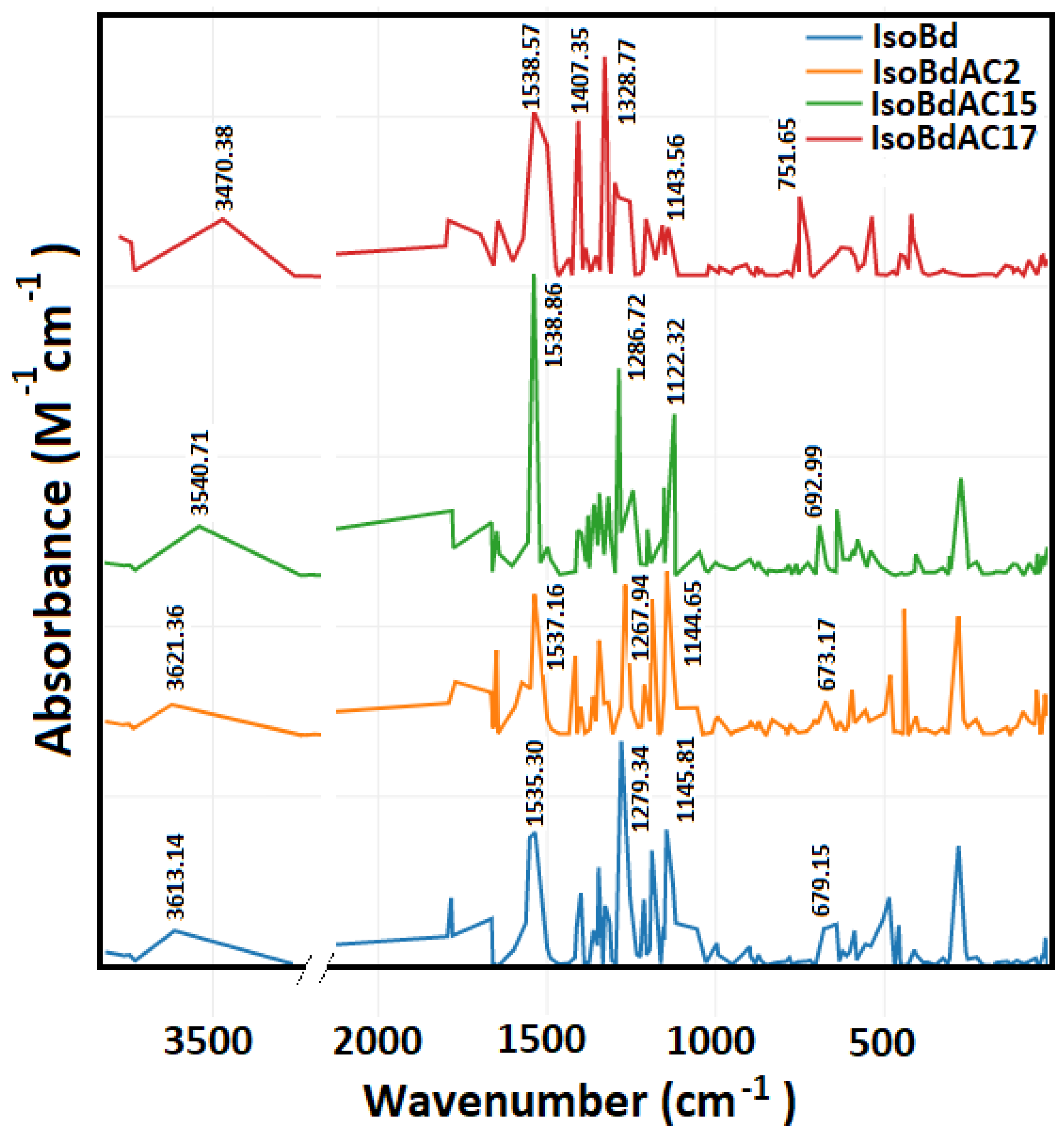
In Figure 2, we can see the IR spectrum of the anions C2COOH, C15COOH, and C17COOH of IsoBd. Intense peaks at 1538.57 (b,s) cm−1, 1538.86 (s) cm−1, and 1537.16 (s) cm−1 belong to C=C stretch mode for C2COOH, C15COOH, and C17COOH complex, this behavior at phenyl compounds with pair of bands only for the parent IsoBd and anion C17COOH has the origin of the degenerate pair of fundamentals in benzene. The overall displacement in benzene results in a change in dipole moment [61]. At 1407.35 (s) cm−1, the C17COOH presents CO stretch mode more attenuated in other anions. The whole of more intense peaks also can see at 1328.77 (s) cm−1, 1286.72 (s) cm−1 and, 1267.94 (s) cm−1 for C17COOH, C15COOH, and C2COOH anions, respectively, belong to ( C−N) asymmetric stretching. The bands observed at 1143.56 (w) cm−1, 1122.32 (m) cm−1, and 1144.65 (m) cm−1 for C17COOH, C15COOH, and C2COOH complex could be attributed to the C=C stretch mode; at C17COOH anion, their intensity is attenuated that could be indicated loss energies vibrations and a reconfiguration of the kinetic. Furthermore, it observed a bathochromic shift for the anion C17COOH in all absorptions frequencies implying an increase in velocity the loss of hydrogen [16,62,63]. This behavior correlated the same outcomes with the Shape Theory and DFT simulations. Out-of-plane ( C−H) bend (aromatic ring), 751 (b,m) cm−1 for C17COOH. The fingerprint area near 1500 cm−1 is a good characterization of the deprotonated (loss hydrogen) change. According to the literature, the range of 1700–1400 cm−1 contains the series of maximum characteristics of COO- and the amine groups [64]. The amide N atoms have a broadband region of 3700 to 3200 cm−1 (NH); with an appropriate deconvolution, it is possible to find peaks corresponding to the coordination of deprotonated peptides of complex or ligands with these characteristics [64,65]
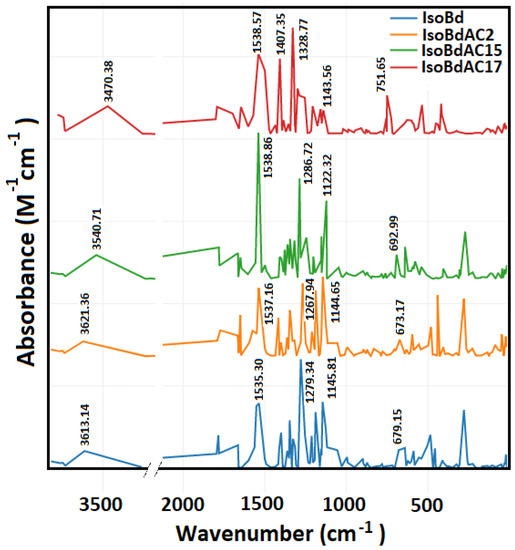
Figure 2.
Parent Isobetanidin (IsoBd) and their anios IsoBdAC2, IsoBdAC15, and IsoBdAC17.
3.4. Shape Space Distances
The molecules studied in the present work showed marked variations the antioxidant capacity. We found that the hydrogen is lose firstly in the C17COOH group in all the studied compounds. Similar researches for Bd, Bn, and IsoBd show that the first deprotonation occurs at C17COOH, C2COOH and C15COOH [38] and in [54] occurs at C17COOH, C15COOH and C2COOH. The last results agree with the present study for the case of Bn and Bd. However, an explicit relation with the geometry of the molecules could be found and a parametrization of chemistry descriptor could be related with the invariant shape theory analysis. Table 2 shows that this is possible. The values of the PA and RD (global and by subspaces) are described for all of the studied compounds and anions. We note that the global DR preserves the order of the PA descriptor. Concerning the PA, the order between each compound and its respective C15-epimer is conserved, indicating that the most stable deprotonated molecule is also the closest to the parent compound.
Table 2.
RD between study compounds and their respective anions and PA values.
When the RDs are compared among clusters of the same molecule, a similar conclusion is obtain. The RDs by subspaces define stages of verification, given that the partial distance inherits the global distance of the complete molecule, which is expect harmonically balanced and optimized in all the group elements.
This surprising result complements the usual analysis defined by the physical and chemical descriptors. The intrinsic Riemannian geometric properties of the optimized molecule are related with the first deprotonated group and the most stable molecule.
In this case, the most consistent RD (global and partial) reveals that C17COOH is the first deprotonated group in all the four compounds. These molecules exhibit the lowest proton affinity and at the same time show the lowest RD of that anion to the corresponding parent molecule.
3.5. New Shape Molecular Docking
This section aims to visualize the possible interactions of the selected compounds in complex with the XO enzyme and to elucidate the potential mechanism of action of this group of molecules in preventing ROS formation. The literature widely reported the antioxidant capacity of betalains [16,39,51,52]. Additionally, other studies [12,13,66] showed that in a reducing half-reaction, the substrate is hydroxylated oxidatively at the molybdenum (Mo) center, whose deprotonation can be facilitated by a conserved glutamate residue (Glu1261 in the bovine enzyme). Xanthine is formed from the hydroxylation of hypoxanthine at C-2 (see Figure S5) and is then converted to uric acid by hydroxylation at C-8 (see Figure S5), which involves the generation of ROS. If the XO enzyme is inhibited, it could be a way to treat or prevent diseases related to uric acid accumulation [67]. The XO inhibitor considered as a reference in this study is the drug Febuxostat, which is not affected by the redox status of the enzyme and the interaction with XO. It is also very potent in antioxidant activity and is very well coupled to the XO pocket that obstructs substrate binding [14]. We explore the probable interactions of the union of the compounds with XO via molecular docking in the binding sites of the protein made up of the amino acid residues within and around the binding pockets of the hypoxanthine and molybdopterin cofactor, framed within the C chain and delimited by the grid box defined to enclose the active site according to the methodology. This complex with XO yielded binding energy (ΔG) of −9.0 kcal/mol. The reference showed hydrogen bonding interactions with the amino acids Asn768, Arg880, and Thr1010. In addition, it presented hydrophobic interaction with amino acids Phe1009, Phe1013, Leu873, Val1011, Leu1014, and Ala1079. Other hydrophobic bonds shown were with Ala1079 and Phe914, respectively. The above agrees with results found in the literature [15,33,67].
Regarding the studied molecules, Bn, Bd, and their respective C15-epimers, the docking results showed that IsoBd and Bn could be more active since their a lower ΔG value, which corresponds to −8.0 kcal/mol and −7.5 kcal/mol, respectively, while IsoBn reached a value of −7.0 kcal/mol and Bd −6.9 kcal/mol. These molecules formed stable complexes with XO. The interaction of the compounds with the target was favorable through nonbond interactions, primarily by hydrogen bonding and in smaller proportion by hydrophobic and electrostatic interactions (Figure 3). For example, for Bn, a hydrogen bond (2.136 Å) was formed between the hydrogen atom of the carboxylic acid of the non-chiral carbon of Bn and the oxygen atom of the residue Glu802. It occurs similarly between the oxygen atom of the same residue with the hydrogen atom of the carboxylic acid of the chiral carbon of Bn (2.934 Å). We observe Hydrogen bonding type interactions between hydrogens of the glucose of Bn and the Glu879 residue with distances of 2.667 Åand 1.971 Å, respectively.
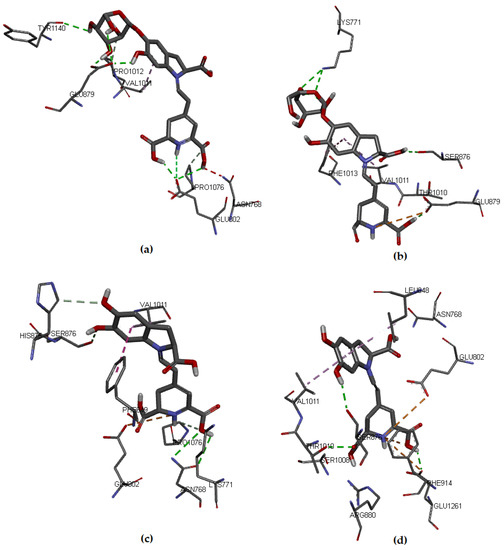
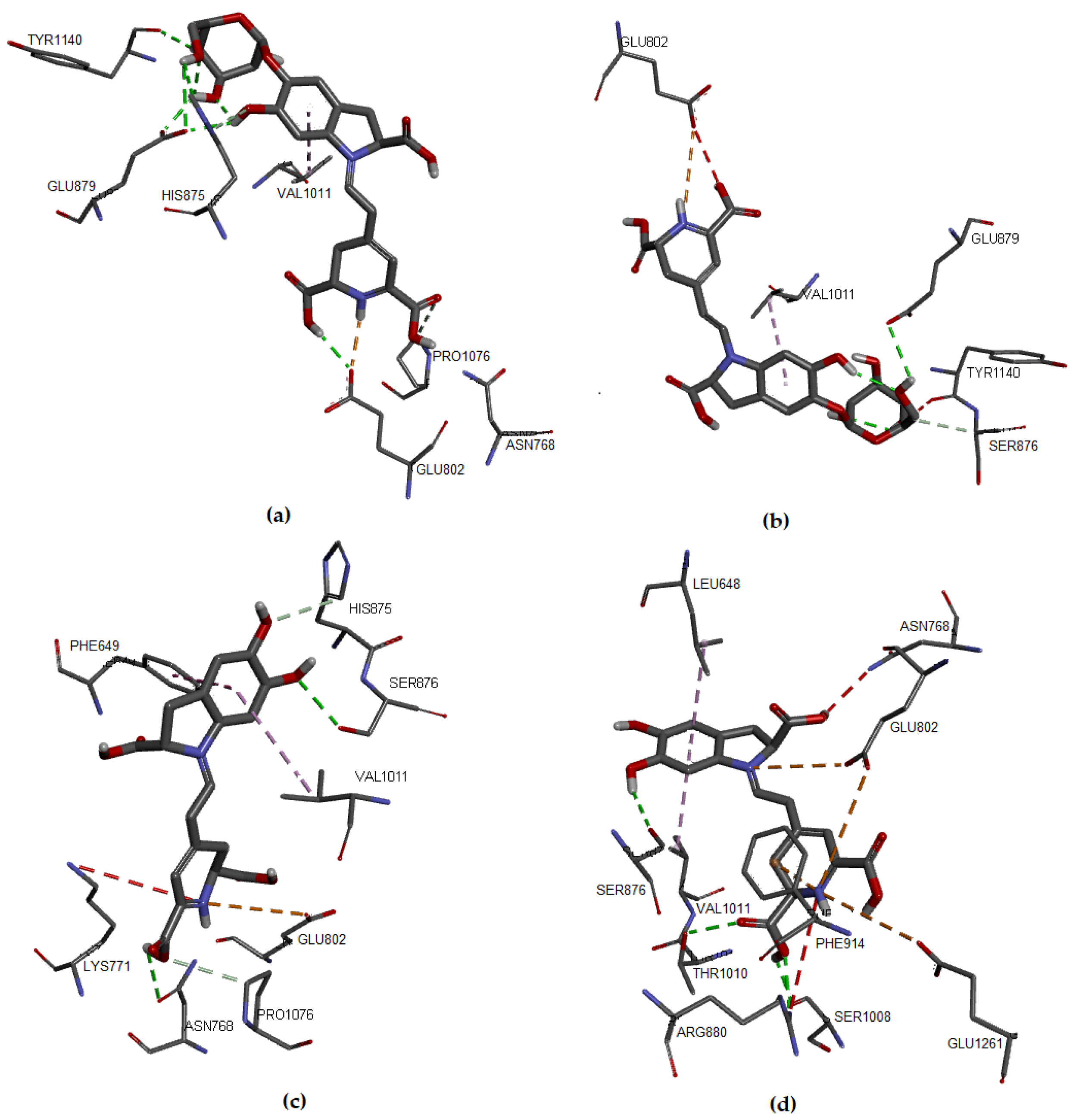
Figure 3.
Molecular models of binding of compounds to active site of xanthine oxidase. A three-dimensional model (Docking): (a) Bn, (b) IsoBn, (c) Bd, and (d) IsoBd.
There is an interaction between the hydrogen corresponding to the hydrogen of the ligand and the oxygen of Glu802 (2.934 Å). In Figure 3a, we present the hydrophobic interaction between the Bn’s aromatic ring and Val1011, distance in Åat 4.357. In IsoBn (see Figure 3b), hydrogen bonds can be observed with residues Lys771 (2.855 Å), Ser876 (2.810 Å), and Glu879 (1.877 Å), in addition to an electrostatic-type bond with residue Glu879 (5.323 Å). Furthermore, there are hydrophobic interactions with Phe1013 (5.292 Å) and Val1011(5.466 Å). Figure 3c shows that the interactions of Bd with the protein pocket are of hydrogen bonding type; the amino acids involved in such interactions are His875 (2.316 Å), Glu879 (2.403 Å), Ser876 (3.711 Å), electrostatic type with residue Glu879 (3.326 Å) and hydrophobic interactions with residues Leu648 (4.848 Å) and Val1011 (4.534 Å). IsoBd (see Figure 3d) exhibited electrostatic interactions with amino acids: Glu802 (5.305 Å), Phe914 (3.431 Å), and Glu1261 (5.211 Å), hydrogen bonding type with Thr1010 (2.510 Å), Glu1261 (2.147 Å) and Ser876 (2.267 Å) and hydrophobic interactions were present with residues Leu648 (4.834 Å) and Val1011 (4.399 Å).
According to the results, Bn and IsoBd interact favorably with the active site of XO through the formation of hydrogen bonds with the C15-hydroxyl group and the C17-hydroxyl group in the case of Bn and an electrostatic interaction for the possibility of IsoBd generating inhibition in the Glu802 residue. We could say that IsoBd can behave as a competitive inhibitor of the XO enzyme since it interacts with Glu1261 in the active site, which is responsible for the deprotonation of the Mo-OH prosthetic group in the enzyme. After deprotonation, the nucleophilic attack on the substrate occurs, promoting the oxidation reaction, generating ROS [14]. In general, the interactions of the this compound with the protein pocket showed favorable interactions, allowing the inhibition of the protein. This situation is critical at the cellular level since oxygen-based radicals are associated with a series of events, including the activation of nicotinamide adenine dinucleotide phosphate oxidase (NADPH), cytochrome catalyzed linoleate peroxidation, xanthine oxidase (XO), which is a form of xanthine oxidoreductase that generates reactive oxygen species, such as radicals superoxide and hydrogen peroxide [13,66].
The values of the binding affinities of the compounds are in a not very wide range (−6.9–−8.0 Kcal/mol, there is not much difference and above Febuxostat. Furthermore, these compounds interact with amino acids Val1011, Phe914, Phe1013, Glu802, Glu1261, or Thr1010, amino acids involved in interactions with the reference ligand Febuxostat and with the control ligand Hypoxanthine reported in the literature [15,33]. These results imply that the molecules studied bind to XO at the active site of the cofactor molybdopterin, allowing the interruption of the catalytic pathway for the activation of the enzymatic action of XO which is to generate the superoxide radical anion.
One of the most common measures to evaluate the condition of the complex along the trajectory is the quantification of the structural formation compared to the initial reference point. This evaluation is performed by the root mean square deviation (RMSD) of the main structure of the protein with the studied ligand, compared to the reference structure. In Figure S6 of the Supplementary Material, the complex formed with IsoBd has high stability during the simulation; the setting does not vary significantly, as in this case, there is an ascent of the RMSD until reaching a point where the values fluctuate around of 0.07 and 0.12 nm of RMSD. After of 10 ns, the structure remains within the parameter that considers the system to be in equilibrium; the complex did not lose its structure during the simulation. We used the VMD graphic viewer to obtain the trajectory (frame) corresponding to the most stable configuration to analyze this complex better. Figure 4 presents the interactions between the IsoBd ligand and XO.
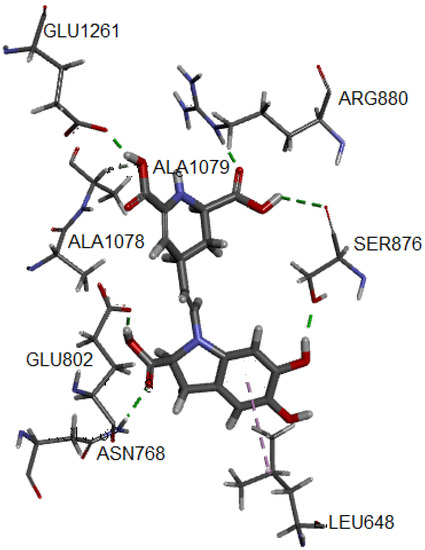
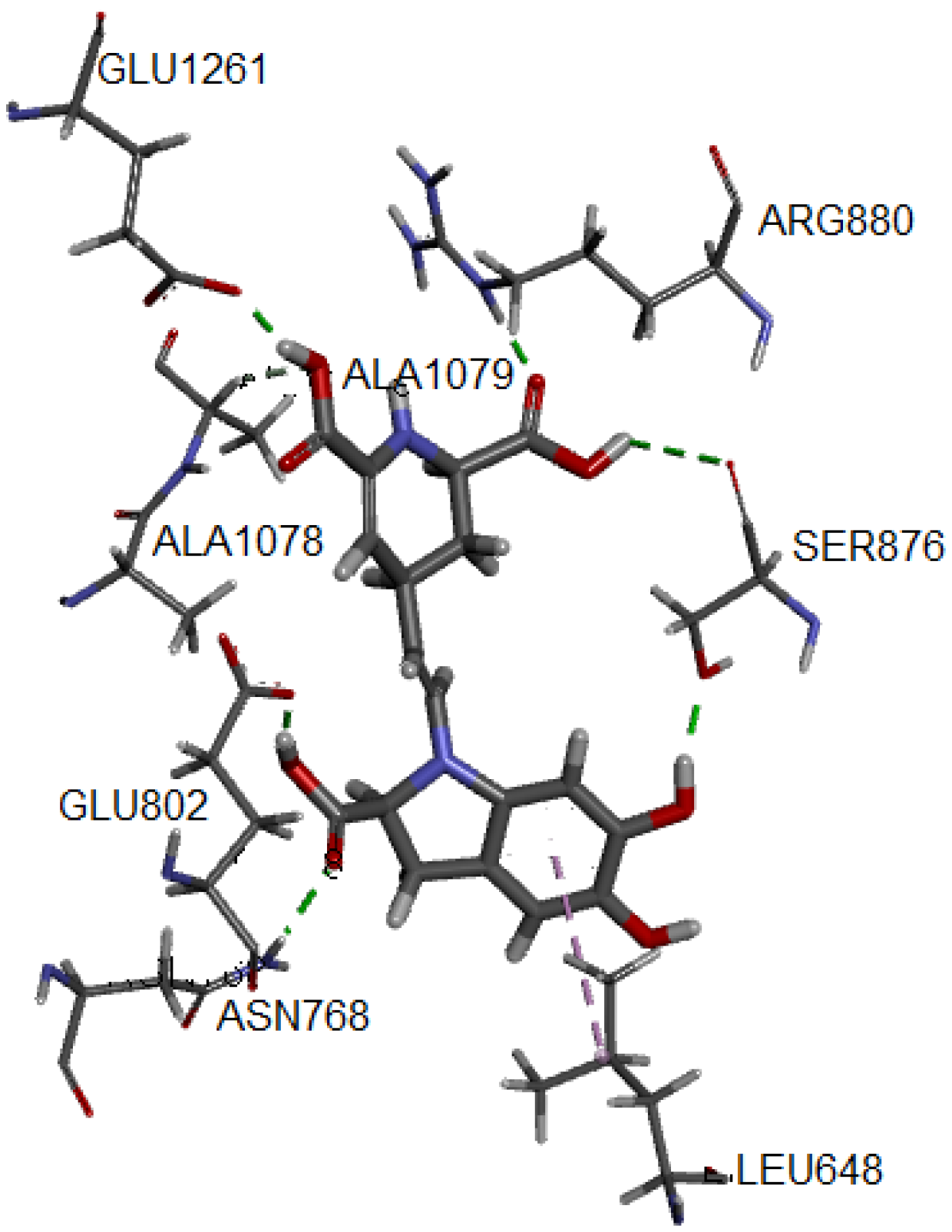
Figure 4.
Hydrogen bonds and alkyl interactions of IsoBd with XO obtained from MD study.
The hydrogen bond correspond to the residues Glu1261 (1.741 Å), Arg880, (1.696 Å), Ala1079 (2.364 Å), Glu802 (1.592 Å), Ser876 (3.055 Å) and Asn768 (2.045 Å). On the other hand, the complex presents an interaction of alkyl type marked by a violet segmented line with the Ala1078 residue. Both the enhanced stability and antiradical of betalains may be explained by the formation of hydrogen bonding, such as an adjacent hydroxyl or amino group and the betalains contain more than one phenolic hydroxy group. The polar nature of the O–H bond of the phenol group results in the formation of hydrogen bonds with other phenol molecules or other H-bonding systems and implies a high solubility in aqueous media. The radical quenching activity of betalain pigments is basically supported by the “intrinsic activity” shared between the imino and the tetrahydropyridine groups [68].
Regarding the implementation of the docking method based on shape theory through the definition of an initial ligand arranged according to the maximum variability along the x-axis and rotated to maintain the remaining variabilities in the y- and z-axes, it observed that the results compete in quality with those obtained by the expert algorithm of the molecular docking software as Autodock Vina. Specifically, one of the method’s main findings consisted of improving or equaling the bond affinities obtained by the software, as mentioned earlier, with the additional advantage that this method is entirely geometric. In addition, we noted that all 10 ligands proposed for each molecule yielded ΔG values consistent in most cases with those obtained by the expert software (see Table 3).
Table 3.
ΔG of compounds under study and reference considering geometry of ligands founded on shape theory and one calculated by expert software.
This method modifies the ligand coordinates, by using its Riemannian geometric properties. The protein (pocket) is not perturbed because it is fixed by an expert. Finally, the Autodock Vina software optimizes the protein-ligand complex according to the corresponding chemical interactions.
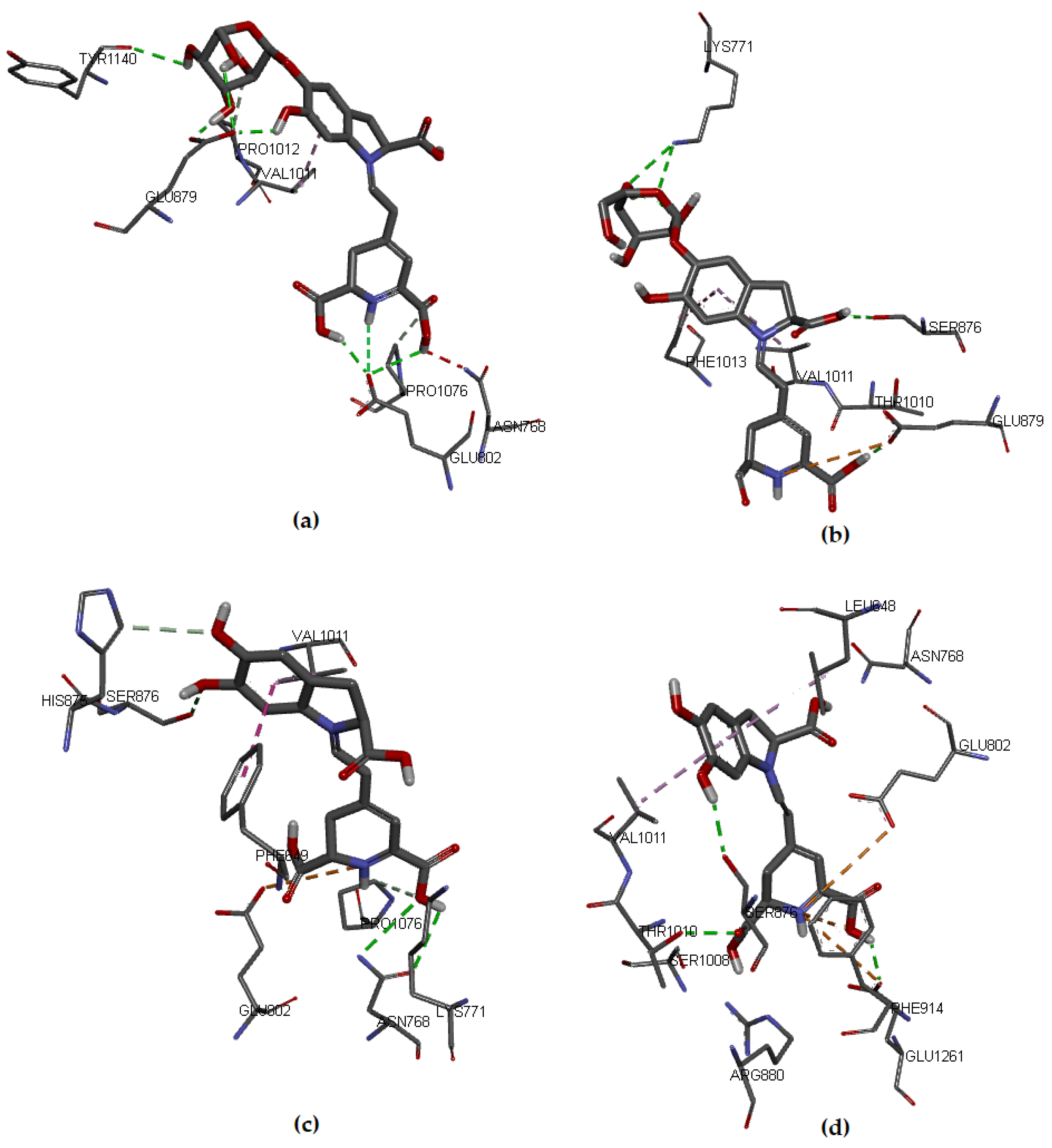
Figure 5 shows the interactions in the protein-ligand complex, where the ligand coordinates were computed by our algorithm.
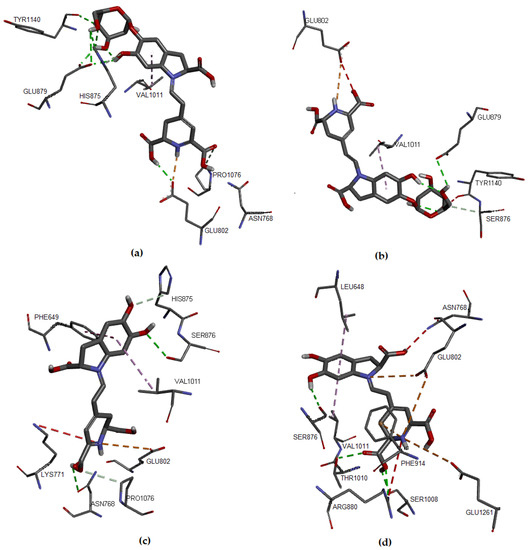
Figure 5.
Molecular models of binding of compounds to active site of xanthine oxidase. A three-dimensional model based on shape theory (a) Bn, (b) IsoBn, (c) Bd, and (d) IsoBd.
The results obtained for binding affinities were very similar to those previously obtained from the Euclidean coordinates of the protein-ligand complex (Table 3), showing that IsoBd and Bn possess the lowest ΔG value, followed by Bd and IsoBn. The interactions with the amino acids of the protein pocket are listed in Table 4, Table 5, Table 6 and Table 7. We highlight that IsoBd presented a more significant number of interactions, which is in agreement with other investigations about antioxidant capacity [11,13,33].
Table 4.
Docking results of binding of Bn to active site of xanthine oxidase.
Table 5.
Docking results of binding of IsoBn to active site of xanthine oxidase.
Table 6.
Docking results of binding of Bd to active site of xanthine oxidase.
Table 7.
Docking results of binding of IsoBd to active site of xanthine oxidase.
The range of values of affinities of all compounds biding is between −6.2 kcal/mol −8.1 kcal/mol and exceeds the binding affinity of the reference ligand Febusxostat (−9.1 kcal/mol) obtained by applying the same method. Regarding the reference interactions, their residues are Arg880, Thr1010, Phe914, Ala1079, Ala1078, Leu873, Val1011, and Leu1014, which are in the same line as those reported in other works [15,33].
4. Conclusions
We analyze the antioxidant activities of some betacyanins (Bn, IsoBn, Bd, and IsoBd) via Shape Theory, Molecular Dynamics, Density Functional Theory, and Infrared Spectroscopy. The antioxidant activity of the compounds considered in this study were analyzed at the B3LYP level of theory and with the basis set 6-31+G(d,p). We find it important to include spin-contamination analysis via UB3LYP, which is often omitted in numerous applications of DFT to catalysis. The values’ descriptors BDE, PA, ETE, IP, and PDE evidenced water’s most likely reaction mechanism. Furthermore, the calculation showed that the SPLET mechanism is more likely than the HAT and SET-PT mechanisms since the PA values are lower than IP and BDE. Additionally, the group that loss the hydrogen first is C17COOH. We constructed our own algorithms based on shape theory. The results conclude that the order of the RDs is consistent with the order of the PA values. In this case, the lowest PA and RD values indicates the optimal mechanism of the antioxidant activity for studied molecules. The assessments of algorithms also establish a consistent results between the global and partial RD for groups of elements. This fact provides a number of checking rule for choosing the best mechanism of antioxidant capacity. The method of shape theory also defines a routine for molecular docking based on desirable invariant properties of the Riemmanian geometric. The new molecular docking studies showed that most of the compounds presented interactions with XO. In addition, molecular dynamics for IsoBd compound showed interactions with a higher number of amino acids belonging to the protein pocket. A similar behavior was found for the Febuxotat, the antioxidant drug used as reference.
About the molecular dynamic, the study of the free radical scavenger via MD is explained by the formation of hydrogen bonding, preserved during the simulation. This characteristic is in accordance with previous results of the reference molecule.
The new method of molecular docking for the ligand shows some correspondence with the results of the molecular docking carried out with the expert software Autodock Vina. As robust strategy, a method that integrates the protein coordinates will be propose in a future work by the authors.
We found in IR spectra from IsoBd a correlation between a bathochromic shift for the anion C17COOH in all absorptions frequencies alone an increase in velocity the loss of hydrogen. We proved the chemical stability of the molecules in regions of the amine at 1700 to 1300 cm−1 and 3700 to 3200 cm−1 that corresponding to the coordination of deprotonated peptides of complex or ligands.
In a future work, we could explore the link of the neural networks under nonlinear models with predictors of preshapes, studied in the Riemannian hyper-sphere. This translation into the well know language of artificial intelligence can include predictors and responses involving the geometric invariances of the Riemannian Shape Theory.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/molecules27062003/s1. Figure S1. Basic structure of Betacyanins. (a): Betalamic Acid, (b): cyclo-DOPA, (c): Betacyanins R = Glc (Betanin), R = H (Betanidin). Figure S2. Epimer at C15 of Betanin and Betanidine. Figure S3. Optimized structures obtained for Betanin (Bn), Isobetanín (IsoBn), Betanidin (Bd) and Isobetanidin (IsoBd). Method used/IEFPCM/ B3LYP /6-31+G(d,p). Solvent: water. Coordinates of the geometry optimized with IEFPCM/ B3LYP /6-31+G(d,p). Solvent: water. Geometry of IsoBd and anions. Figure S4. Parent molecules for Betanine (Bn), Isobetanina (IsoBn), Betanidine (Bd), and Isobetanidine (IsoBd). Figure S5. Chemical structures and numbering scheme of xanthine and hypoxanthine. Table S1. IR data for Bn, IsoBn, Bd, and IsoBd. Figure S6. RMSD of the protein backbone during the simulation of DM trajectories.
Author Contributions
Date curation, I.M.R.-V., E.V., A.B.-C. and F.J.C.-L.; Investigation, I.M.R.-V., A.B.-C. and F.J.C.-L.; Analysis formal, F.J.C.-L. Writing—original draft, I.M.R.-V., E.V., A.B.-C. and F.J.C.-L.; Writing, revision and editing, I.M.R.-V., E.V., A.B.-C. and F.J.C.-L. All authors contributed the final manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This work was funded by Instituto Tecnologico Metropolitano (ITM) in Colombia through grant No. P21112.
Institutional Review Board Statement
Not applicable.
Data Availability Statement
Ramirez Velásquez, Iliana (2021), “Antioxidant activities of betanin, betanidin and respective C15-epimers via DFT methods, shape theory and molecular docking”, Mendeley Data, V1, doi:10.17632/vvvxj44s9y.1.
Acknowledgments
E.V. would like to acknowledge the use of computational resources provided by the Institute for Biocomputation and the Physics of Complex Systems (BIFI)–Universidad de Zaragoza -Spain. The authors are grateful for the support of the ITM for the publication of this study. The authors also thanks to Doctorate in Modeling and Scientific Computing at University of Medellin.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| Affine | is a geometric transformation that preserves lines and parallelism |
| BDE | bond dissociation enthalpy |
| Bd | betanidin |
| Bn | betanin |
| B3LYP | Becke, 3-parameter, Lee–Yang–Parr |
| DFT | Density Functional Theory |
| DNA | Deoxyribonucleic acid |
| DPPH | 2,2-Diphenyl-1-Picrylhydrazyl Assay |
| ETE | the electron transfer enthalpy |
| FAD | flavin adenine dinucleotid |
| FRAP | Ferric Reducing Antioxidant Power Assay |
| HAT | hydrogen atom transfer mechanism |
| HOMO | highest occupied molecular orbital |
| IC50 | The maximum mean inhibitory concentration |
| IEFPCM | Integral-Equation-Formalism Polarizable Continuum Model |
| IP | ionization potential |
| IR | infrared spectra |
| IsoBd | isobetanidin |
| IsoBn | isobetanin |
| LUMO | lowest unoccupied molecular orbital |
| MD | Molecular Dynamic |
| Mo-pt | Molybdopterin |
| NADPH | nicotinamide adenine dinucleotide dinucleotide phosphate oxidase |
| ORAC | Oxygen Radical Absorbance Capacity Assay |
| PA | proton affinity |
| PDE | proton dissociation enthalpy |
| QR | is a A = QR of an orthogonal matrix Q and an upper triangular matrix R |
| RD | Riemann distance |
| RNS | Reactive Nitrogen Species |
| ROS | Reactive Oxygen Species |
| SET-PT | sequential electron proton transfer mechanism |
| SLEPT | sequential proton-loss electron transfer mechanism |
| SVD | singular value decomposition |
| TEAC | the Trolox Equivalent Antioxidant Capacity |
| VMD | visual molecular dynamics |
| XO | Xanthine oxidse |
References
- Apak, R. Current Issues in Antioxidant Measurement. J. Agric. Food Chem. 2019, 67, 9187–9202. [Google Scholar] [CrossRef] [PubMed]
- Kapadia, G.J.; Rao, G.S. Anticancer Effects of Red Beet Pigments. In Red Beet Biotechnology; Springer: Berlin/Heidelberg, Germany, 2012; pp. 125–154. [Google Scholar] [CrossRef]
- Gandía-Herrero, F.; García-Carmona, F. Biosynthesis of betalains: Yellow and violet plant pigments. Trends Plant Sci. 2013, 18, 334–343. [Google Scholar] [CrossRef] [PubMed]
- Slavov, A.; Karagyozov, V.; Denev, P.; Kratchanova, M.; Kratchanov, C. Antioxidant activity of red beet juices obtained after microwave and thermal pretreatments. Czech J. Food Sci. 2013, 31, 139–147. [Google Scholar] [CrossRef] [Green Version]
- Mereddy, R.; Chan, A.; Fanning, K.; Nirmal, N.; Sultanbawa, Y. Betalain rich functional extract with reduced salts and nitrate content from red beetroot (Beta vulgaris L.) using membrane separation technology. Food Chem. 2017, 215, 311–317. [Google Scholar] [CrossRef] [Green Version]
- Cortez, R.; Luna-Vital, D.A.; Margulis, D.; de Mejia, E.G. Natural Pigments: Stabilization Methods of Anthocyanins for Food Applications. Compr. Rev. Food Sci. Food Saf. 2016, 16, 180–198. [Google Scholar] [CrossRef]
- Strack, D.; Vogt, T.; Schliemann, W. Recent advances in betalain research. Phytochemistry 2003, 62, 247–269. [Google Scholar] [CrossRef]
- Esatbeyoglu, T.; Wagner, A.E.; Schini-Kerth, V.B.; Rimbach, G. Betanin-A food colorant with biological activity. Mol. Nutr. Food Res. 2014, 59, 36–47. [Google Scholar] [CrossRef]
- Kanner, J.; Harel, S.; Granit, R. BetalainsA New Class of Dietary Cationized Antioxidants. J. Agric. Food Chem. 2001, 49, 5178–5185. [Google Scholar] [CrossRef]
- Prieto-Santiago, V.; Cavia, M.M.; Alonso-Torre, S.R.; Carrillo, C. Relationship between color and betalain content in different thermally treated beetroot products. J. Food Sci. Technol. 2020, 57, 3305–3313. [Google Scholar] [CrossRef]
- Ardjani, A.T.E.; Mekelleche, S.M. Analysis of the antioxidant activity of 4-(5-chloro-2-hydroxyphenylamino)-4-oxobut-2-enoic acid derivatives using quantum-chemistry descriptors and molecular docking. J. Mol. Model. 2016, 22. [Google Scholar] [CrossRef]
- Cao, H.; Pauff, J.M.; Hille, R. Substrate Orientation and Catalytic Specificity in the Action of Xanthine Oxidase. J. Biol. Chem. 2010, 285, 28044–28053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, H.C.; Tsai, S.H.; Chen, C.S.; Chang, Y.C.; Lee, C.M.; Lai, Z.Y.; Lin, C.M. Structure–activity relationship of coumarin derivatives on xanthine oxidase-inhibiting and free radical-scavenging activities. Biochem. Pharmacol. 2008, 75, 1416–1425. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, K.; Eger, B.T.; Nishino, T.; Kondo, S.; Pai, E.F.; Nishino, T. An Extremely Potent Inhibitor of Xanthine Oxidoreductase. J. Biol. Chem. 2003, 278, 1848–1855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujishima, M.T.; Silva, N.; Ramos, R.; Ferreira, E.B.; Santos, K.; Silva, C.; Silva, J.; Rosa, J.C.; Santos, C. An Antioxidant Potential, Quantum-Chemical and Molecular Docking Study of the Major Chemical Constituents Present in the Leaves of Curatella americana Linn. Pharmaceuticals 2018, 11, 72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gliszczyńska-Świgło, A.; Szymusiak, H.; Malinowska, P. Betanin, the main pigment of red beet: Molecular origin of its exceptionally high free radical-scavenging activity. Food Addit. Contam. 2006, 23, 1079–1087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dryden, I.L.; Mardia, K.V. Statistical Shape Analysis, with Applications in R; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2016. [Google Scholar] [CrossRef]
- Goodall, C.R.; Mardia, K.V. Multivariate Aspects of Shape Theory. Ann. Stat. 1993, 21, 848–866. [Google Scholar] [CrossRef]
- Caro-Lopera, F.J.; Díaz-García, J.A.; González-Farías, G. Noncentral elliptical configuration density. J. Multivar. Anal. 2010, 101, 32–43. [Google Scholar] [CrossRef] [Green Version]
- Díaz-García, J.A.; Caro-Lopera, F.J. Estimation of mean form and mean form difference under elliptical laws. Electron. J. Stat. 2017, 11, 2424–2460. [Google Scholar] [CrossRef]
- Díaz-García, J.A.; Caro-Lopera, F.J. Elliptical affine shape distributions for real normed division algebras. J. Multivar. Anal. 2016, 144, 139–149. [Google Scholar] [CrossRef]
- Díaz-García, J.A.; Caro-Lopera, F.J. Generalised Shape Theory Via Pseudo-Wishart Distribution. Sankhya A 2013, 75, 253–276. [Google Scholar] [CrossRef]
- Díaz-García, J.A.; Caro-Lopera, F.J. Statistical theory of shape under elliptical models and singular value decompositions. J. Multivar. Anal. 2012, 103, 77–92. [Google Scholar] [CrossRef] [Green Version]
- Díaz-García, J.A.; Caro-Lopera, F.J. Generalised shape theory via SV decomposition I. Metrika 2010, 75, 541–565. [Google Scholar] [CrossRef]
- Díaz-García, J.A.; Caro-Lopera, F.J. Statistical theory of shape under elliptical models via QR decomposition. Statistics 2013, 48, 456–472. [Google Scholar] [CrossRef]
- Villarreal-Rios, A.L.; Bedoya-Calle, Á.H.; Caro-Lopera, F.J.; Ortiz-Méndez, U.; García-Méndez, M.; Pérez-Ramírez, F.O. Ultrathin tunable conducting oxide films for near-IR applications: An introduction to spectroscopy shape theory. SN Appl. Sci. 2019, 1, 1553. [Google Scholar] [CrossRef] [Green Version]
- Quintero, J.; Mariño, A.; Šiller, L.; Restrepo-Parra, E.; Caro-Lopera, F. Rocking curves of gold nitride species prepared by arc pulsed - physical assisted plasma vapor deposition. Surf. Coatings Technol. 2017, 309, 249–257. [Google Scholar] [CrossRef]
- Arias, E.; Caro-Lopera, F.J.; Flórez, E.; Pérez-Torres, J.F. Two Novel Approaches Based on the Thompson Theory and Shape Analysis for Determination of Equilibrium Structures of Nanoclusters: Cu8, Ag8 and Ag18 as study cases. J. Physic. Conf. Ser. 2019, 1247, 012008. [Google Scholar] [CrossRef]
- Valencia, G.M.; Anaya, J.A.; Velásquez, É.A.; Ramo, R.; Caro-Lopera, F.J. About Validation-Comparison of Burned Area Products. Remote. Sens. 2020, 12, 3972. [Google Scholar] [CrossRef]
- Nath, D.; Laik, R.; Meena, V.S.; Pramanick, B.; Singh, S.K. Can mid-infrared (mid-IR) spectroscopy evaluate soil conditions by predicting soil biological properties? Soil Secur. 2021, 100008. [Google Scholar] [CrossRef]
- Elderderi, S.; Leman-Loubiere, C.; Wils, L.; Henry, S.; Bertrand, D.; Byrne, H.J.; Chourpa, I.; Enguehard-Gueiffier, C.; Munnier, E.; Elbashir, A.A.; et al. ATR-IR spectroscopy for rapid quantification of water content in deep eutectic solvents. J. Mol. Liq. 2020, 311, 113361. [Google Scholar] [CrossRef]
- Velkov, Z.; Traykov, M.; Trenchev, I.; Saso, L.; Tadjer, A. Topology-Dependent Dissociation Mode of the O–H Bond in Monohydroxycoumarins. J. Phys. Chem. A 2019, 123, 5106–5113. [Google Scholar] [CrossRef]
- Yusuff, O.K.; Raheem, M.A.O.A.; Mukadam, A.A.; Sulaimon, R.O. Kinetics and Mechanism of the Antioxidant Activities of C. olitorius and V. amygdalina by Spectrophotometric and DFT Methods. ACS Omega 2019, 4, 13671–13680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, Y.; Zheng, Y.; An, L.; Dou, Y.; Liu, Y. Density functional theory study of the structure–antioxidant activity of polyphenolic deoxybenzoins. Food Chem. 2014, 151, 198–206. [Google Scholar] [CrossRef] [PubMed]
- Deepha, V.; Praveena, R.; Sadasivam, K. DFT studies on antioxidant mechanisms, electronic properties, spectroscopic (FT-IR and UV) and NBO analysis of C-glycosyl flavone, an isoorientin. J. Mol. Struct. 2015, 1082, 131–142. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09. 2016. Available online: https://gaussian.com/g09citation/ (accessed on 16 December 2021).
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem in 2021: New data content and improved web interfaces. Nucleic Acids Res. 2020, 49, D1388–D1395. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, S.A.; Baumgartner, M.T. Betanidin pKa Prediction Using DFT Methods. ACS Omega 2020, 5, 13751–13759. [Google Scholar] [CrossRef]
- Klein, E.; Lukeš, V.; Ilčin, M. DFT/B3LYP study of tocopherols and chromans antioxidant action energetics. Chem. Phys. 2007, 336, 51–57. [Google Scholar] [CrossRef]
- Cancès, E.; Mennucci, B.; Tomasi, J. A new integral equation formalism for the polarizable continuum model: Theoretical background and applications to isotropic and anisotropic dielectrics. J. Chem. Phys. 1997, 107, 3032–3041. [Google Scholar] [CrossRef]
- Cancès, E.; Mennucci, B.; Tomasi, J. Analytical derivatives for geometry optimization in solvation continuum models. II. Numerical applications. J. Chem. Phys. 1998, 109, 260–266. [Google Scholar] [CrossRef]
- Shireen, P.A.; Muraleedharan, K.; Mujeeb, V.A. Theoretical studies on anti-oxidant potential of alpinetin. Mater. Today Proc. 2018, 5, 8908–8915. [Google Scholar] [CrossRef]
- Miller, F.A. Spectra of X-H Systems (with Emphasis on O-H and N-H Groups). In Course Notes on the Interpretation of Infrared and Raman Spectra; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2004; pp. 163–178. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2018. [Google Scholar]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2009, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Enroth, C.; Eger, B.T.; Okamoto, K.; Nishino, T.; Nishino, T.; Pai, E.F. Crystal structures of bovine milk xanthine dehydrogenase and xanthine oxidase: Structure-based mechanism of conversion. Proc. Natl. Acad. Sci. USA 2000, 97, 10723–10728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Lee, J.; Cheng, X.; Swails, J.M.; Yeom, M.S.; Eastman, P.K.; Lemkul, J.A.; Wei, S.; Buckner, J.; Jeong, J.C.; Qi, Y.; et al. CHARMM-GUI input generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM simulations using the CHARMM36 additive force field. J. Chem. Theory Comput. 2016, 12, 405–413. [Google Scholar] [CrossRef]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef]
- Gandía-Herrero, F.; Escribano, J.; García-Carmona, F. The Role of Phenolic Hydroxy Groups in the Free Radical Scavenging Activity of Betalains. J. Nat. Prod. 2009, 72, 1142–1146. [Google Scholar] [CrossRef]
- Gandía-Herrero, F.; Escribano, J.; García-Carmona, F. Structural implications on color, fluorescence, and antiradical activity in betalains. Planta 2010, 232, 449–460. [Google Scholar] [CrossRef]
- Musialik, M.; Kuzmicz, R.; Pawłowski, T.S.; Litwinienko, G. Acidity of Hydroxyl Groups: An Overlooked Influence on Antiradical Properties of Flavonoids. J. Org. Chem. 2009, 74, 2699–2709. [Google Scholar] [CrossRef]
- Oprea, C.I.; Dumbravă, A.; Enache, I.; Georgescu, A.; Gîrţu, M.A. A combined experimental and theoretical study of natural betalain pigments used in dye-sensitized solar cells. J. Photochem. Photobiol. A Chem. 2012, 240, 5–13. [Google Scholar] [CrossRef]
- Qin, C.; Clark, A.E. DFT characterization of the optical and redox properties of natural pigments relevant to dye-sensitized solar cells. Chem. Phys. Lett. 2007, 438, 26–30. [Google Scholar] [CrossRef]
- Munteanu, I.G.; Apetrei, C. Analytical Methods Used in Determining Antioxidant Activity: A Review. Int. J. Mol. Sci. 2021, 22, 3380. [Google Scholar] [CrossRef] [PubMed]
- Craft, B.D.; Kerrihard, A.L.; Amarowicz, R.; Pegg, R.B. Phenol-Based Antioxidants and the In Vitro Methods Used for Their Assessment. Compr. Rev. Food Sci. Food Saf. 2012, 11, 148–173. [Google Scholar] [CrossRef]
- Miller, N.J.; Rice-Evans, C.; Davies, M.J.; Gopinathan, V.; Milner, A. A Novel Method for Measuring Antioxidant Capacity and its Application to Monitoring the Antioxidant Status in Premature Neonates. Clin. Sci. 1993, 84, 407–412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smeriglio, A.; Francesco, C.D.; Denaro, M.; Trombetta, D. Prickly Pear Betalain-Rich Extracts as New Promising Strategy for Intestinal Inflammation: Plant Complex vs. Main Isolated Bioactive Compounds. Front. Pharmacol. 2021, 12. [Google Scholar] [CrossRef] [PubMed]
- Calogero, G.; Bartolotta, A.; Di Marco, G.; Di Carlo, A.; Bonaccorso, F. Vegetable-based dye-sensitized solar cells. Chem. Soc. Rev. 2015, 44, 3244–3294. [Google Scholar] [CrossRef]
- Mayo, D.W. Survey of Infrared and Raman Group Frequencies. In Course Notes on the Interpretation of Infrared and Raman Spectra; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2004; pp. 355–398. [Google Scholar] [CrossRef]
- Estrada–delaVega, A.; Bedoya–Calle, Á.H.; Diabb, J.; Villarreal–Rios, A.L.; Elías–Zuniga, A.; López–Cuellar, E.M.; Ortiz–Méndez, U. Preparation and characterization of lanthanum nitrate nanoparticles by a polyol method. Mater. Res. Express 2019, 6, 095030. [Google Scholar] [CrossRef]
- Sheppard, N. 50 Years in Vibrational Spectroscopy at the Gas/Solid Interface. In Surface Chemistry and Catalysis; Springer: Berlin/Heidelberg, Germany, 2002; pp. 27–54. [Google Scholar] [CrossRef]
- Kolev, T.; Ivanova, B.B.; Zareva, S.Y. An Au (III) complex of glycyl-S-serine: A linear polarized IR and 1H-and 13C-NMR investigation. J. Coord. Chem. 2007, 60, 109–115. [Google Scholar] [CrossRef]
- Mistry, B. A Handbook of Spectroscopic Data CHEMISTRY (UV, JR, PMR, JJCNMR and Mass Spectroscopy); Oxford Book Company: Oxford, UK, 2009. [Google Scholar]
- Yang, J.; Dong, C.; Kirk, M.L. Xanthine oxidase–product complexes probe the importance of substrate/product orientation along the reaction coordinate. Dalton Trans. 2017, 46, 13242–13250. [Google Scholar] [CrossRef]
- Malik, U.Z.; Hundley, N.J.; Romero, G.; Radi, R.; Freeman, B.A.; Tarpey, M.M.; Kelley, E.E. Febuxostat inhibition of endothelial-bound XO: Implications for targeting vascular ROS production. Free Radic. Biol. Med. 2011, 51, 179–184. [Google Scholar] [CrossRef] [Green Version]
- Slimen, I.B.; Najar, T.; Abderrabba, M. Chemical and Antioxidant Properties of Betalains. J. Agric. Food Chem. 2017, 65, 675–689. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).