
Natural Products with Antitumor Potential Targeting the MYB-C/EBPβ-p300 Transcription Module



Abstract
:1. The MYB-C/EBPβ-p300 Transcription Machinery: Its Physiological and Pathophysiological Roles and Its Potential as a Target in Cancer
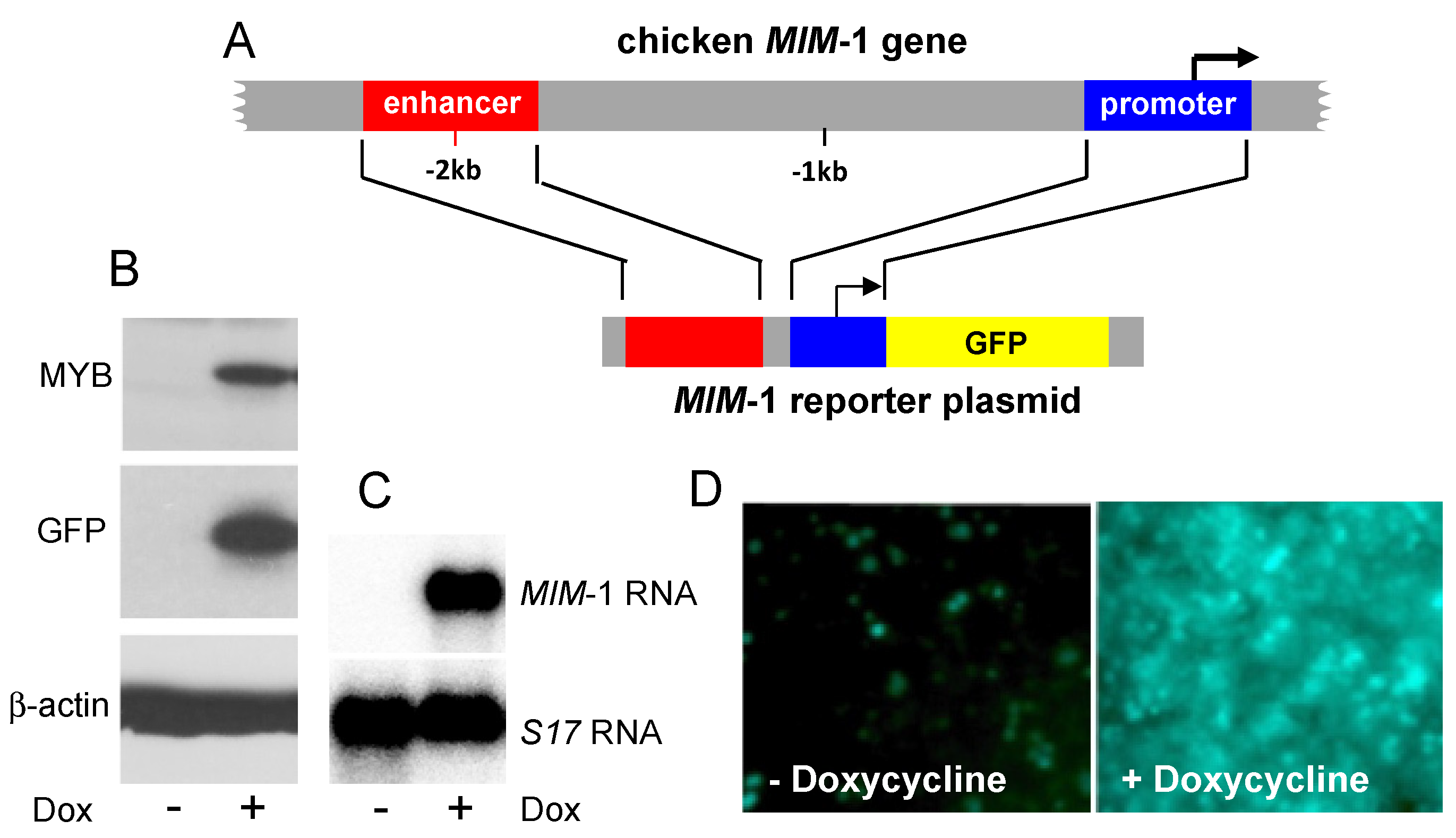
2. Development of a Fluorescence-Based MYB Reporter Cell-Line
3. Natural Products as Inhibitors of MYB-Related Transcription in the Fluorescence-Based Reporter Cell Line
4. Sesquiterpene Lactones
5. Withanolides: Withaferin A
6. Quinone Methide Triterpenes: Celastrol
7. Naphthoquinones: Plumbagin
8. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ramsay, R.G.; Gonda, T.J. MYB function in normal and cancer cells. Nat. Rev. Cancer 2008, 8, 523–534. [Google Scholar] [CrossRef] [PubMed]
- Cicirò, Y.; Sala, A. MYB oncoproteins: Emerging players and potential therapeutic targets in human cancer. Oncogenesis 2021, 10, 19. [Google Scholar] [CrossRef] [PubMed]
- Mucenski, M.L.; McLain, K.; Kier, A.B.; Swerdlow, S.H.; Schreiner, C.M.; Miller, T.A.; Pietryga, D.W.; Scott, W.J., Jr.; Potter, S.S. A functional c-myb gene is required for normal murine fetal hepatic hematopoiesis. Cell 1991, 65, 677–689. [Google Scholar] [CrossRef]
- Emambokus, N.; Vegiopoulos, A.; Harman, B.; Jenkinson, E.; Anderson, G.; Frampton, J. Progression through key stages of haemopoiesis is dependent on distinct threshold levels of c-Myb. EMBO J. 2003, 22, 4478–4488. [Google Scholar] [CrossRef] [Green Version]
- Bender, T.P.; Kremer, C.S.; Kraus, M.; Buch, T.; Rajewsky, K. Critical functions for c-Myb at three checkpoints during thymocyte development. Nat. Immunol. 2004, 5, 721–729. [Google Scholar] [CrossRef]
- Carpinelli, M.R.; Hilton, D.J.; Metcalf, D.; Antonchuk, J.L.; Hyland, C.D.; Mifsud, S.L.; Di Rago, L.; Hilton, A.A.; Willson, T.A.; Roberts, A.W.; et al. Suppressor screen in Mpl-/- mice: C-Myb mutation causes supraphysiological production of platelets in the absence of thrombopoietin signaling. Proc. Natl. Acad. Sci. USA 2004, 101, 6553–6558. [Google Scholar] [CrossRef] [Green Version]
- Sandberg, M.L.; Sutton, S.E.; Pletcher, M.T.; Wiltshire, T.; Tarantino, L.M.; Hogenesch, J.B.; Cooke, M.P. c-Myb and p300 regulate hematopoietic stem cell proliferation and differentiation. Dev. Cell 2005, 8, 153–166. [Google Scholar] [CrossRef]
- Thomas, M.D.; Kremer, C.S.; Ravichandran, K.S.; Rajewsky, K.; Bender, T.P. c-Myb is critical for B cell development and maintenance of follicular B cells. Immunity 2005, 23, 275–286. [Google Scholar] [CrossRef] [Green Version]
- Sitzmann, J.; Noben-Trauth, K.; Klempnauer, K.-H. Expression of mouse c-myb during embryonic development. Oncogene 1995, 11, 2273–2279. [Google Scholar]
- Malaterre, J.; Carpinelli, M.; Ernst, M.; Alexander, W.; Cooke, M.; Sutton, S.; Dworkin, S.; Heath, J.K.; Frampton, J.; McArthur, G.; et al. c-Myb is required for progenitor cell homeostasis in colonic crypts. Proc. Natl. Acad. Sci. USA 2007, 104, 3829–3834. [Google Scholar] [CrossRef] [Green Version]
- Ness, S.A.; Marknell, A.; Graf, T. The v-myb oncogene product binds to and activates the promyelocyte-specific mim-1 gene. Cell 1989, 59, 1115–1125. [Google Scholar] [CrossRef]
- Chayka, O.; Kintscher, J.; Braas, D.; Klempnauer, K.-H. v-Myb mediates cooperation of a cell-specific enhancer with the mim-1 promoter. Mol. Cell. Biol. 2005, 25, 499–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burk, O.; Mink, S.; Ringwald, M.; Klempnauer, K.-H. Synergistic activation of the chicken mim-1 gene by v-myb and C/EBP transcription factors. EMBO J. 1993, 12, 2027–2038. [Google Scholar] [CrossRef] [PubMed]
- Ness, S.A.; Kowentz-Leutz, E.; Casini, T.; Graf, T.; Leutz, A. Myb and NF-M: Combinatorial activators of myeloid genes in heterologous cell types. Genes Dev. 1993, 7, 749–759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mink, S.; Kerber, U.; Klempnauer, K.-H. Interaction of C/EBPbeta and v-Myb is required for synergistic activation of the mim-1 gene. Mol. Cell. Biol. 1996, 16, 1316–1325. [Google Scholar] [CrossRef] [Green Version]
- Plachetka, A.; Chayka, O.; Wilczek, C.; Melnik, S.; Bonifer, C.; Klempnauer, K.-H. C/EBPβ induces chromatin opening at a cell-type-specific enhancer. Mol. Cell. Biol. 2008, 28, 2102–2112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rushton, J.J.; Davis, L.M.; Lei, W.; Mo, X.; Leutz, A.; Ness, S.A. Distinct changes in gene expression induced by A-Myb, B-Myb and c-Myb proteins. Oncogene 2003, 22, 308–313. [Google Scholar] [CrossRef] [Green Version]
- Liu, F.; Lei, W.; O’Rourke, J.P.; Ness, S.A. Oncogenic mutations cause dramatic, qualitative changes in the transcriptional activity of c-Myb. Oncogene 2006, 2, 5795–5805. [Google Scholar]
- Zhao, L.; Glazov, E.A.; Pattabiraman, D.R.; Al-Owaidi, F.; Zhang, P.; Brown, M.A.; Leo, P.J.; Gonda, T.J. Integrated genome-wide chromatin occupancy and expression analyses identify key myeloid pro-differentiation transcription factors repressed by Myb. Nucleic Acids Res. 2011, 39, 4664–4679. [Google Scholar] [CrossRef]
- Oelgeschläger, M.; Janknecht, R.; Krieg, J.; Schreek, S.; Lüscher, B. Interaction of the co-activator CBP with Myb proteins: Effects on Myb-specific transactivation and on the cooperativity with NF-M. EMBO J. 1996, 15, 2771–2780. [Google Scholar] [CrossRef]
- Dai, P.; Akimaru, H.; Tanaka, Y.; Hou, D.X.; Yasukawa, T.; Kanei-Ishii, C.; Takahashi, T.; Ishii, S. CBP as a transcriptional coactivator of c-Myb. Genes Dev. 1996, 10, 528–540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zor, T.; De Guzman, R.N.; Dyson, H.J.; Wright, P.E. Solution structure of the KIX domain of CBP bound to the transactivation domain of c-Myb. J. Mol. Biol. 2004, 337, 521–534. [Google Scholar] [CrossRef] [PubMed]
- Kasper, L.H.; Boussouar, F.; Ney, P.A.; Jackson, C.W.; Rehg, J.; van Deursen, J.M.; Brindle, P.K. A transcription-factor-binding surface of coactivator p300 is required for haematopoiesis. Nature 2002, 419, 738–743. [Google Scholar] [CrossRef] [PubMed]
- Pattabiraman, D.R.; Sun, J.; Dowhan, D.H.; Ishii, S.; Gonda, T.J. Mutations in multiple domains of c-Myb disrupt interaction with CBP/p300 and abrogate myeloid transforming ability. Mol. Cancer Res. 2009, 7, 1477–1486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pattabiraman, D.R.; McGirr, C.; Shakhbazov, K.; Barbier, V.; Krishnan, K.; Mukhopadhyay, P.; Hawthorne, P.; Trezise, A.; Ding, J.; Grimmond, S.M.; et al. Interaction of c-Myb with p300 is required for the induction of acute myeloid leukemia (AML) by human AML oncogenes. Blood 2014, 123, 2682–2690. [Google Scholar] [CrossRef] [Green Version]
- Klempnauer, K.-H.; Gonda, T.J.; Bishop, J.M. Nucleotide sequence of the retroviral leukemia gene v-myb and its cellular progenitor c-myb: The architecture of a transduced oncogene. Cell 1982, 31, 453–463. [Google Scholar] [CrossRef]
- Lahortiga, I.; De Keersmaecker, K.; Van Vlierberghe, P.; Graux, C.; Cauwelier, B.; Lambert, F.; Mentens, N.; Beverloo, H.B.; Pieters, R.; Speleman, F.; et al. Duplication of the MYB oncogene in T cell acute lymphoblastic leukemia. Nat. Genet. 2007, 39, 593–595. [Google Scholar] [CrossRef]
- Clappier, E.; Cuccuini, W.; Kalota, A.; Crinquette, A.; Cayuela, J.M.; Dik, W.A.; Langerak, A.W.; Montpellier, B.; Nadel, B.; Walrafen, P.; et al. The C-MYB locus is involved in chromosomal translocation and genomic duplications in human T-cell acute leukemia (T-ALL), the translocation defining a new T-ALL subtype in very young children. Blood 2007, 110, 1251–1261. [Google Scholar] [CrossRef] [Green Version]
- O’Neil, J.; Tchinda, J.; Gutierrez, A.; Moreau, L.; Maser, R.S.; Wong, K.K.; Li, W.; McKenna, K.; Liu, X.S.; Feng, B.; et al. Alu elements mediate MYB gene tandem duplication in human T-ALL. J. Exp. Med. 2007, 204, 3059–3066. [Google Scholar] [CrossRef]
- Mansour, M.R.; Abraham, B.J.; Anders, L.; Berezovskaya, A.; Gutierrez, A.; Durbin, A.D.; Etchin, J.; Lawton, L.; Sallan, S.E.; Silverman, L.B.; et al. Oncogene regulation. An oncogenic super-enhancer formed through somatic mutation of a noncoding intergenic element. Science 2014, 346, 1373–1377. [Google Scholar] [CrossRef] [Green Version]
- Rahman, S.; Magnussen, M.; León, T.E.; Farah, N.; Li, Z.; Abraham, B.J.; Alapi, K.Z.; Mitchell, R.J.; Naughton, T.; Fielding, A.K.; et al. Activation of the LMO2 oncogene through a somatically acquired neomorphic promoter in T-cell acute lymphoblastic leukemia. Blood 2017, 129, 3221–3226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hess, J.L.; Bittner, C.B.; Zeisig, D.T.; Bach, C.; Fuchs, U.; Borkhardt, A.; Frampton, J.; Slany, R.K. Myb is an essential downstream target for homeobox-mediated transformation of hematopoietic cells. Blood 2006, 108, 297–304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Somervaille, T.C.; Matheny, C.J.; Spencer, G.J.; Iwasaki, M.; Rinn, J.L.; Witten, D.M.; Chang, H.Y.; Shurtleff, S.A.; Downing, J.R.; Cleary, M.L. Hierarchical maintenance of MLL myeloid leukemia stem cells employs a transcriptional program shared with embryonic rather than adult stem cells. Cell Stem Cell 2009, 4, 129–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuber, J.; Rappaport, A.R.; Luo, W.; Wang, E.; Chen, C.; Vaseva, A.V.; Shi, J.; Weissmueller, S.; Fellmann, C.; Taylor, M.J.; et al. An integrated approach to dissecting oncogene addiction implicates a Myb-coordinated self-renewal program as essential for leukemia maintenance. Genes Dev. 2011, 25, 1628–1640. [Google Scholar] [CrossRef] [Green Version]
- Drabsch, Y.; Hugo, H.; Zhang, R.; Dowhan, D.H.; Miao, Y.R.; Gewirtz, A.M.; Barry, S.C.; Ramsay, R.G.; Gonda, T.J. Mechanism of and requirement for estrogen-regulated MYB expression in estrogen-receptor-positive breast cancer cells. Proc. Natl. Acad. Sci. USA 2007, 104, 13762–13767. [Google Scholar] [CrossRef] [Green Version]
- Biroccio, A.; Benassi, B.; D’Agnano, I.; D’Angelo, C.; Buglioni, S.; Mottolese, M.; Ricciotti, A.; Citro, G.; Cosimelli, M.; Ramsay, R.G.; et al. Myb and Bcl-x overexpression predicts poor prognosis in colorectal cancer: Clinical and experimental findings. Am. J. Pathol. 2001, 158, 1289–1299. [Google Scholar] [CrossRef]
- Hugo, H.; Cures, A.; Suraweera, N.; Drabsch, Y.; Purcell, D.; Mantamadiotis, T.; Phillips, W.; Dobrovic, A.; Zupi, G.; Gonda, T.J.; et al. Mutations in the MYB intron I regulatory sequence increase transcription in colon cancers. Genes Chromosomes Cancer 2006, 45, 1143–1154. [Google Scholar] [CrossRef]
- Persson, M.; Andrén, Y.; Mark, J.; Horlings, H.M.; Persson, F.; Stenman, G. Recurrent fusion of MYB and NFIB transcription factor genes in carcinomas of the breast and head and neck. Proc. Natl. Acad. Sci. USA 2009, 106, 18740–18744. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Wu, G.; Miller, C.P.; Tatevossian, R.G.; Dalton, J.D.; Tang, B.; Orisme, W.; Punchihewa, C.; Parker, M.; Qaddoumi, I.; et al. Whole-genome sequencing identifies genetic alterations in pediatric low-grade gliomas. Nat. Genet. 2013, 45, 602–612. [Google Scholar]
- Bandopadhayay, P.; Ramkissoon, L.A.; Jain, P.; Bergthold, G.; Wala, J.; Zeid, R.; Schumacher, S.E.; Urbanski, L.; O’Rourke, R.; Gibson, W.J.; et al. MYB-QKI rearrangements in angiocentric glioma drive tumorigenicity through a tripartite mechanism. Nat. Genet. 2016, 48, 273–282. [Google Scholar] [CrossRef]
- Drier, Y.; Cotton, M.J.; Williamson, K.E.; Gillespie, S.M.; Ryan, R.J.; Kluk, M.J.; Carey, C.D.; Rodig, S.J.; Sholl, L.M.; Afrogheh, A.H.; et al. An oncogenic MYB feedback loop drives alternate cell fates in adenoid cystic carcinoma. Nat. Genet. 2016, 48, 265–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sebastian, T.; Johnson, P.F. Stop and go: Anti-proliferative and mitogenic functions of the transcription factor C/EBPbeta. Cell Cycle 2006, 5, 953–957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nerlov, C. The C/EBP family of transcription factors: A paradigm for interaction between gene expression and proliferation control. Trends Cell Biol. 2007, 17, 318–324. [Google Scholar] [CrossRef] [PubMed]
- Nerlov, C. C/EBPs: Recipients of extracellular signals through proteome modulation. Curr. Opin. Cell Biol. 2008, 20, 180–185. [Google Scholar] [CrossRef] [PubMed]
- Descombes, P.; Schibler, U. A liver-enriched transcriptional activator protein, LAP, and a transcriptional inhibitory protein, LIP, are translated from the same mRNA. Cell 1991, 67, 569–579. [Google Scholar] [CrossRef]
- Calkhoven, C.F.; Müller, C.; Leutz, A. Translational control of C/EBPα and C/EBPβ isoform expression. Genes Dev. 2000, 14, 1920–1932. [Google Scholar] [CrossRef]
- Kowenz-Leutz, E.; Twamley, G.; Ansieau, S.; Leutz, A. Novel mechanism of C/EBP beta (NF-M) transcriptional control: Activation through derepression. Genes Dev. 1994, 8, 2781–2791. [Google Scholar] [CrossRef] [Green Version]
- Buck, M.; Poli, V.; van der Geer, P.; Chojkier, M.; Hunter, T. Phosphorylation of rat serine 105 or mouse threonine 217 in C/EBPβ is required for hepatocyte proliferation induced by TGF. Mol. Cell 1999, 4, 1087–1092. [Google Scholar] [CrossRef]
- Eaton, E.M.; Sealy, L. Modification of CCAAT/enhancer-binding protein-beta by the small ubiquitin-like modifier (SUMO) family members, SUMO-2 and SUMO-3. J. Biol. Chem. 2003, 278, 33416–33421. [Google Scholar] [CrossRef] [Green Version]
- Joo, M.; Park, G.Y.; Wright, J.G.; Blackwell, T.S.; Atchison, M.L.; Christman, J.W. Transcriptional regulation of the cyclooxygenase-2 gene in macrophages by PU.1. J. Biol. Chem. 2004, 279, 6658–6665. [Google Scholar] [CrossRef] [Green Version]
- Steinmann, S.; Coulibaly, A.; Ohnheiser, J.; Jakobs, A.; Klempnauer, K.-H. Interaction and cooperation of the CCAAT-box enhancer binding protein β (C/EBPβ) with the homeo-domain interacting protein kinase 2 (Hipk2). J. Biol. Chem. 2013, 288, 22257–22269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ceseña, T.I.; Cui, T.X.; Subramanian, L.; Fulton, C.T.; Iñiguez-Lluhi, J.A.; Kwok, R.P.; Schwartz, J. Acetylation and deace- tylation regulate CCAAT/enhancer binding protein beta at K39 in mediating gene transcription. Mol. Cell. Endocrinol. 2008, 289, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Pless, O.; Kowenz-Leutz, E.; Knoblich, M.; Lausen, J.; Beyermann, M.; Walsh, M.J.; Leutz, A. G9a-mediated lysine methylation alters the function of CCAAT/enhancer-binding protein-beta. J. Biol. Chem. 2008, 283, 26357–26363. [Google Scholar] [CrossRef] [Green Version]
- Kowenz-Leutz, E.; Pless, O.; Dittmar, G.; Knoblich, M.; Leutz, A. Crosstalk between C/EBPbeta phosphorylation, arginine methylation, and SWI/SNF/Mediator implies an indexing transcription factor code. EMBO J. 2010, 29, 1105–1115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mink, S.; Haenig, B.; Klempnauer, K.-H. Interaction and functional collaboration of p300 and C/EBPβ. Mol. Cell. Biol. 1997, 17, 6609–66017. [Google Scholar] [CrossRef] [Green Version]
- Kowenz-Leutz, E.; Leutz, A. A C/EBPβ isoform recruits the SWI/SNF complex to activate myeloid genes. Mol. Cell 1999, 4, 735–743. [Google Scholar] [CrossRef]
- Dittmar, G.; Hernandez, D.P.; Kowenz-Leutz, E.; Kirchner, M.; Kahlert, G.; Wesolowski, R.; Baum, K.; Knoblich, M.; Hofstätter, M.; Muller, A.; et al. PRISMA: Protein interaction screen on peptide matrix reveals interaction footprints and modifications-dependent interactome of intrinsically disordered C/ EBPβ. iScience 2019, 13, 351–370. [Google Scholar] [CrossRef] [Green Version]
- Rask, K.; Thörn, M.; Pontén, F.; Kraaz, W.; Sundfeldt, K.; Hedin, L.; Enerbäck, S. Increased expression of the transcription factors CCAAT-enhancer binding protein-beta (C/EBPbeta) and C/EBPzeta (CHOP) correlate with invasiveness of human colorectal cancer. Int. J. Cancer 2000, 86, 337–343. [Google Scholar] [CrossRef]
- Kim, M.H.; Minton, A.Z.; Agarwal, V. C/EBPbeta regulates metastatic gene expression and confers TNF-alpha resistance to prostate cancer cells. Prostate 2009, 69, 1435–1447. [Google Scholar] [CrossRef]
- Regalo, G.; Förster, S.; Resende, C.; Bauer, B.; Fleige, B.; Kemmner, W.; Schlag, P.M.; Meyer, T.F.; Machado, J.C.; Leutz, A. C/EBPβ regulates homeostatic and oncogenic gastric cell proliferation. J. Mol. Med. 2016, 94, 1385–1395. [Google Scholar] [CrossRef] [Green Version]
- Aguilar-Morante, D.; Morales-Garcia, J.A.; Santos, A.; Perez-Castillo, A. CCAAT/enhancer binding protein beta induces motility and invasion of glioblastome cells through transcriptional regulation of the calcium binding protein S100A4. Oncotarget 2015, 6, 4369–4384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Homma, J.; Yamanaka, R.; Yajima, N.; Tsuchiya, N.; Genkai, N.; Sano, M.; Tanaka, R. Increased expression of CCAAT/enhancer binding protein beta correlates with prognosis in glioma patients. Oncol. Rep. 2006, 15, 595–601. [Google Scholar] [PubMed]
- Carro, M.S.; Lim, W.K.; Alvarez, M.J.; Bollo, R.J.; Zhao, X.; Snyder, E.Y.; Sulman, E.P.; Anne, S.L.; Doetsch, F.; Colman, H.; et al. The transcriptional network for mesenchymal transformation of brain tumours. Nature 2010, 463, 318–327. [Google Scholar] [CrossRef] [PubMed]
- Piva, R.; Pellegrino, E.; Mattioli, M.; Agnelli, L.; Lombardi, L.; Boccalatte, F.; Costa, G.; Ruggeri, B.A.; Cheng, M.; Chiarle, R.; et al. Functional validation of the anaplastic lymphoma kinase signature identifies CEBPB and BCL2A1 as critical target genes. J. Clin. Investig. 2006, 116, 3171–3182. [Google Scholar] [CrossRef]
- Anastasov, N.; Bonzheim, I.; Rudelius, M.; Klier, M.; Dau, T.; Angermeier, D.; Duyster, J.; Pittaluga, S.; Fend, F.; Raffeld, M.; et al. C/EBPβ expression in ALK-positive anaplastic large cell lymphomas is required for cell proliferation and is induced by the STAT3 signaling pathway. Haematologica 2010, 95, 760–767. [Google Scholar] [CrossRef] [Green Version]
- Yusenko, M.V.; Trentmann, A.; Casolari, D.A.; Abdel Ghani, L.; Lenz, M.; Horn, M.; Dörner, W.; Klempnauer, S.; Mootz, H.D.; Arteaga, M.F.; et al. C/EBPβ is a pro-leukemogenic factor and promising drug target in acute myeloid leukemia. Oncogene 2021, 40, 4746–4758. [Google Scholar] [CrossRef]
- Klempnauer, K.-H. C/EBPβ sustains the oncogenic program of AML cells by cooperating with MYB and co-activator p300 in a transcriptional module. Exp. Hematol. 2022. Online ahead of print. [Google Scholar] [CrossRef]
- Bujnicki, T.; Wilczek, C.; Schomburg, C.; Feldmann, F.; Schlenke, P.; Müller-Tidow, C.; Schmidt, T.J.; Klempnauer, K.-H. Inhibition of Myb-dependent gene expression by the sesquiterpene lactone mexicanin-I. Leukemia 2012, 26, 615–622. [Google Scholar] [CrossRef]
- Newman, D.J.; Cragg, G.M. Natural Products as Sources of New Drugs over the Nearly Four Decades from 01/1981 to 09/2019. J. Nat. Prod. 2020, 83, 770–803. [Google Scholar] [CrossRef]
- Picman, A.K. Biological Activities of Sesquiterpene Lactones. Biochem. Syst. Ecol. 1986, 14, 255–281. [Google Scholar] [CrossRef]
- Schmidt, T.J. Toxic Activities of Sesquiterpene Lactones—Structural and Biochemical Aspects. Current Org. Chem. 1999, 3, 577–605. [Google Scholar]
- Schmidt, T.J. Structure-Activity Relationships of Sesquiterpene Lactones. In Studies in Natural Products Chemistry; Atta ur, R., Ed.; Elsevier Science: Amsterdam, The Netherlands, 2006; Volume 33, Part M, pp. 309–392. [Google Scholar]
- Merfort, I. Perspectives on Sesquiterpene Lactones in Inflammation and Cancer. Curr. Drug Targets 2011, 12, 1560–1573. [Google Scholar] [CrossRef] [PubMed]
- Quintana, J.; Estévez, F. Recent Advances on Cytotoxic Sesquiterpene Lactones. Curr. Pharm. Des. 2018, 24, 4355–4361. [Google Scholar] [CrossRef] [PubMed]
- Schomburg, C. Naturstoffe als Inhibitoren c-Myb-Abhängiger Transkriptionsprozesse. Ph.D. Thesis, University of Münster, Münster, Germany, 2013. [Google Scholar]
- Seaman, F.C. Sesquiterpene lactones as taxonomic characters in the Asteraceae. Bot. Rev. 1982, 48, 121–594. [Google Scholar] [CrossRef]
- Schomburg, C.; Schuehly, W.; Da Costa, F.B.; Klempnauer, K.H.; Schmidt, T.J. Natural sesquiterpene lactones as inhibitors of Myb-dependent gene expression: Structure-activity relationships. Eur. J. Med. Chem. 2013, 63, 313–320. [Google Scholar] [CrossRef]
- Schmidt, T.J. Helenanolide Type Sesquiterpene Lactones. III. Rates and Stereochemistry in the Reaction of Helenalin and Related Helenanolides with Sulfhydryl Containing Biomolecules. Bioorg. Med. Chem. 1997, 5, 645–653. [Google Scholar] [CrossRef]
- Schmidt, T.J.; Lyß, G.; Pahl, H.L.; Merfort, I. Helenanolide Type Sesquiterpene Lactones-V. The Role of Glutathione Addition under Physiological Conditions. Bioorg. Med. Chem. 1999, 7, 2849–2855. [Google Scholar] [CrossRef]
- LoPachin, R.M.; Gavin, T.; DeCaprio, A.; Barber, D.S. Application of the Hard and Soft, Acids and Bases (HSAB) Theory to Toxicant-Target Interactions. Chem. Res. Toxicol. 2012, 25, 239–251. [Google Scholar] [CrossRef]
- Jakobs, A.; Uttarkar, S.; Schomburg, C.; Steinmann, S.; Coulibaly, A.; Schlenke, P.; Berdel, W.; Müller-Tidow, C.; Schmidt, T.J.; Klempnauer, K.-H. An isoform-specific C/EBPβ inhibitor targets acute myeloid leukemia cells. Leukemia 2016, 30, 1612–1615. [Google Scholar] [CrossRef]
- Jakobs, A.; Steinmann, S.; Henrich, S.M.; Schmidt, T.J.; Klempnauer, K.-H. Helenalin acetate, a natural sesquiterpene lactone with anti-inflammatory and anti-cancer activity, disrupts the cooperation of CCAAT-box/enhancer-binding protein beta (C/EBPβ) and co-activator p300. J. Biol. Chem. 2016, 291, 26098–26108. [Google Scholar] [CrossRef] [Green Version]
- Abdel Ghani, L.; Yusenko, M.V.; Frank, D.; Moorthy, R.; Widen, J.C.; Dörner, W.; Khandanpour, C.; Harki, D.A.; Klempnauer, K.-H. A synthetic covalent ligand of the C/EBPβ transactivation domain inhibits acute myeloid leukemia cells. Cancer Lett. 2022, 530, 170–180. [Google Scholar] [CrossRef]
- Schmidt, T.J. Helenanolide Type Sesquiterpene Lactones. I. Conformations and Molecular Dynamics of Helenalin, its Esters and 11,13-Dihydro-Derivatives. J. Mol. Struct. 1996, 385, 99–112. [Google Scholar] [CrossRef]
- Ichinose, Y.; Wakamatsu, K.; Nozaki, K.; Birbaum, J.-L.; Oshima, K.; Utimoto, K. Et3B Induced Radical Addition of Thiols to Acetylenes. Chem. Lett. 1987, 16, 1647–1650. [Google Scholar] [CrossRef]
- Lowe, A.B.; Hoyle, C.E.; Bowman, C.N. Thiol-yne click chemistry: A powerful and versatile methodology for materials synthesis. J. Mater. Chem. 2010, 20, 4745–4750. [Google Scholar] [CrossRef]
- Petri, L.; Ábrányi-Balogh, P.; Varga, P.R.; Imre, T.; Keserű, G.M. Comparative reactivity analysis of small-molecule thiol surrogates. Bioorg. Med. Chem. 2020, 28, 115357. [Google Scholar] [CrossRef]
- Drogosz, J.; Janecka, A. Helenalin—A Sesquiterpene Lactone with Multidirectional Activity. Curr. Drug Targets 2019, 20, 444–452. [Google Scholar] [CrossRef]
- Roe, J.-S.; Mercan, F.; Rivera, K.; Pappin, D.J.; Vakoc, C.R. BET bromodomain inhibition suppresses the function of hematopoietic transcription factors in acute myeloid leukemia. Mol. Cell 2015, 58, 1028–1039. [Google Scholar] [CrossRef] [Green Version]
- Lyss, G.; Knorre, A.; Schmidt, T.J.; Pahl, H.L.; Merfort, I. The Anti-Inflammatory Sesquiterpene Lactone Helenalin Inhibits the Transcription Factor NF-kappaB by directly targeting p65. J. Biol. Chem. 1998, 273, 33508–33516. [Google Scholar] [CrossRef] [Green Version]
- Rüngeler, P.; Castro, V.; Mora, G.; Gören, N.; Vichnewski, W.; Pahl, H.L.; Merfort, I.; Schmidt, T.J. Inhibition of Transcription Factor NF-kappa B by Sesquiterpene Lactones—A Proposed Molecular Mechanism of Action. Bioorg. Med. Chem. 1999, 7, 2343–2352. [Google Scholar] [CrossRef]
- Garcia-Pineres, A.J.; Castro, V.; Mora, G.; Schmidt, T.J.; Strunck, E.; Pahl, H.L.; Merfort, I. Cysteine 38 in p65/NF-κB Plays a Crucial Role in DNA Binding Inhibition by Sesquiterpene Lactones*. J. Biol. Chem. 2001, 276, 39713–39720. [Google Scholar] [CrossRef] [Green Version]
- Willuhn, G. Arnicae flos. In Herbal Drugs and Phytopharmaceuticals: A Handbook for Practice on a Scientific Basis, 3rd ed.; Wichtl, M., Ed.; Medpharm: Stuttgart, Germany; CRC Press: Boca Raton, FL, USA, 2004; pp. 54–59. [Google Scholar]
- Sticher, O.; Heilmann, J.; Zündorf, I. Hänsel/Sticher Pharmakognosie Phytopharmazie, 10th ed.; Wissenschaftliche Verlagsgesellschaft: Stuttgart, Germany, 2015. [Google Scholar]
- Natsuka, S.; Akira, S.; Nishio, Y.; Hashimoto, S.; Sugita, T.; Isshiki, H.; Kishimoto, T. Macrophage differentiation-specific expression of NF-IL6, a transcription factor for interleukin-6. Blood 1992, 79, 460–466. [Google Scholar] [CrossRef] [Green Version]
- Davydov, I.V.; Krammer, P.H.; Li-Weber, M. Nuclear factor-IL6 activates the human IL-4 promoter in T cells. J. Immunol. 1995, 155, 5273–5279. [Google Scholar] [PubMed]
- van Dijk, T.B.; Baltus, B.; Raaijmakers, J.A.; Lammers, J.W.; Koenderman, L.; de Groot, R.P. A composite C/EBP binding site is essential for the activity of the promoter of the IL-3/IL-5/granulocyte-macrophage colony-stimulating factor receptor beta c gene. J. Immunol. 1999, 163, 2674–2680. [Google Scholar] [PubMed]
- Greenwel, P.; Tanaka, S.; Penkov, D.; Zhang, W.; Olive, M.; Moll, J.; Vinson, C.; Di Liberto, M.; Ramirez, F. Tumor Necrosis Factor Alpha Inhibits Type I Collagen Synthesis through Repressive CCAAT/Enhancer-Binding Proteins. Mol. Cell. Biol. 2000, 20, 912–918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huber, R.; Pietsch, D.; Panterodt, T.; Brand, K. Regulation of C/EBPβ and resulting functions in cells of the monocytic lineage. Cell Signal. 2012, 24, 1287–1296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyss, G.; Schmidt, T.J.; Pahl, H.L.; Merfort, I. Studies on the anti-inflammatory activity of Arnica tincture using the transcription factor NF-kappa B as molecular target. Pharm. Pharmacol. Lett. 1999, 9, 5–8. [Google Scholar]
- Chen, L.-X.; He, H.; Qiu, F. Natural withanolides: An overview. Nat. Prod. Rep. 2011, 28, 705–740. [Google Scholar] [CrossRef] [PubMed]
- Xia, G.-Y.; Cao, S.-J.; Chen, L.-X.; Qiu, F. Natural withanolides, an update. Nat. Prod. Rep. 2021. [Google Scholar] [CrossRef]
- Falkenberg, K.D.; Jakobs, A.; Matern, J.C.; Dörner, W.; Uttarkar, S.; Trentmann, A.; Steinmann, S.; Coulibaly, A.; Schomburg, C.; Mootz, H.D.; et al. A natural compound with anti-tumor activity, is a potent inhibitor of transcription factor C/EBPβ. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 1349–1358. [Google Scholar] [CrossRef]
- Shi, L.H.; Wu, X.J.; Liu, J.S.; Gao, Y.B. Withaferin A activates stress signalling proteins in high risk acute lymphoblastic leukemia. Int. J. Clin. Exp. Pathol. 2015, 8, 15652–15660. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4730047/ (accessed on 21 March 2022).
- Salminen, A.; Lehtonen, M.; Paimela, T.; Kaarniranta, K. Celastrol: Molecular targets of Thunder God Vine. Biochem. Biophys. Res. Commun. 2010, 394, 439–442. [Google Scholar] [CrossRef]
- Uttarkar, S.; Dassé, E.; Coulibaly, A.; Steinmann, S.; Jakobs, A.; Schomburg, C.; Trentmann, A.; Jose, J.; Schlenke, P.; Berdel, W.E.; et al. Targeting acute myeloid leukemia with a small molecule inhibitor of the Myb/p300 Interaction. Blood 2016, 127, 1173–1182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coulibaly, A.; Haas, A.; Steinmann, S.; Jakobs, A.; Schmidt, T.J.; Klempnauer, K.-H. The natural anti-tumor compound Celastrol targets a Myb-C/EBPβ-p300 transcriptional module implicated in myeloid gene expression. PLoS ONE 2018, 13, e0190934. [Google Scholar] [CrossRef] [Green Version]
- Bhaumik, P.; Davis, J.; Tropea, J.E.; Cherry, S.; Johnson, P.F.; Miller, M. Structural insights into interactions of C/EBP transcriptional activators with the Taz2 domain of p300. Acta Crystallogr. D Biol. Crystallogr. 2014, 70, 1914–1921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uttarkar, S.; Piontek, T.; Dukare, S.; Schomburg, C.; Schlenke, P.; Berdel, W.E.; Müller-Tidow, C.; Schmidt, T.J.; Klempnauer, K.H. Small-Molecule Disruption of the Myb/p300 Cooperation Targets Acute Myeloid Leukemia Cells. Mol. Cancer Ther. 2016, 15, 2905–2915. [Google Scholar] [CrossRef] [Green Version]
- Qiu, H.-Y.; Wang, P.-F.; Lin, H.-Y.; Tang, C.-Y.; Zhu, H.-L.; Yang, Y.-H. Naphthoquinones: A continuing source for discovery of therapeutic antineoplastic agents. Chem. Biol. Drug Des. 2018, 91, 681–690. [Google Scholar] [CrossRef]
- Ellendorf, T. Entdeckung Bioaktiver Naturstoffe unter Verwendung der Multivariaten Datenanalyse (MVDA). Ph.D. Thesis, University of Münster, Münster, Germany, 2014. [Google Scholar]
- Ravindra, K.C.; Selvi, B.R.; Arif, M.; Reddy, B.A.; Thanuja, G.R.; Agrawal, S.; Pradhan, S.K.; Nagashayana, N.; Dasgupta, D.; Kundu, T.K. Inhibition of lysine acetyltransferase KAT3B/p300 activity by a naturally occurring hydroxynaphthoquinone, plumbagin. J. Biol. Chem. 2009, 284, 24453–24464. [Google Scholar] [CrossRef] [Green Version]
- Olgen, S. Overview on Anticancer Drug Design and Development. Curr. Med. Chem. 2018, 25, 1704–1719. [Google Scholar] [CrossRef]
- Anighoro, A.; Bajorath, J.; Rastelli, G. Polypharmacology: Challenges and Opportunities in Drug Discovery. J. Med. Chem. 2014, 57, 7874–7887. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | NP Class a | MYB | ±SD | MTS | ±SD | SI |
|---|---|---|---|---|---|---|
| Shikonin | NQ | 0.30 | ±0.06 | 16.11 | ±8.32 | 53.7 |
| Plumbagin | NQ | 0.56 | ±0.18 | 11.45 | ±5.30 | 20.4 |
| Goyazensolide | STL | 0.63 | ±0.19 | 10.05 | ±3.96 | 16.0 |
| Helenalin acetate | STL | 0.71 | ±0.03 | 7.34 | ±0.95 | 10.3 |
| Pristimerin | QMT | 0.73 | ±0.15 | 6.24 | ±2.53 | 8.5 |
| Celastrol | QMT | 0.85 | ±0.10 | 6.62 | ±2.35 | 7.8 |
| Juglone | NQ | 0.98 | ±0.03 | >30 | >30 | |
| Warburganal | SDA | 1.54 | ±0.97 | 12.80 | ±5.27 | 8.3 |
| 4,15-iso-Atriplicolide tiglate | STL | 1.57 | ±0.47 | 12.07 | ±6.46 | 7.7 |
| Withaferin A | SEL | 1.77 | ±0.16 | 15.67 | ±11.09 | 8.9 |
| Mexicanin I | STL | 1.80 | ±0.06 | 21.13 | ±2.19 | 11.7 |
| 4,15-iso-Atriplicolide methacrylate | STL | 2.21 | ±0.32 | 10.38 | ±3.96 | 4.7 |
| Helenalin | STL | 2.37 | ±0.39 | 21.11 | ±2.63 | 8.9 |
| Budlein A | STL | 2.39 | ±0.25 | 9.00 | ±2.96 | 3.8 |
| Helenalin isobutyrate | STL | 2.54 | ±0.28 | >30 | >12 | |
| Primin | BQ | 2.73 | ±0.21 | 22.11 | ±4.89 | 8.1 |
| Tagitinin C | STL | 2.92 | ±1.08 | 30.91 | ±10.93 | 10.6 |
| Enhydrin | STL | 2.95 | ±1.23 | 7.49 | ±0.29 | 2.5 |
| Uvedalin | STL | 3.83 | ±1.22 | 11.42 | ±4.19 | 3.0 |
| Perezone | BQ | 4.57 | ±0.29 | 30.0 | ±13.81 | 6.6 |
| Melcanthin C | STL | 4.68 | ±1.63 | 24.80 | ±4.36 | 5.3 |
| Nobilin | STL | 4.73 | ±1.40 | >30 | >6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schmidt, T.J.; Klempnauer, K.-H. Natural Products with Antitumor Potential Targeting the MYB-C/EBPβ-p300 Transcription Module. Molecules 2022, 27, 2077. https://doi.org/10.3390/molecules27072077
Schmidt TJ, Klempnauer K-H. Natural Products with Antitumor Potential Targeting the MYB-C/EBPβ-p300 Transcription Module. Molecules. 2022; 27(7):2077. https://doi.org/10.3390/molecules27072077
Chicago/Turabian StyleSchmidt, Thomas J., and Karl-Heinz Klempnauer. 2022. "Natural Products with Antitumor Potential Targeting the MYB-C/EBPβ-p300 Transcription Module" Molecules 27, no. 7: 2077. https://doi.org/10.3390/molecules27072077
APA StyleSchmidt, T. J., & Klempnauer, K.-H. (2022). Natural Products with Antitumor Potential Targeting the MYB-C/EBPβ-p300 Transcription Module. Molecules, 27(7), 2077. https://doi.org/10.3390/molecules27072077