
Neuroprotective Effects of Green Tea Seed Isolated Saponin Due to the Amelioration of Tauopathy and Alleviation of Neuroinflammation: A Therapeutic Approach to Alzheimer’s Disease

 ,
,
Abstract
:
1. Introduction
2. Results
2.1. MTT Assay
2.2. Effects of Saponins on the Activities of Various Kinases and Phosphatases
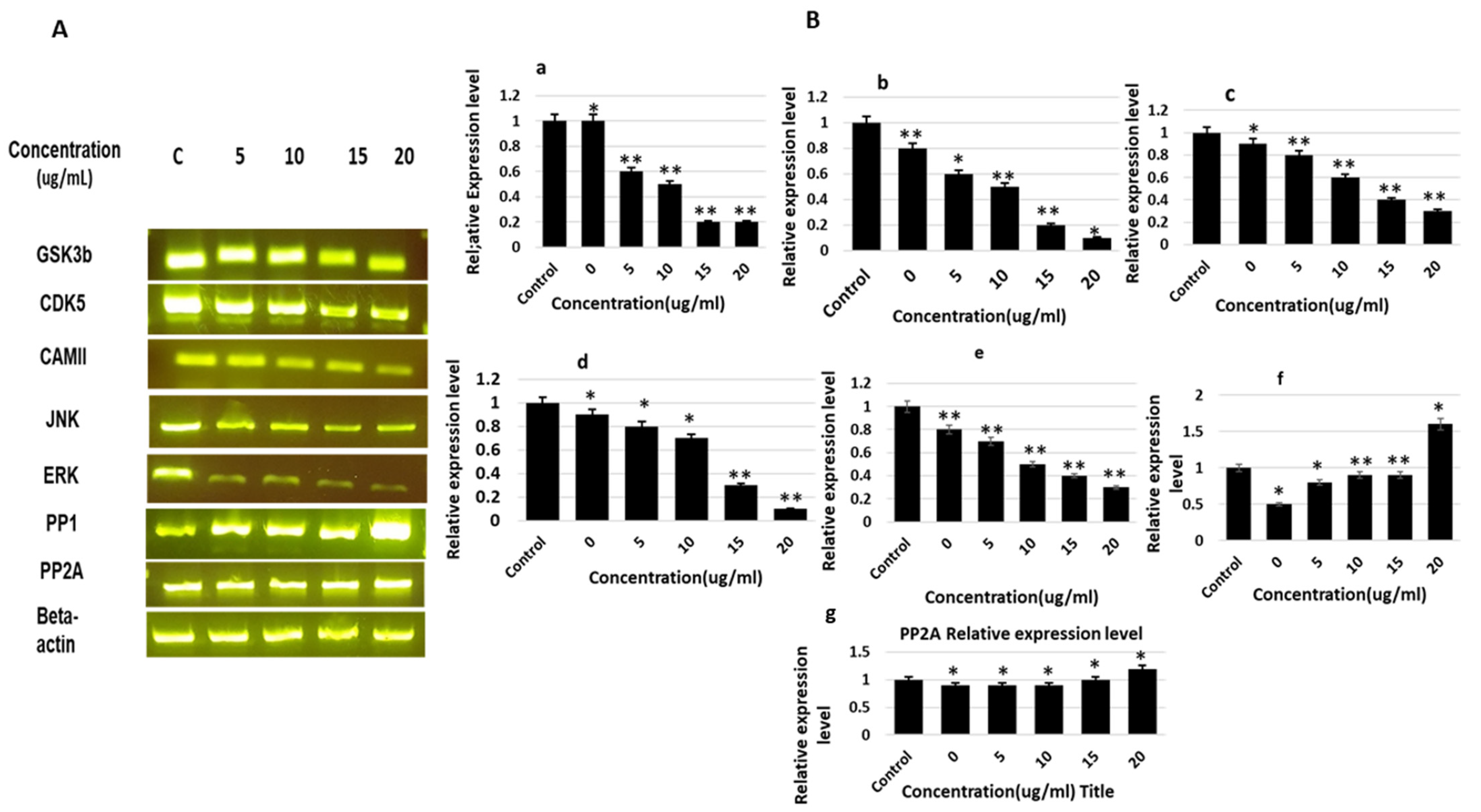
2.3. Gene Expression of Various Kinases and Phosphatases
2.4. Effects of Theasaponin E1 on the Expression of Genes Involved in AD Pathology
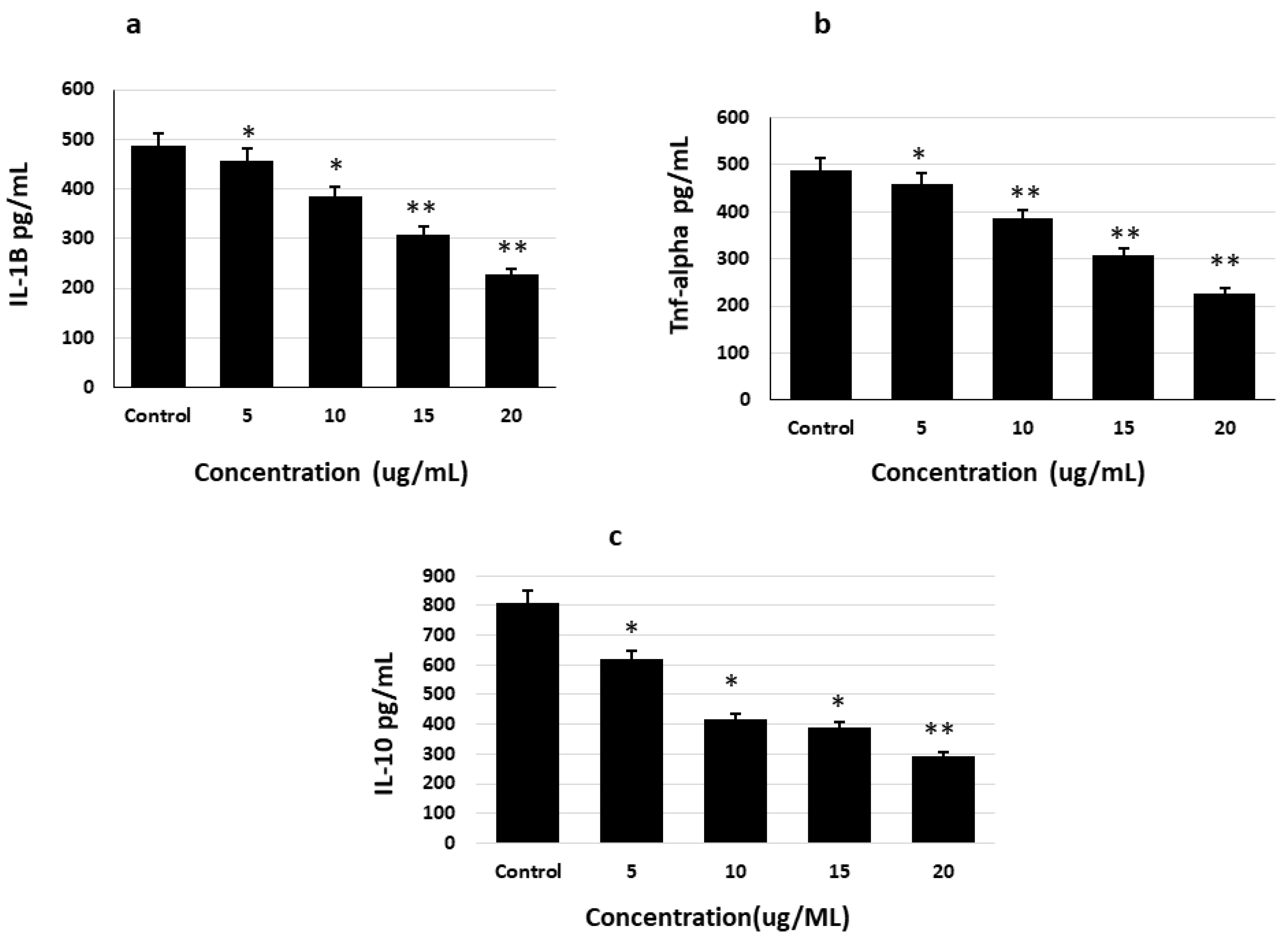
2.5. Effects of Saponins on Proinflammatory Cytokines
2.6. Determination of Aβ and P-Tau Levels Using ELISA
2.7. Quantification of P-Tau in SH-SY5Y Cells Using Western Blotting
3. Discussion
4. Materials and Methods
4.1. Process of Theasaponin E1 from Isolation and Purification
4.2. MTT Assay
4.3. qRT-PCR Analysis
4.4. Quantification of the In Vitro Activities of Kinases and Phosphatases
4.5. Effects of Saponins on Proinflammatory Cytokines and Related Pathways in Glial Cells
4.6. Determination of the Levels of Aβ and Phosphorylated Tau (P-Tau) Using ELISA
4.7. Quantification of Phosphorylated Tau (P-Tau) in SH-SY5Y Cells Using Western Blotting
4.8. Quantification of Proinflammatory Cytokines in Glial Cells
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Mayeux, R. Epidemiology of neurodegeneration. Annu. Rev. Neurosci. 2003, 26, 81–104. [Google Scholar] [CrossRef] [PubMed]
- Ferri, C.P.; Prince, M.; Brayne, C.; Brodaty, H.; Fratiglioni, L.; Ganguli, M.; Hall, K.; Hasegawa, K.; Hendrie, H.; Huang, Y.; et al. Global prevalence of dementia: A Delphi consensus study. Lancet 2005, 366, 2112–2117. [Google Scholar] [CrossRef]
- Kukull, W.A.; Higdon, R.; Bowen, J.D.; McCormick, W.C.; Teri, L.; Schellenberg, G.D.; van Belle, G.; Jolley, L.; Larson, E.B. Dementia and Alzheimer disease incidence: A prospective cohort study. Arch. Neurol. 2002, 59, 1737–1746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bird, T.D. Genetic aspects of Alzheimer disease. Genet. Med. 2008, 10, 231–239. [Google Scholar] [CrossRef] [Green Version]
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef] [Green Version]
- Serrano-Pozo, A.; Frosch, M.P.; Masliah, E.; Hyman, B.T. Neuropathological alterations in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2011, 1, a006189. [Google Scholar] [CrossRef]
- Holtzman, D.M.; Morris, J.C.; Goate, A.M. Alzheimer’s disease: The challenge of the second century. Sci. Transl. Med. 2011, 3, 77sr1. [Google Scholar] [CrossRef] [Green Version]
- Garcia, M.L.; Cleveland, D.W. Going new places using an old MAP: Tau, microtubules and human neurodegenerative disease. Curr. Opin. Cell Biol. 2001, 13, 41–48. [Google Scholar] [CrossRef]
- Binder, L.I.; Guillozet-Bongaarts, A.L.; Garcia-Sierra, F.; Berry, R.W. Tau, tangles, and Alzheimer’s disease. Biochim. Biophys. Acta 2005, 1739, 216–223. [Google Scholar] [CrossRef] [Green Version]
- Cuchillo-Ibanez, I.; Seereeram, A.; Byers, H.L.; Leung, K.Y.; Ward, M.A.; Anderton, B.H.; Hanger, D. Phosphorylation of tau regulates its axonal transport by controlling its binding to kinesin. FASEB J. 2008, 22, 3186–3195. [Google Scholar] [CrossRef]
- Trinczek, B.; Ebneth, A.; Mandelkow, E.M.; Mandelkow, E. Tau regulates the attachment/detachment but not the speed of motors in microtubule-dependent transport of single vesicles and organelles. J. Cell Sci. 1999, 112 Pt 14, 2355–2367. [Google Scholar] [CrossRef]
- Drubin, D.G.; Kirschner, M.W. Tau protein function in living cells. J. Cell Biol. 1986, 103 Pt 2, 2739–2746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grundke-Iqbal, I.; Iqbal, K.; Tung, Y.C.; Quinlan, M.; Wisniewski, H.M.; Binder, L.I. Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc. Natl. Acad. Sci. USA 1986, 83, 4913–4917. [Google Scholar] [CrossRef] [Green Version]
- Brion, J.P.; Couck, A.M.; Passareiro, E.; Flament-Durand, J. Neurofibrillary tangles of Alzheimer’s disease: An immunohistochemical study. J. Submicrosc. Cytol. 1985, 17, 89–96. [Google Scholar] [PubMed]
- Singh, T.J.; Haque, N.; Grundke-Iqbal, I.; Iqbal, K. Rapid Alzheimer-like phosphorylation of tau by the synergistic actions of non-proline-dependent protein kinases and GSK-3. FEBS Lett. 1995, 358, 267–272. [Google Scholar] [CrossRef] [Green Version]
- Cohen, P.; Frame, S. The renaissance of GSK3. Nat. Rev. Mol. Cell Biol. 2001, 2, 769–776. [Google Scholar] [CrossRef] [PubMed]
- Beurel, E.; Grieco, S.F.; Jope, R.S. Glycogen synthase kinase-3 (GSK3): Regulation, actions, and diseases. Pharmacol. Ther. 2015, 148, 114–131. [Google Scholar] [CrossRef] [Green Version]
- Li, T.; Paudel, H.K. Glycogen synthase kinase 3beta phosphorylates Alzheimer’s disease-specific Ser396 of microtubule-associated protein tau by a sequential mechanism. Biochemistry 2006, 45, 3125–3133. [Google Scholar] [CrossRef]
- Stancu, I.C.; Ris, L.; Vasconcelos, B.; Marinangeli, C.; Goeminne, L.; Laporte, V.; Haylani, L.E.; Couturier, J.; Schakman, O.; Gailly, P.; et al. Tauopathy contributes to synaptic and cognitive deficits in a murine model for Alzheimer’s disease. FASEB J. 2014, 28, 2620–2631. [Google Scholar] [CrossRef]
- Sudhakar, V.; Richardson, R.M. Gene Therapy for Neurodegenerative Diseases. Neurotherapeutics 2019, 16, 166–175. [Google Scholar] [CrossRef] [Green Version]
- Shaftel, S.S.; Kyrkanides, S.; Olschowka, J.A.; Miller, J.N.; Johnson, R.E.; O’Banion, M.K. Sustained hippocampal IL-1 beta overexpression mediates chronic neuroinflammation and ameliorates Alzheimer plaque pathology. J. Clin. Investig. 2007, 117, 1595–1604. [Google Scholar] [CrossRef]
- Griffin, W.S.; Stanley, L.C.; Ling, C.; White, L.; MacLeod, V.; Perrot, L.J.; White, C.L., 3rd; Araoz, C. Brain interleukin 1 and S-100 immunoreactivity are elevated in Down syndrome and Alzheimer disease. Proc. Natl. Acad. Sci. USA 1989, 86, 7611–7615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perlmutter, L.S.; Barron, E.; Chui, H.C. Morphologic association between microglia and senile plaque amyloid in Alzheimer’s disease. Neurosci. Lett. 1990, 119, 32–36. [Google Scholar] [CrossRef]
- Yuan, P.; Condello, C.; Keene, C.D.; Wang, Y.; Bird, T.D.; Paul, S.M.; Luo, W.; Colonna, M.; Baddeley, D.; Grutzendler, J. TREM2 Haplodeficiency in Mice and Humans Impairs the Microglia Barrier Function Leading to Decreased Amyloid Compaction and Severe Axonal Dystrophy. Neuron 2016, 90, 724–739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmid, C.D.; Sautkulis, L.N.; Danielson, P.E.; Cooper, J.; Hasel, K.W.; Hilbush, B.S.; Sutcliffe, J.G.; Carson, M.J. Heterogeneous expression of the triggering receptor expressed on myeloid cells-2 on adult murine microglia. J. Neurochem. 2002, 83, 1309–1320. [Google Scholar] [CrossRef] [Green Version]
- Jay, T.R.; von Saucken, V.E.; Landreth, G.E. TREM2 in Neurodegenerative Diseases. Mol. Neurodegener. 2017, 12, 56. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Cella, M.; Mallinson, K.; Ulrich, J.D.; Young, K.L.; Robinette, M.L.; Gilfillan, S.; Krishnan, G.M.; Sudhakar, S.; Zinselmeyer, B.; et al. TREM2 lipid sensing sustains the microglial response in an Alzheimer’s disease model. Cell 2015, 160, 1061–1071. [Google Scholar] [CrossRef] [Green Version]
- Atagi, Y.; Liu, C.C.; Painter, M.M.; Chen, X.F.; Verbeeck, C.; Zheng, H.; Li, X.; Rademakers, R.; Kang, S.S.; Xu, H.; et al. Apolipoprotein E Is a Ligand for Triggering Receptor Expressed on Myeloid Cells 2 (TREM2). J. Biol. Chem. 2015, 290, 26043–26050. [Google Scholar] [CrossRef] [Green Version]
- Bailey, C.C.; DeVaux, L.B.; Farzan, M. The Triggering Receptor Expressed on Myeloid Cells 2 Binds Apolipoprotein E. J. Biol. Chem. 2015, 290, 26033–26042. [Google Scholar] [CrossRef] [Green Version]
- Yeh, F.L.; Wang, Y.; Tom, I.; Gonzalez, L.C.; Sheng, M. TREM2 Binds to Apolipoproteins, Including APOE and CLU/APOJ, and Thereby Facilitates Uptake of Amyloid-Beta by Microglia. Neuron 2016, 91, 328–340. [Google Scholar] [CrossRef]
- Zhao, Y.; Wu, X.; Li, X.; Jiang, L.L.; Gui, X.; Liu, Y.; Sun, Y.; Zhu, B.; Piña-Crespo, J.; Zhang, M.; et al. TREM2 Is a Receptor for β-Amyloid that Mediates Microglial Function. Neuron 2018, 97, 1023–1031.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, L.; Wang, Z.; Wang, D.; Wang, Z.; Martens, Y.A.; Wu, L.; Xu, Y.; Wang, K.; Li, J.; Huang, R.; et al. Amyloid-beta modulates microglial responses by binding to the triggering receptor expressed on myeloid cells 2 (TREM2). Mol. Neurodegener. 2018, 13, 15. [Google Scholar] [CrossRef] [PubMed]
- Gratuze, M.; Leyns, C.E.G.; Holtzman, D.M. New insights into the role of TREM2 in Alzheimer’s disease. Mol. Neurodegener. 2018, 13, 66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filipello, F.; Morini, R.; Corradini, I.; Zerbi, V.; Canzi, A.; Michalski, B.; Erreni, M.; Markicevic, M.; Starvaggi-Cucuzza, C.; Otero, K.; et al. The Microglial Innate Immune Receptor TREM2 Is Required for Synapse Elimination and Normal Brain Connectivity. Immunity 2018, 48, 979.e978–991.e978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ulland, T.K.; Song, W.M.; Huang, S.C.; Ulrich, J.D.; Sergushichev, A.; Beatty, W.L.; Loboda, A.A.; Zhou, Y.; Cairns, N.J.; Kambal, A.; et al. TREM2 Maintains Microglial Metabolic Fitness in Alzheimer’s Disease. Cell 2017, 170, 649.e613–663.e613. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Jia, L.; Liu, C.C.; Rong, Z.; Zhong, L.; Yang, L.; Chen, X.F.; Fryer, J.D.; Wang, X.; Zhang, Y.W.; et al. TREM2 Promotes Microglial Survival by Activating Wnt/β-Catenin Pathway. J. Neurosci. 2017, 37, 1772–1784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, L.; Chen, X.F.; Zhang, Z.L.; Wang, Z.; Shi, X.Z.; Xu, K.; Zhang, Y.W.; Xu, H.; Bu, G. DAP12 Stabilizes the C-terminal Fragment of the Triggering Receptor Expressed on Myeloid Cells-2 (TREM2) and protects against LPS-induced Pro-inflammatory Response. J. Biol. Chem. 2015, 290, 15866–15877. [Google Scholar] [CrossRef] [Green Version]
- Jay, T.R.; Miller, C.M.; Cheng, P.J.; Graham, L.C.; Bemiller, S.; Broihier, M.L.; Xu, G.; Margevicius, D.; Karlo, J.C.; Sousa, G.L.; et al. TREM2 deficiency eliminates TREM2+ inflammatory macrophages and ameliorates pathology in Alzheimer’s disease mouse models. J. Exp. Med. 2015, 212, 287–295. [Google Scholar] [CrossRef]
- Feuerbach, D.; Schindler, P.; Barske, C.; Joller, S.; Beng-Louka, E.; Worringer, K.A.; Kommineni, S.; Kaykas, A.; Ho, D.J.; Ye, C.; et al. ADAM17 is the main sheddase for the generation of human triggering receptor expressed in myeloid cells (hTREM2) ectodomain and cleaves TREM2 after Histidine 157. Neurosci. Lett. 2017, 660, 109–114. [Google Scholar] [CrossRef]
- Schlepckow, K.; Kleinberger, G.; Fukumori, A.; Feederle, R.; Lichtenthaler, S.F.; Steiner, H.; Haass, C. An Alzheimer-associated TREM2 variant occurs at the ADAM cleavage site and affects shedding and phagocytic function. EMBO Mol. Med. 2017, 9, 1356–1365. [Google Scholar] [CrossRef]
- Thornton, P.; Sevalle, J.; Deery, M.J.; Fraser, G.; Zhou, Y.; Ståhl, S.; Franssen, E.H.; Dodd, R.B.; Qamar, S.; Gomez Perez-Nievas, B.; et al. TREM2 shedding by cleavage at the H157-S158 bond is accelerated for the Alzheimer’s disease-associated H157Y variant. EMBO Mol. Med. 2017, 9, 1366–1378. [Google Scholar] [CrossRef]
- Suárez-Calvet, M.; Kleinberger, G.; Araque Caballero, M.; Brendel, M.; Rominger, A.; Alcolea, D.; Fortea, J.; Lleó, A.; Blesa, R.; Gispert, J.D.; et al. sTREM2 cerebrospinal fluid levels are a potential biomarker for microglia activity in early-stage Alzheimer’s disease and associate with neuronal injury markers. EMBO Mol. Med. 2016, 8, 466–476. [Google Scholar] [CrossRef] [PubMed]
- Heslegrave, A.; Heywood, W.; Paterson, R.; Magdalinou, N.; Svensson, J.; Johansson, P.; Öhrfelt, A.; Blennow, K.; Hardy, J.; Schott, J.; et al. Increased cerebrospinal fluid soluble TREM2 concentration in Alzheimer’s disease. Mol. Neurodegener. 2016, 11, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swardfager, W.; Lanctôt, K.; Rothenburg, L.; Wong, A.; Cappell, J.; Herrmann, N. A meta-analysis of cytokines in Alzheimer’s disease. Biol. Psychiatry 2010, 68, 930–941. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.S.; West, A.P.; Ghosh, S. NF-kappa B and the immune response. Oncogene 2006, 25, 6758–6780. [Google Scholar] [CrossRef] [Green Version]
- Kaltschmidt, B.; Uherek, M.; Volk, B.; Baeuerle, P.A.; Kaltschmidt, C. Transcription factor NF-kappaB is activated in primary neurons by amyloid beta peptides and in neurons surrounding early plaques from patients with Alzheimer disease. Proc. Natl. Acad. Sci. USA 1997, 94, 2642–2647. [Google Scholar] [CrossRef] [Green Version]
- Iqbal, K.; Liu, F.; Gong, C.X.; Alonso Adel, C.; Grundke-Iqbal, I. Mechanisms of tau-induced neurodegeneration. Acta Neuropathol. 2009, 118, 53–69. [Google Scholar] [CrossRef] [Green Version]
- Alonso, A.; Zaidi, T.; Novak, M.; Grundke-Iqbal, I.; Iqbal, K. Hyperphosphorylation induces self-assembly of tau into tangles of paired helical filaments/straight filaments. Proc. Natl. Acad. Sci. USA 2001, 98, 6923–6928. [Google Scholar] [CrossRef] [Green Version]
- Alonso, A.C.; Grundke-Iqbal, I.; Iqbal, K. Alzheimer’s disease hyperphosphorylated tau sequesters normal tau into tangles of filaments and disassembles microtubules. Nat. Med. 1996, 2, 783–787. [Google Scholar] [CrossRef]
- Alonso Adel, C.; Mederlyova, A.; Novak, M.; Grundke-Iqbal, I.; Iqbal, K. Promotion of hyperphosphorylation by frontotemporal dementia tau mutations. J. Biol. Chem. 2004, 279, 34873–34881. [Google Scholar] [CrossRef] [Green Version]
- Grundke-Iqbal, I.; Iqbal, K.; Quinlan, M.; Tung, Y.C.; Zaidi, M.S.; Wisniewski, H.M. Microtubule-associated protein tau. A component of Alzheimer paired helical filaments. J. Biol. Chem. 1986, 261, 6084–6089. [Google Scholar] [CrossRef]
- Sengupta, A.; Kabat, J.; Novak, M.; Wu, Q.; Grundke-Iqbal, I.; Iqbal, K. Phosphorylation of tau at both Thr 231 and Ser 262 is required for maximal inhibition of its binding to microtubules. Arch. Biochem. Biophys. 1998, 357, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Baudier, J.; Cole, R.D. Interactions between the microtubule-associated tau proteins and S100b regulate tau phosphorylation by the Ca2+/calmodulin-dependent protein kinase II. J. Biol. Chem. 1988, 263, 5876–5883. [Google Scholar] [CrossRef]
- Drewes, G.; Ebneth, A.; Preuss, U.; Mandelkow, E.M.; Mandelkow, E. MARK, a novel family of protein kinases that phosphorylate microtubule-associated proteins and trigger microtubule disruption. Cell 1997, 89, 297–308. [Google Scholar] [CrossRef]
- Drewes, G.; Lichtenberg-Kraag, B.; Döring, F.; Mandelkow, E.M.; Biernat, J.; Goris, J.; Dorée, M.; Mandelkow, E. Mitogen activated protein (MAP) kinase transforms tau protein into an Alzheimer-like state. EMBO J. 1992, 11, 2131–2138. [Google Scholar] [CrossRef] [PubMed]
- Hanger, D.P.; Hughes, K.; Woodgett, J.R.; Brion, J.P.; Anderton, B.H. Glycogen synthase kinase-3 induces Alzheimer’s disease-like phosphorylation of tau: Generation of paired helical filament epitopes and neuronal localisation of the kinase. Neurosci. Lett. 1992, 147, 58–62. [Google Scholar] [CrossRef]
- Litersky, J.M.; Johnson, G.V. Phosphorylation by cAMP-dependent protein kinase inhibits the degradation of tau by calpain. J. Biol. Chem. 1992, 267, 1563–1568. [Google Scholar] [CrossRef]
- Noble, W.; Olm, V.; Takata, K.; Casey, E.; Mary, O.; Meyerson, J.; Gaynor, K.; LaFrancois, J.; Wang, L.; Kondo, T.; et al. Cdk5 is a key factor in tau aggregation and tangle formation in vivo. Neuron 2003, 38, 555–565. [Google Scholar] [CrossRef] [Green Version]
- Gong, C.X.; Grundke-Iqbal, I.; Iqbal, K. Dephosphorylation of Alzheimer’s disease abnormally phosphorylated tau by protein phosphatase-2A. Neuroscience 1994, 61, 765–772. [Google Scholar] [CrossRef]
- Liu, R.; Zhou, X.W.; Tanila, H.; Bjorkdahl, C.; Wang, J.Z.; Guan, Z.Z.; Cao, Y.; Gustafsson, J.A.; Winblad, B.; Pei, J.J. Phosphorylated PP2A (tyrosine 307) is associated with Alzheimer neurofibrillary pathology. J. Cell Mol. Med. 2008, 12, 241–257. [Google Scholar] [CrossRef] [Green Version]
- Pei, J.J.; An, W.L.; Zhou, X.W.; Nishimura, T.; Norberg, J.; Benedikz, E.; Götz, J.; Winblad, B. P70 S6 kinase mediates tau phosphorylation and synthesis. FEBS Lett. 2006, 580, 107–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, X.W.; Gustafsson, J.A.; Tanila, H.; Bjorkdahl, C.; Liu, R.; Winblad, B.; Pei, J.J. Tau hyperphosphorylation correlates with reduced methylation of protein phosphatase 2A. Neurobiol. Dis. 2008, 31, 386–394. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.W.; Winblad, B.; Guan, Z.; Pei, J.J. Interactions between glycogen synthase kinase 3beta, protein kinase B, and protein phosphatase 2A in tau phosphorylation in mouse N2a neuroblastoma cells. J. Alzheimers Dis. 2009, 17, 929–937. [Google Scholar] [CrossRef] [PubMed]
- Caccamo, A.; Oddo, S.; Billings, L.M.; Green, K.N.; Martinez-Coria, H.; Fisher, A.; LaFerla, F.M. M1 receptors play a central role in modulating AD-like pathology in transgenic mice. Neuron 2006, 49, 671–682. [Google Scholar] [CrossRef] [Green Version]
- Fonseca, M.I.; Ager, R.R.; Chu, S.H.; Yazan, O.; Sanderson, S.D.; LaFerla, F.M.; Taylor, S.M.; Woodruff, T.M.; Tenner, A.J. Treatment with a C5aR antagonist decreases pathology and enhances behavioral performance in murine models of Alzheimer’s disease. J. Immunol. 2009, 183, 1375–1383. [Google Scholar] [CrossRef] [Green Version]
- Sundaram, R.; Naresh, R.; Shanthi, P.; Sachdanandam, P. Modulatory effect of green tea extract on hepatic key enzymes of glucose metabolism in streptozotocin and high fat diet induced diabetic rats. Phytomedicine 2013, 20, 577–584. [Google Scholar] [CrossRef]
- Kim, J.D.; Khan, M.I.; Shin, J.H.; Lee, M.G.; Seo, H.J.; Shin, T.S.; Kim, M.Y. HPLC fractionation and pharmacological assessment of green tea seed saponins for antimicrobial, anti-angiogenic and hemolytic activities. Biotechnol. Bioprocess Eng. 2015, 20, 1035–1043. [Google Scholar] [CrossRef]
- Khan, M.I.; Ahhmed, A.; Shin, J.H.; Baek, J.S.; Kim, M.Y.; Kim, J.D. Green Tea Seed Isolated Saponins Exerts Antibacterial Effects against Various Strains of Gram Positive and Gram-Negative Bacteria, a Comprehensive Study In Vitro and In Vivo. Evid. Based Complement Alternat. Med. 2018, 2018, 3486106. [Google Scholar] [CrossRef] [Green Version]
- Johnson, J.J.; Bailey, H.H.; Mukhtar, H. Green tea polyphenols for prostate cancer chemoprevention: A translational perspective. Phytomedicine 2010, 17, 3–13. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, H.; Tanaka, Y.; Asagiri, K.; Asakawa, T.; Tanikawa, K.; Kage, M.; Yagi, M. The antioxidant effect of green tea catechin ameliorates experimental liver injury. Phytomedicine 2010, 17, 197–202. [Google Scholar] [CrossRef]
- Mancini, E.; Beglinger, C.; Drewe, J.; Zanchi, D.; Lang, U.E.; Borgwardt, S. Green tea effects on cognition, mood, and human brain function: A systematic review. Phytomedicine 2017, 34, 26–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, M.I.; Shin, J.H.; Kim, M.Y.; Shin, T.S.; Kim, J.D. Green Tea Seed Isolated Theasaponin E1 Ameliorates AD Promoting Neurotoxic Pathogenesis by Attenuating Aβ Peptide Levels in SweAPP N2a Cells. Molecules 2020, 25, 2334. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primers | Forward | Reverse |
|---|---|---|
| GSK-3β | 5′-GGAACTCCAACAAGGGAGCA-3′ | 5′-TTCGGGGTCGGAAGACCTTA-3′ |
| CDK5 | 5′-CGCCGCGATGCAGAAATACGAGAA-3′ | 5′-TGGCCCCAAAGAGGACATC-3′ |
| JNK1 | 5’-AACTCTTTGACGCTGCTTGC-3’ | 5’-TGAAGCACTGTGCCTTTACC-3’ |
| MAPK | 5′-CCAACTCCTGCCTCCGCTCTA-3′ | 5′-CCGCCAAAATAACCGATGTGATAC-3′ |
| ERK1/MARK | 5’-CGCTTCCGCCATGAGAATGTC-3′ | 5’-CAGGTCAGTCTCCATCAGGTCCTG-3’ |
| CaMKIIα | 5′-AGGAGGAAACTGAAGGGAG-3′ | 5′-CAGGGTCGCACATCTTCGTG-3′ |
| PP2A | 5′-GAGGGTACTACTCTGTGGAGAC-3′ | 5′-CCGGCTTTCGTGATTTCCT-3′ |
| PP-1 | 5′-TCCATGGAGCAGATTAGACG-3′ | 5′-GCTTTGGCAGAATTGCGG-3′ |
| APP | 5′-TCAGTTTCCTCGGCAGCG-3′ | 5′-GCACCAGTTCTGGATGGTCA-3′ |
| PESN1 | 5′-GCACCGTTGTCCTACTTCCA-3′ | 5′-CCATGCAGAGAGTCACAGGG-3′ |
| PESN2 | 5′-GCGGCAGAGCAGGCATTT-3′ | 5′-AGGTGAAGAGGAACAGCAGC-3′ |
| EPOE4(E4) | 5′-GGATGGGGAGATAAGAGAAGAC-3′ | 5′-CGCAGGTAATCCCAAAAGCG-3′ |
| IDE | 5′-CAAGCAGGAAGCGTTTGCG-3′ | 5′-CAACCTGGTAGTTCCCACACA-3′ |
| TREM2 | 5′- TTCCCACCCACTTCCATCCTT-3′ | 5′-AGCAGTGTTCAGGCAGAGTAG-3′ |
| IL-1B | 5′- AACAGGCTGCTCTGGGATTC-3′ | 5′-TTTGGTCCCTCCCAGGAAGA-3′ |
| PICALM | 5′-GCAGCTGCCTGTTCCTCTTA-3′ | 5′-GCAGCTGCCTGTTCCTCTTA-3′ |
| β-actin | 5′-CCTCGCCTTTGCCGATCC-3′ | 5′-GGATCTTCATGAGGTAGTGAGTC-3′ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khan, M.I.; Khan, M.Z.; Shin, J.H.; Shin, T.S.; Lee, Y.B.; Kim, M.Y.; Kim, J.D. Neuroprotective Effects of Green Tea Seed Isolated Saponin Due to the Amelioration of Tauopathy and Alleviation of Neuroinflammation: A Therapeutic Approach to Alzheimer’s Disease. Molecules 2022, 27, 2079. https://doi.org/10.3390/molecules27072079
Khan MI, Khan MZ, Shin JH, Shin TS, Lee YB, Kim MY, Kim JD. Neuroprotective Effects of Green Tea Seed Isolated Saponin Due to the Amelioration of Tauopathy and Alleviation of Neuroinflammation: A Therapeutic Approach to Alzheimer’s Disease. Molecules. 2022; 27(7):2079. https://doi.org/10.3390/molecules27072079
Chicago/Turabian StyleKhan, Muhammad Imran, Muhammad Zubair Khan, Jin Hyuk Shin, Tai Sun Shin, Young Bok Lee, Min Yong Kim, and Jong Deog Kim. 2022. "Neuroprotective Effects of Green Tea Seed Isolated Saponin Due to the Amelioration of Tauopathy and Alleviation of Neuroinflammation: A Therapeutic Approach to Alzheimer’s Disease" Molecules 27, no. 7: 2079. https://doi.org/10.3390/molecules27072079
APA StyleKhan, M. I., Khan, M. Z., Shin, J. H., Shin, T. S., Lee, Y. B., Kim, M. Y., & Kim, J. D. (2022). Neuroprotective Effects of Green Tea Seed Isolated Saponin Due to the Amelioration of Tauopathy and Alleviation of Neuroinflammation: A Therapeutic Approach to Alzheimer’s Disease. Molecules, 27(7), 2079. https://doi.org/10.3390/molecules27072079