
Design, Synthesis, Pharmacodynamic and In Silico Pharmacokinetic Evaluation of Some Novel Biginelli-Derived Pyrimidines and Fused Pyrimidines as Calcium Channel Blockers

 ,
,  ,
,  and
and
Abstract
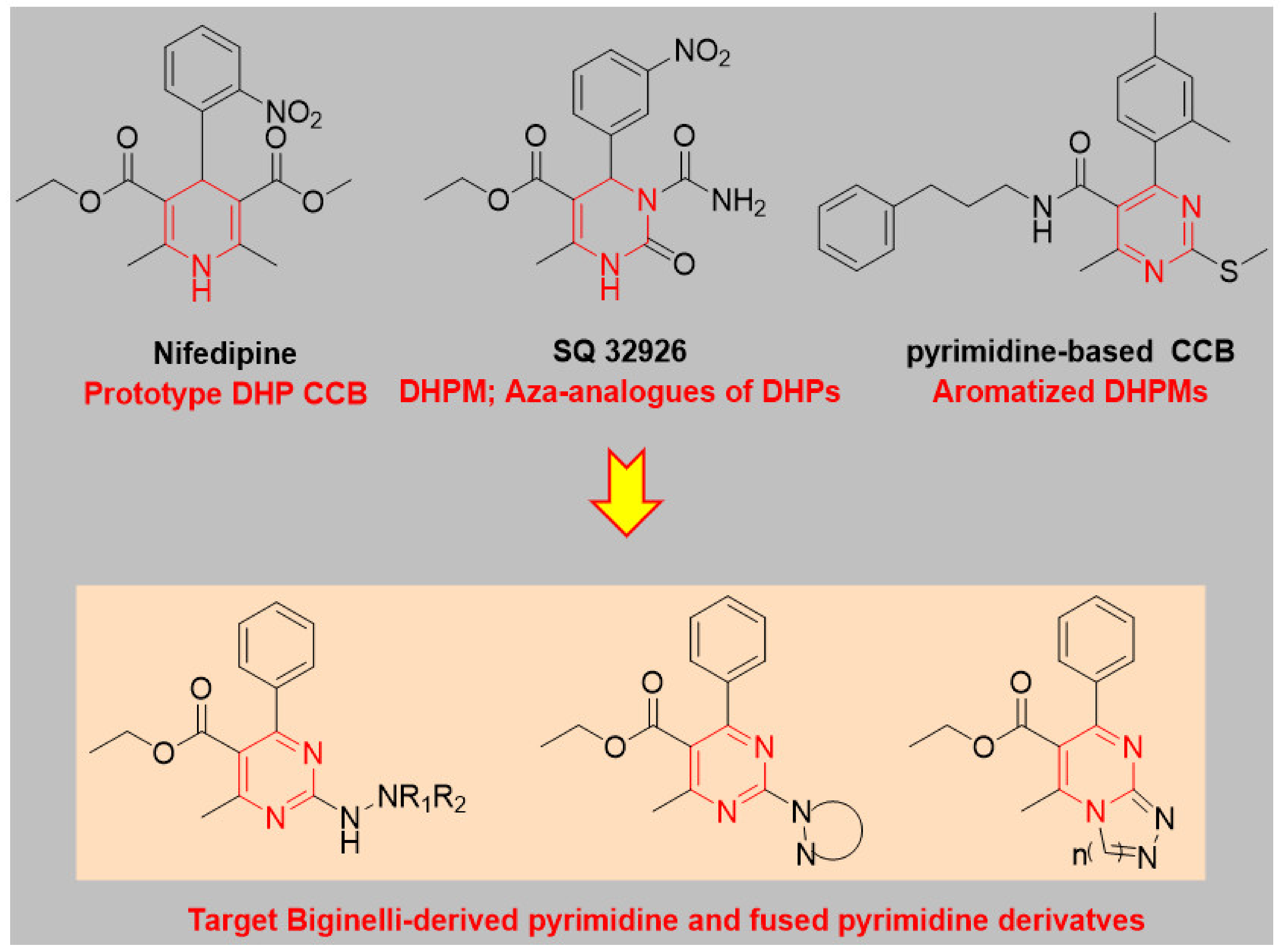
:1. Introduction
2. Results and Discussion
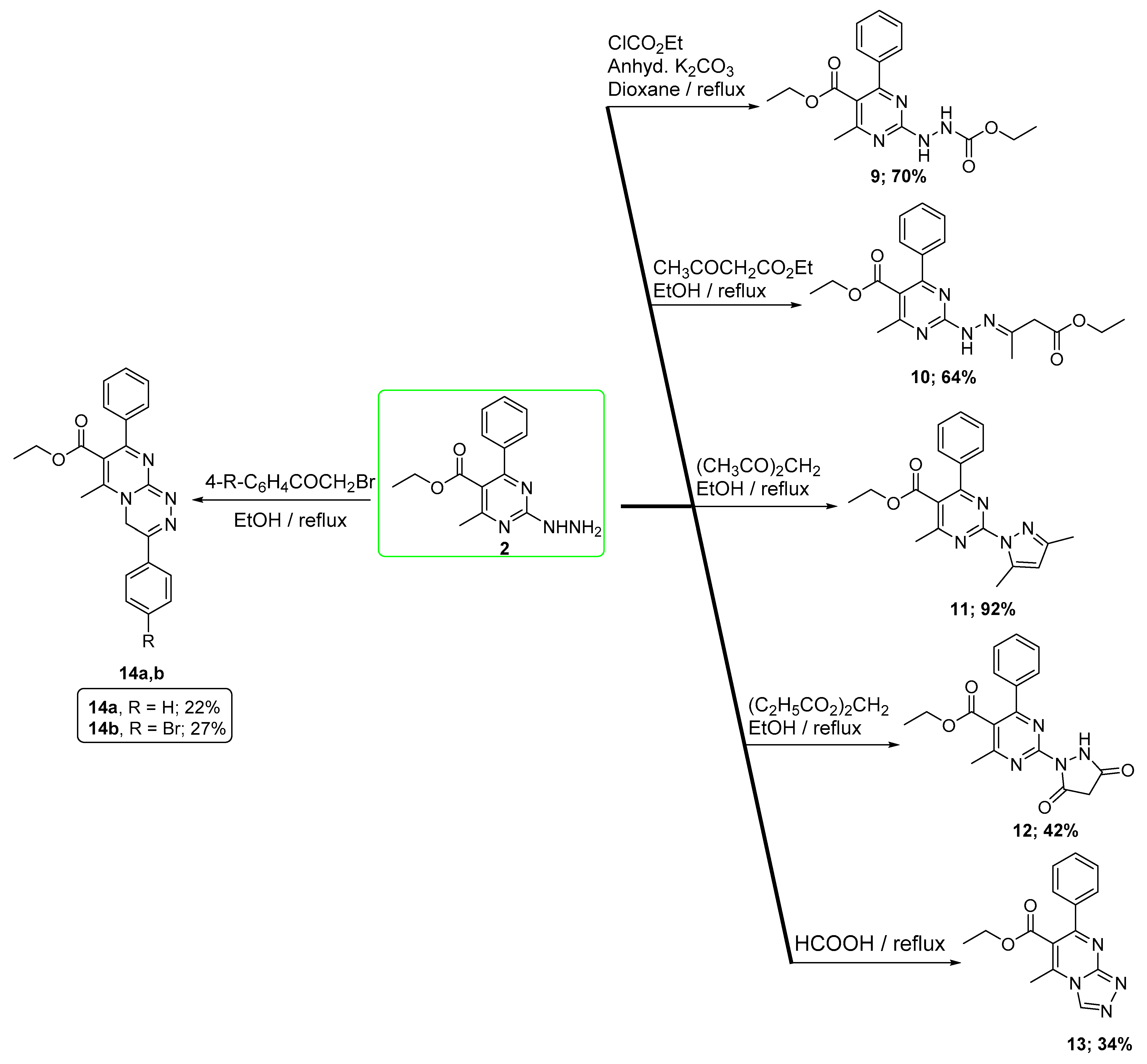
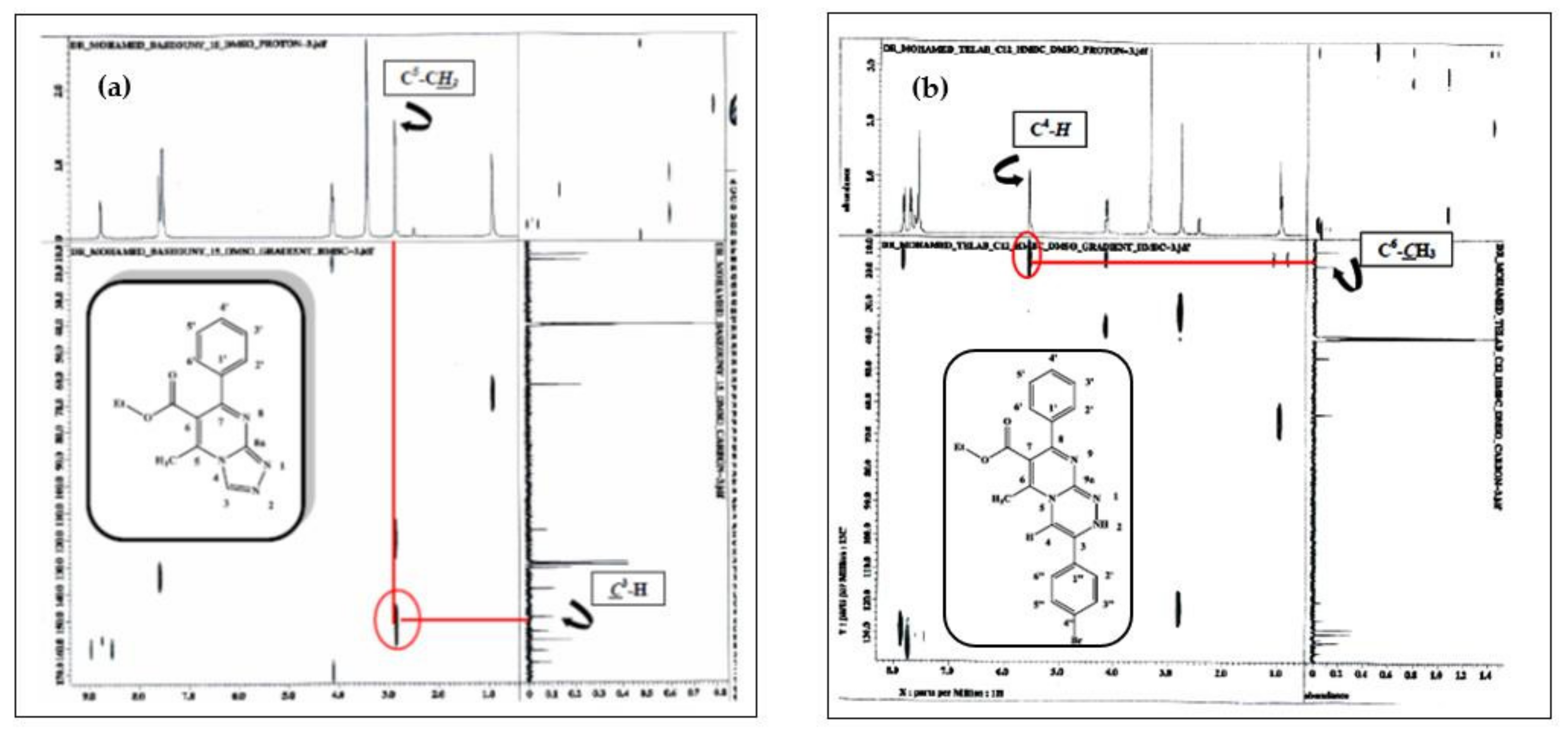
2.1. Chemistry
2.2. Biological Evaluation
2.3. Molecular Modeling
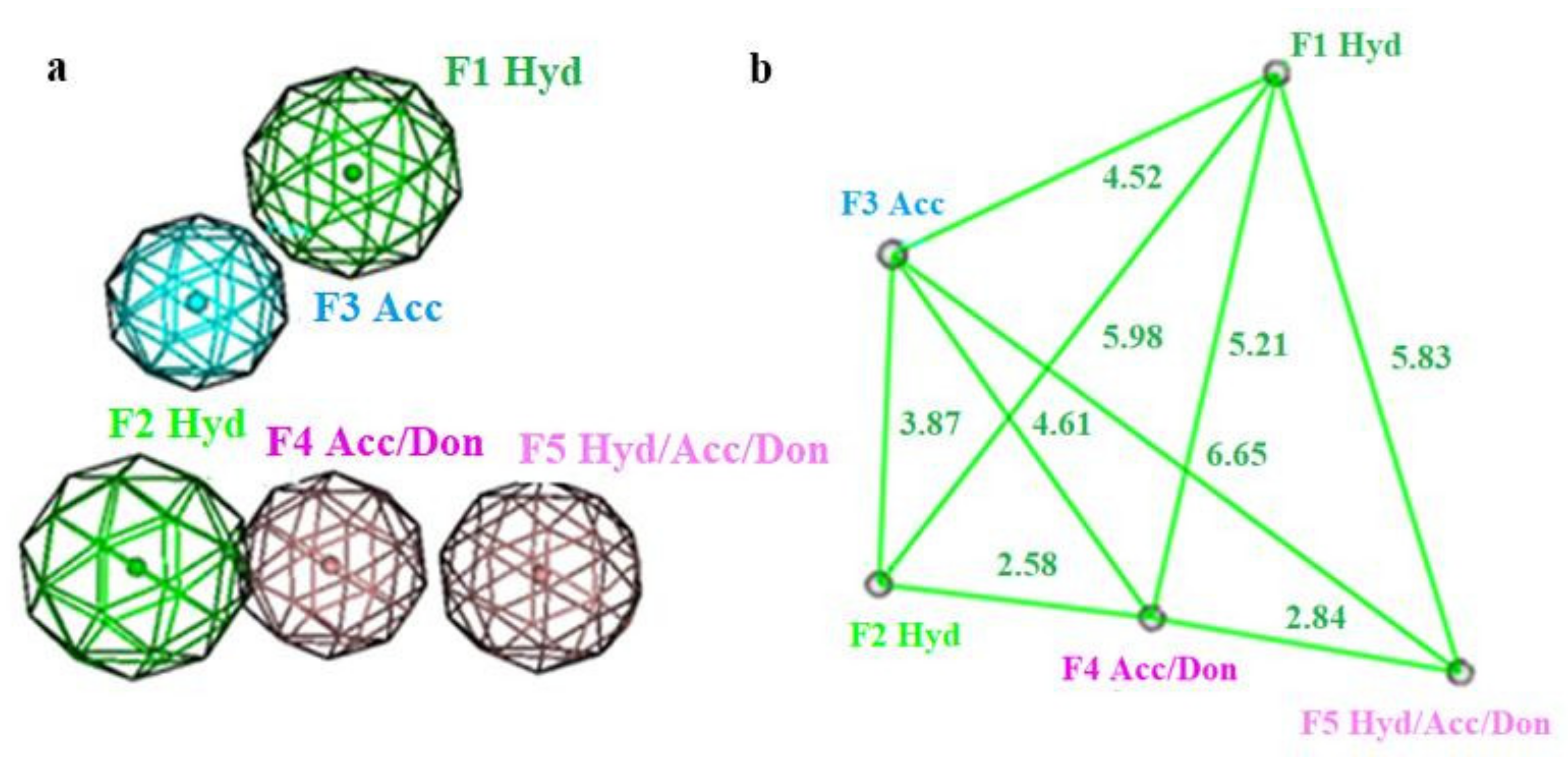
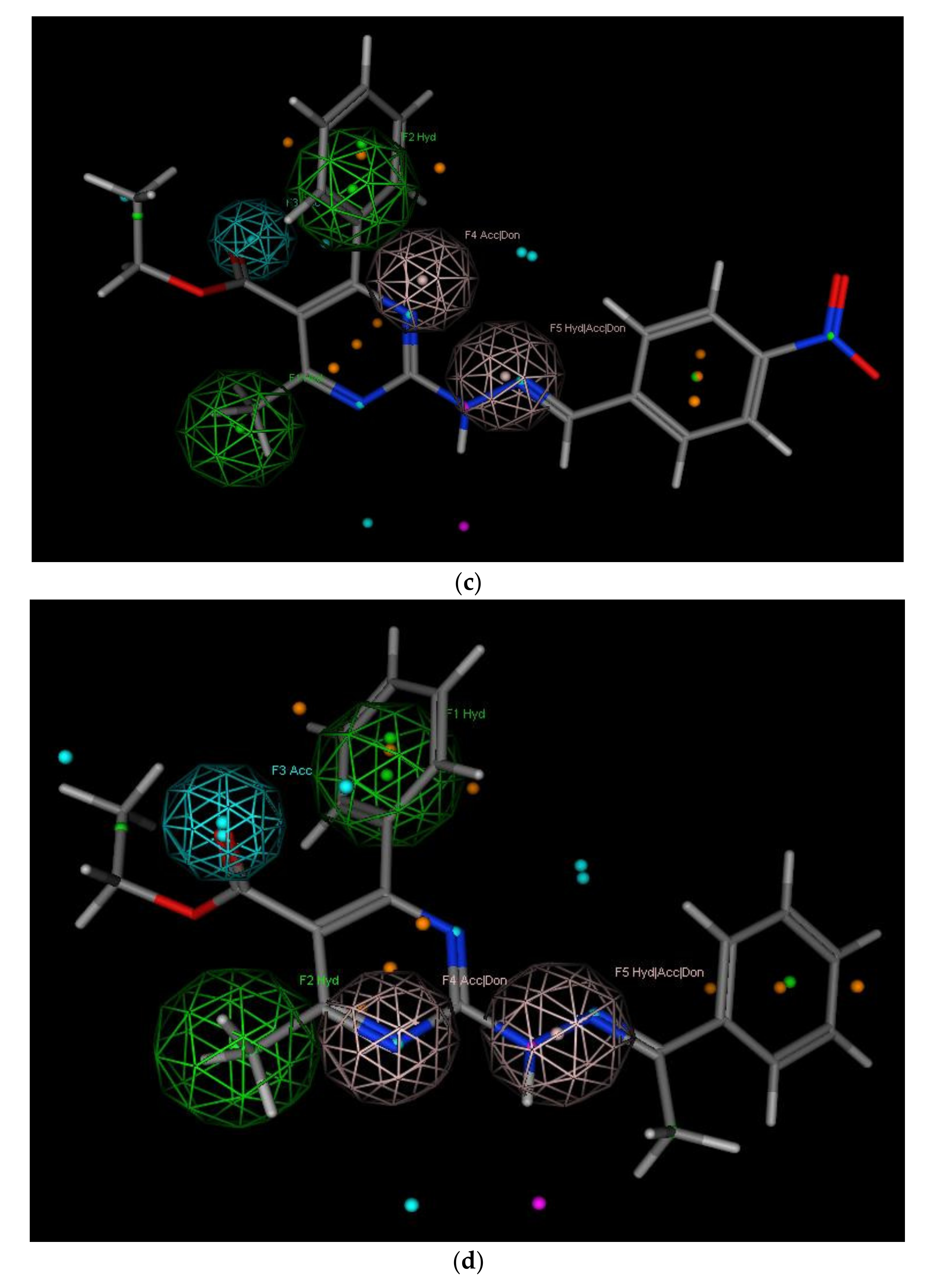
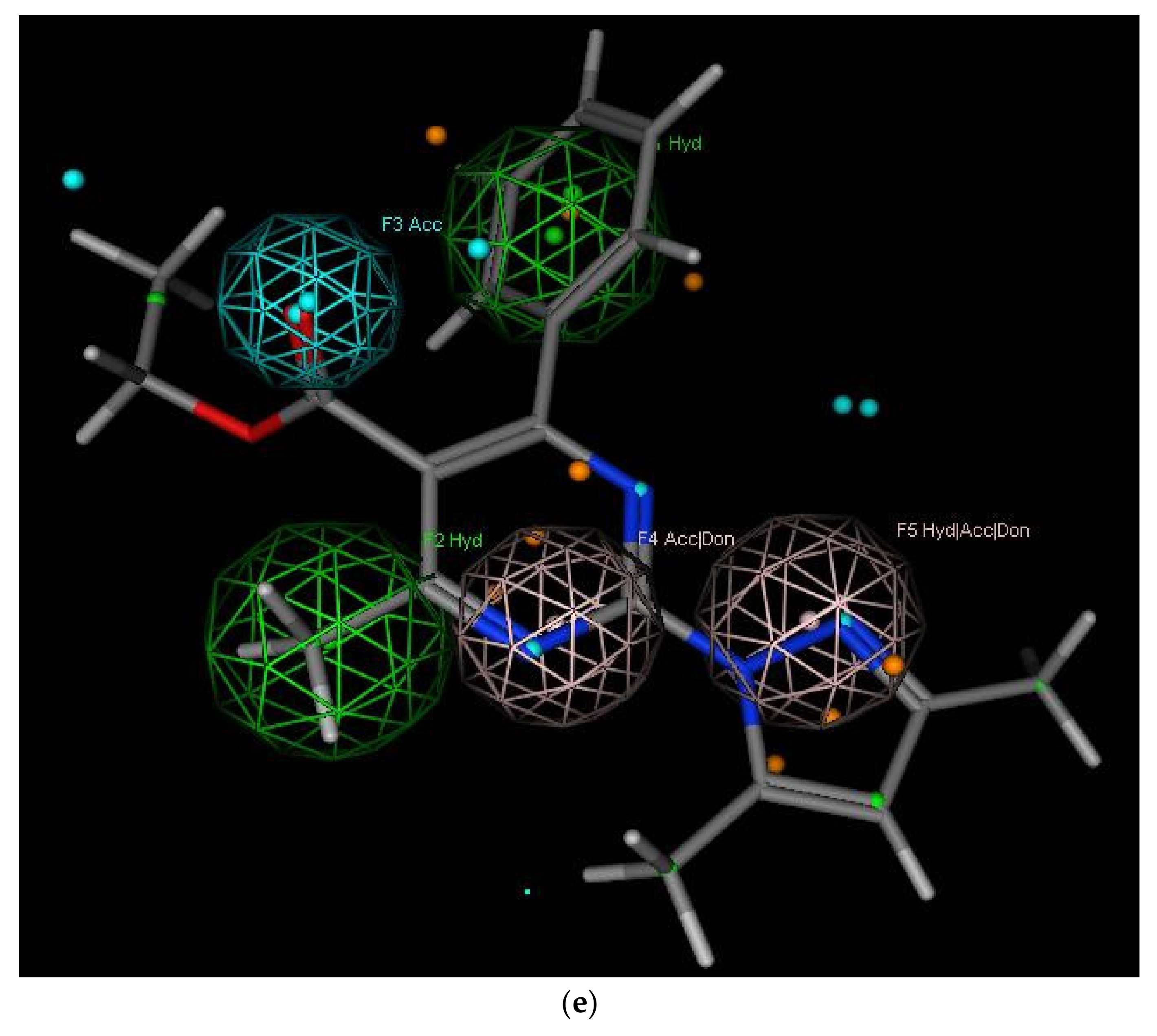
2.3.1. Pharmacophore Modeling
- Hydrophobic center (green sphere) involving C4 phenyl ring (F1: Hyd).
- Hydrophobic center (green sphere) involving C6 methyl group (F2: Hyd).
- Hydrogen bond acceptor function (cyan sphere) involving C5 carbonyl group (F3: Acc).
- Hydrogen bond acceptor/donor function (pink sphere) involving ring N (F4: Acc/Don).
- Hydrophobic center with H-bond acceptor or donor function (pink sphere) involving C2 substitutions (F5: Hyd/Acc/Don).
2.3.2. In Silico Physicochemical Properties, Drug-Likeness and ADME
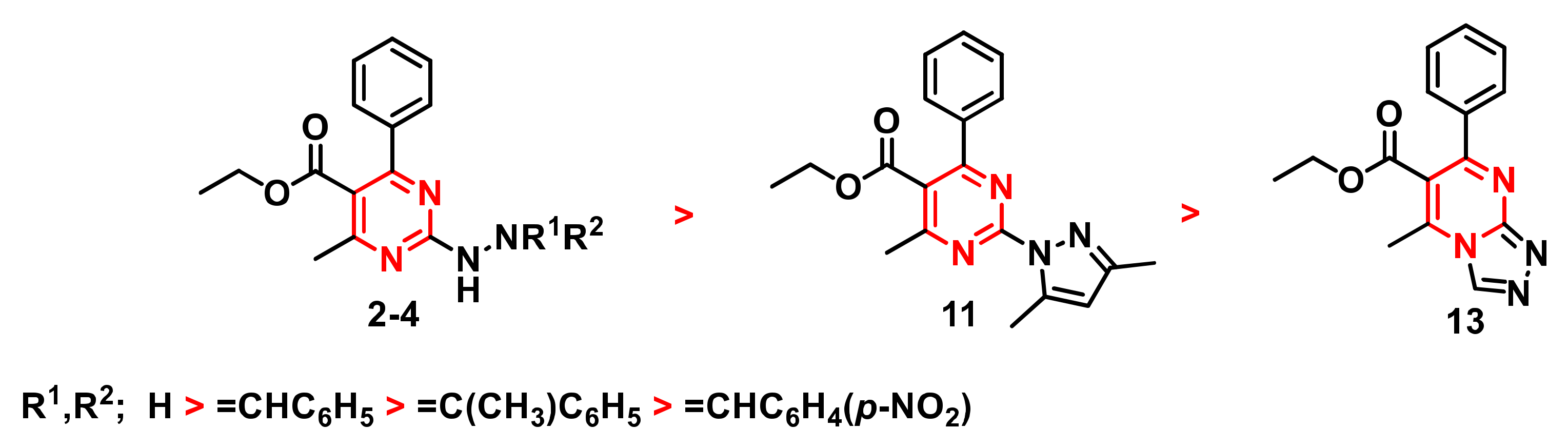
2.4. Structure–Activity Relationship
3. Materials and Methods
3.1. Chemistry
3.2. Biological Evaluation
3.2.1. Experimental Animals
3.2.2. Data Recoding
3.2.3. Statistical Analysis and Data Interpretation
3.2.4. In Vitro Calcium Channel Blocking Activity
Rat Colon
Rabbit Jejunum
3.2.5. In Vivo Hypotensive Activity on Normotensive Anesthetized Dogs
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mozaffarian, D.; Benjamin, E.J.; Go, A.S.; Arnett, D.K.; Blaha, M.J.; Cushman, M.; de Ferranti, S.; Després, J.P.; Fullerton, H.J.; Howard, V.J.; et al. Heart disease and stroke statistics—2015 update: A report from the American Heart Association. Circulation 2015, 131, e29–e322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- World Health Organization. Global Atlas on Cardiovascular Disease Prevention and Control; World Health Organization: Geneva, Switzerland, 2011. [Google Scholar]
- Williams, B.; Mancia, G.; Spiering, W.; Agabiti Rosei, E.; Azizi, M.; Burnier, M.; Clement, D.L.; Coca, A.; De Simone, G.; Dominiczak, A.; et al. 2018 ESC/ESH Guidelines for the management of arterial hypertension: The Task Force for the management of arterial hypertension of the European Society of Cardiology (ESC) and the European Society of Hypertension (ESH). Eur. Heart J. 2018, 39, 3021–3104. [Google Scholar] [CrossRef] [PubMed]
- Nakov, R.; Pfarr, E.; Eberle, S. Darusentan: An effective endothelinA receptor antagonist for treatment of hypertension. Am. J. Hypertens. 2002, 15, 583–589. [Google Scholar] [CrossRef] [Green Version]
- Enseleit, F.; Luscher, T.F.; Ruschitzka, F. Darusentan, a selective endothelin A receptor antagonist, for the oral treatment of resistant hypertension. Ther. Adv. Cardiovasc. Dis. 2010, 111, 231–240. [Google Scholar] [CrossRef] [Green Version]
- Aggarwal, R.K.; Showkathali, R. Rosuvastatin calcium in acute coronary syndromes. Expert Opin. Pharmacother. 2013, 14, 1215–1227. [Google Scholar] [CrossRef]
- Selvam, T.P.; James, C.R.; Dniandev, P.V.; Valzita, S.K. A mini review of pyrimidine and fused pyrimidine marketed drugs. Res. Pharm. 2012, 2, 1–9. [Google Scholar]
- Nicolai, E.; Cure, G.; Goyard, J.; Kirchner, M.; Teulon, J.M.; Versigny, A.; Cazes, M.; Caussade, F.; Virone-Oddos, A.; Cloarec, A. Synthesis and SAR Studies of novel triazolopyrimidine derivatives as potent, orally active angiotensin II receptor antagonists. J. Med. Chem. 1994, 37, 2371–2386. [Google Scholar] [CrossRef]
- Ohno, S.; Otani, K.; Niwa, S.; Iwayama, S.; Takahara, A.; Koganei, H.; Ono, Y.; Fujita, S.; Takeda, T.; Hagihara, M.; et al. Pyrimidine Derivatives and New Pyridine Derivatives. International Patent WO/2002/022588, 21 March 2002. [Google Scholar]
- Biginelli, P. Synthesis of 3,4-dihydropyrimidin-2(1H)-ones. Gazz. Chim. Ital. 1893, 23, 360–416. [Google Scholar]
- Kappe, C.O. Biologically active dihydropyrimidones of the Biginelli-type-a literature survey. Eur. J. Med. Chem. 2000, 35, 1043–1052. [Google Scholar] [CrossRef]
- Teleb, M.; Zhang, F.X.; Farghaly, A.M.; Wafa, O.M.; Fronczek, F.R.; Zamponi, G.W.; Fahmy, H. Synthesis of new N3-substituted dihydropyrimidine derivatives as L-/T-type calcium channel blockers. Eur. J. Med. Chem. 2017, 134, 52–61. [Google Scholar] [CrossRef]
- Teleb, M.; Zhang, F.X.; Junting, H.; Vinicius, M.G.; Farghaly, A.M.; Wafa, O.M.; Zamponi, G.W.; Fahmy, H. Synthesis and biological evaluation of novel N3-substituted dihydropyrimidine derivatives as T-type calcium channel blockers and their efficacy as analgesics in mouse models of inflammatory pain. Bioorg. Med. Chem. 2017, 25, 1926–1938. [Google Scholar] [CrossRef] [PubMed]
- Teleb, M.; Rizk, O.H.; Zhang, F.X.; Fronczek, F.R.; Zamponi, G.W.; Fahmy, H. Design, synthesis and pharmacological evaluation of some substituted dihydropyrimidines with L-/T-type calcium channel blocking activities. Bioorg. Chem. 2019, 83, 354–366. [Google Scholar] [CrossRef] [PubMed]
- Teleb, M.; Rizk, O.H.; Zhang, F.X.; Fronczek, F.R.; Zamponi, G.W.; Fahmy, H. Synthesis of some new C2 substituted dihydropyrimidines and their electrophysiological evaluation as L-/T-type calcium channel blockers. Bioorg. Chem. 2019, 88, 102915. [Google Scholar] [CrossRef]
- Bezencon, O.; Heidmann, B.; Siegrist, R.; Stamm, S.; Richard, S.; Pozzi, D.; Corminboeuf, O.; Roch, C.; Kessler, M.; Ertel, E.A.; et al. Discovery of a potent, selective T-type calcium channel blocker as a drug candidate for the treatment of generalized epilepsies. J. Med. Chem. 2017, 60, 9769–9789. [Google Scholar] [CrossRef] [PubMed]
- Remen, L.; Bezencon, O.; Simons, L.; Gaston, R.; Downing, D.; Gatfield, J.; Roch, C.; Kessler, M.; Mosbacher, J.; Pfeifer, T.; et al. Preparation, antiepileptic activity, and cardiovascular safety of dihydropyrazoles as brain-penetrant T-type calcium channel blockers. J. Med. Chem. 2016, 59, 8398–8411. [Google Scholar] [CrossRef]
- Han, M.; Nam, K.D.; Shin, D.; Jeong, N.; Hahn, H.G. Exploration of novel 2-alkylimino-1,3-thiazolines: T-type calcium channel inhibitory activity. J. Comb. Chem. 2010, 12, 518–530. [Google Scholar] [CrossRef]
- Matloobi, M.; Kappe, C.O. Microwave-assisted solution-and solid-phase synthesis of 2-amino-4-arylpyrimidine derivatives. J. Comb. Chem. 2007, 9, 275–284. [Google Scholar] [CrossRef]
- El-Wakil, M.; Teleb, M.; Abu-Serie, M.A.; Huang, S.; Zamponi, G.; Fahmy, H. Structural optimization, synthesis and in vitro synergistic anticancer activities of combinations of new N3-substituted dihydropyrimidine calcium channel blockers with cisplatin and etoposide. Bioorg. Chem. 2021, 115, 105262. [Google Scholar] [CrossRef]
- Bonde, C.G.; Gaikwad, N.J. Synthesis and preliminary evaluation of some pyrazine containing thiazolines and thiazolidinones as antimicrobial agents. Bioorg. Med. Chem. 2004, 12, 2151–2161. [Google Scholar] [CrossRef]
- Kamal, A.; Khan, M.N.; Reddy, K.S.; Rohini, K. Synthesis of a new class of 2-anilino substituted nicotinyl arylsulfonylhydrazides as potential anticancer and antibacterial agents. Bioorg. Med. Chem. 2007, 15, 1004–1013. [Google Scholar] [CrossRef]
- Kumar, Y.; Green, R.; Borysko, K.Z.; Wise, D.S.; Wotring, L.L.; Townsend, L.B. Synthesis of 2,4-disubstituted thiazoles and selenazoles as potential antitumor and antifilarial agents. 1. Methyl 4-(isothiocyanatomethyl) thiazole-2-carbamates,-selenazole-2-carbamates, and related derivatives. J. Med. Chem. 1993, 36, 3843–3848. [Google Scholar] [CrossRef] [PubMed]
- Kamal, A.M.; Radwan, S.M.; Zaki, R.M. Synthesis and biological activity of pyrazolothienotetrahydroisoquinoline and [1,2, 4]triazolo[3,4-a]thienotetrahydroisoquinoline derivatives. Eur. J. Med. Chem. 2011, 46, 567–578. [Google Scholar] [CrossRef] [PubMed]
- Nishigaki, S.; Ichiba, M.; Sato, J.; Senga, K.; Noguchi, M.; Yoneda, F. Synthesis of pyrimido[4,5-c]pyridazine derivatives. Heterocycles 1978, 9, 11. [Google Scholar]
- Rauwald, H.W.; Brehm, O.; Odenthal, K.-P. Screening of nine vasoactive medicinal plants for their possible calcium antagonistic activity. Strategy of selection and isolation for the active principles of Olea europaea and Peucedanum ostruthium. Phytother. Res. 1994, 8, 135–140. [Google Scholar] [CrossRef]
- Hsu, W.H.; Lu, Z.-X.; Hembrough, F.B. Effect of amitraz on heart rate and aortic blood pressure in conscious dogs: Influence of atropine, prazosin, tolazoline, and yohimbine. Toxicol. Appl. Pharmacol. 1986, 84, 418–422. [Google Scholar] [CrossRef]
- Atwal, K.S.; Rovnyak, G.C.; Schwartz, J.; Moreland, S.; Hedberg, A.; Gougoutas, J.Z.; Malley, M.F.; Floyd, D.M. Dihydropyrimidine calcium channel blockers: 2-heterosubstituted 4-aryl-1,4-dihydro-6-methyl-5-pyrimidinecarboxylic acid esters as potent mimics of dihydropyridines. J. Med. Chem. 1990, 33, 1510–1515. [Google Scholar] [CrossRef]
- Atwal, K.S.; Rovnyak, G.C.; Kimball, S.D.; Floyd, D.M.; Moreland, S.; Swanson, B.N.; Gougoutas, J.Z.; Schwartz, J.; Smillie, K.M.; Malley, M.F. Dihydropyrimidine calcium channel blockers. II. 3-Substituted-4-aryl-1,4-dihydro-6-methyl-5-pyrimidinecarboxylic acid esters as potent mimics of dihydropyridines. J. Med. Chem. 1990, 33, 2629–2635. [Google Scholar] [CrossRef]
- Atwal, K.S.; Swanson, B.N.; Unger, S.E.; Floyd, D.M.; Moreland, S.; Hedberg, A.; O’Reilly, B.C. Dihydropyrimidine calcium channel blockers. 3. 3-Carbamoyl-4-aryl-1,2,3,4-tetrahydro-6-methyl-5-pyrimidinecarboxylic acid esters as orally effective antihypertensive agents. J. Med. Chem. 1991, 34, 806–811. [Google Scholar] [CrossRef]
- Molecular Operating Environment (MOE), Chemical Computing Group, Montreal, Canada. Available online: https://www.chemcomp.com (accessed on 11 December 2019).
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2012, 64, 4–17. [Google Scholar] [CrossRef]
- Molinspiration Cheminformatics. Available online: https://www.molinspiration.com/ (accessed on 1 August 2019).
- Zhao, Y.; Abraham, M.H.; Lee, J.; Hersey, A.; Luscombe, N.C.; Beck, G.; Sherborne, B.; Cooper, I. Rate-limited steps of human oral absorption and QSAR studies. Pharm. Res. 2002, 19, 1446–1457. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.-Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef] [PubMed]
- Polinsky, A.; Shaw, G.B. High-speed chemistry libraries: Assessment of drug-likeness. In Practice of Medicinal Chemistry, 2nd ed.; Wermuth, C.G., Ed.; Elsevier: London, UK, 2003; pp. 147–157. [Google Scholar]
- Ertl, P.; Rohde, B.; Selzer, P. Fast calculation of molecular polar surface area as a sum of fragment-based contributions and its application to the prediction of drug transport properties. J. Med. Chem. 2000, 43, 3714–3717. [Google Scholar] [CrossRef] [PubMed]
- Molsoft LLC. Drug-Likeness and Molecular Property Prediction. Available online: http://molsoft.com/mprop/ (accessed on 1 August 2019).
- PreADMET. Available online: https://preadmet.bmdrc.kr/adme/ (accessed on 1 August 2019).
- Iqbal, N.; Akula, M.R.; Vo, D.; Matowe, W.C.; McEwen, C.A.; Wolowyk, M.W.; Knaus, E.E. Synthesis, rotamer orientation, and calcium channel modulation activities of alkyl and 2-phenethyl-1,4-dihydro-2,6-dimethyl-3-nitro-4-(3-or 6-substituted-2-pyridyl)-5-pyridinecarboxylates. J. Med. Chem. 1998, 41, 1827–1837. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.; Ueda, M.; Shima, K.; Mizuno, A.; Hayashimatsu, M.; Ohnaka, Y.; Takeuchi, Y.; Hamaguchi, M.; Aisaka, K.; Hidaka, T.; et al. Novel calcium antagonists with potent and long-lasting vasodilative and anti-hypertensive activity. J. Med. Chem. 1989, 32, 2399–2406. [Google Scholar] [CrossRef] [PubMed]
- Arrowsmith, J.E.; Campbell, S.F.; Cross, P.E.; Stubbs, J.K.; Burges, R.A.; Gardiner, D.G.; Blackburn, K.J. Long-acting dihydropyridine calcium antagonists. 1. 2-Alkoxymethyl derivatives incorporating basic substituents. J. Med. Chem. 1986, 29, 1696–1702. [Google Scholar] [CrossRef]
- Zhorov, B.S.; Folkman, E.V.; Ananthanarayanan, V.S. Homology model of dihydropyridine receptor: Implications for L-type Ca2+ channel modulation by agonists and antagonists. Arch. Biochem. Biophys. 2001, 393, 22–41. [Google Scholar] [CrossRef]
- GraphPad Prism, Version 3.02 for Windows; GraphPad Software: La Jolla, CA, USA. Available online: http://www.graphpad.com(accessed on 10 June 2019).




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cpd No. | Rat Colon | Rabbit Jejunum | Cpd No. | Rat Colon | Rabbit Jejunum |
|---|---|---|---|---|---|
| 2 | + | + | 8b | - | - |
| 3a | + | + | 9 | - | - |
| 3b | + | + | 10 | - | - |
| 4 | + | + | 11 | + | + |
| 5a | - | - | 12 | - | - |
| 5b | - | - | 13 | + | + |
| 6 | - | - | 14a | - | - |
| 7 | - | - | 14b | - | - |
| 8a | - | - | Nifedipine | + | + |
| Compound No. | % Inhibition of KCl-Induced Contractions | ||
|---|---|---|---|
| 2 × 10−5 M | 4 × 10−5 M | 6 × 10−5 M | |
| 2 | 44.45 ± 26.06 | 66.67 ± 33.33 | 100 ± 0 |
| 3a | 25.00 ± 14.43 | 62.50 ± 23.94 | 72.33 ± 14.68 |
| 3b | 17.00 ± 9.82 | 89.00 ± 11.00 | 100 ± 0 |
| 4 | 11.33 ± 9.81 | 33.45 ± 16.78 | 55.56 ± 24.22 |
| 11 | 15.55 ± 4.45 | 50.00 ± 30.00 | 70.00 ± 15.28 |
| 13 | 25.09 ± 16.03 | 55.09 ± 17.94 | 12.50 ± 7.98 |
| Nifedipine | 100 ± 0 | ||
| Compound No. | IC50 (µM) | pIC50 |
|---|---|---|
| 2 | 0.96 | 6.017 |
| 3a | 1.089 | 5.962 |
| 3b | 2.82 | 5.549 |
| 4 | 1.889 | 5.723 |
| 11 | 2.594 | 5.586 |
| 13 | 3.199 | 5.494 |
| Nifedipine | 6.279 (nM) | 8.202 |
| Compound No. | Decrease in MAP (mmHg) as Mean ± SE | ||
|---|---|---|---|
| 6 mg/kg | 12 mg/kg | 24 mg/kg | |
| Control 2 | 3 ± 1.29 9.6 ± 0.81 | 2.75 ± 1.60 15.4 ± 2.39 | 12.6 ± 3.57 35.4 ± 1.60 |
| 11 | 24.4 ± 2.56 | 28.75 ± 6.01 | 34 ± 4.16 |
| Compound No. | 2 | 3a | 3b | 4 | 11 |
|---|---|---|---|---|---|
| RMSD(Å) | 0.5694 | 0.8392 | 0.7738 | 0.5678 | 0.6054 |
| Cpd. No. | LogP a | M.Wt b | HBA c | HBD d | Lipinski’s Violation | TPSA e | %ABS f | Volumes (A)3 | S g (mg/L) | Drug-Likeness Model Score | CaCo2 h | MDCK i | HIA j | BBB k | PPB l | CYP3A4 Inhibition | CYP2D6 Inhibition |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 2 | 1.82 | 272.31 | 6 | 3 | 0 | 90.14 | 77.90 | 248.72 | 77.22 | 0.16 | 20.44 | 77.38 | 92.60 | 0.67 | 69.58 | Non | Non |
| 11 | 3.17 | 336.39 | 6 | 0 | 0 | 69.92 | 84.87 | 310.77 | 3.06 | 0.06 | 33.73 | 18.32 | 98.72 | 1.58 | 88.16 | inhibitor | Non |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Farghaly, A.M.; Rizk, O.H.; Darwish, I.; Hamza, M.; Altowyan, M.S.; Barakat, A.; Teleb, M. Design, Synthesis, Pharmacodynamic and In Silico Pharmacokinetic Evaluation of Some Novel Biginelli-Derived Pyrimidines and Fused Pyrimidines as Calcium Channel Blockers. Molecules 2022, 27, 2240. https://doi.org/10.3390/molecules27072240
Farghaly AM, Rizk OH, Darwish I, Hamza M, Altowyan MS, Barakat A, Teleb M. Design, Synthesis, Pharmacodynamic and In Silico Pharmacokinetic Evaluation of Some Novel Biginelli-Derived Pyrimidines and Fused Pyrimidines as Calcium Channel Blockers. Molecules. 2022; 27(7):2240. https://doi.org/10.3390/molecules27072240
Chicago/Turabian StyleFarghaly, Ahmed M., Ola H. Rizk, Inas Darwish, Manal Hamza, Mezna Saleh Altowyan, Assem Barakat, and Mohamed Teleb. 2022. "Design, Synthesis, Pharmacodynamic and In Silico Pharmacokinetic Evaluation of Some Novel Biginelli-Derived Pyrimidines and Fused Pyrimidines as Calcium Channel Blockers" Molecules 27, no. 7: 2240. https://doi.org/10.3390/molecules27072240
APA StyleFarghaly, A. M., Rizk, O. H., Darwish, I., Hamza, M., Altowyan, M. S., Barakat, A., & Teleb, M. (2022). Design, Synthesis, Pharmacodynamic and In Silico Pharmacokinetic Evaluation of Some Novel Biginelli-Derived Pyrimidines and Fused Pyrimidines as Calcium Channel Blockers. Molecules, 27(7), 2240. https://doi.org/10.3390/molecules27072240