3.2. Synthetic Procedures
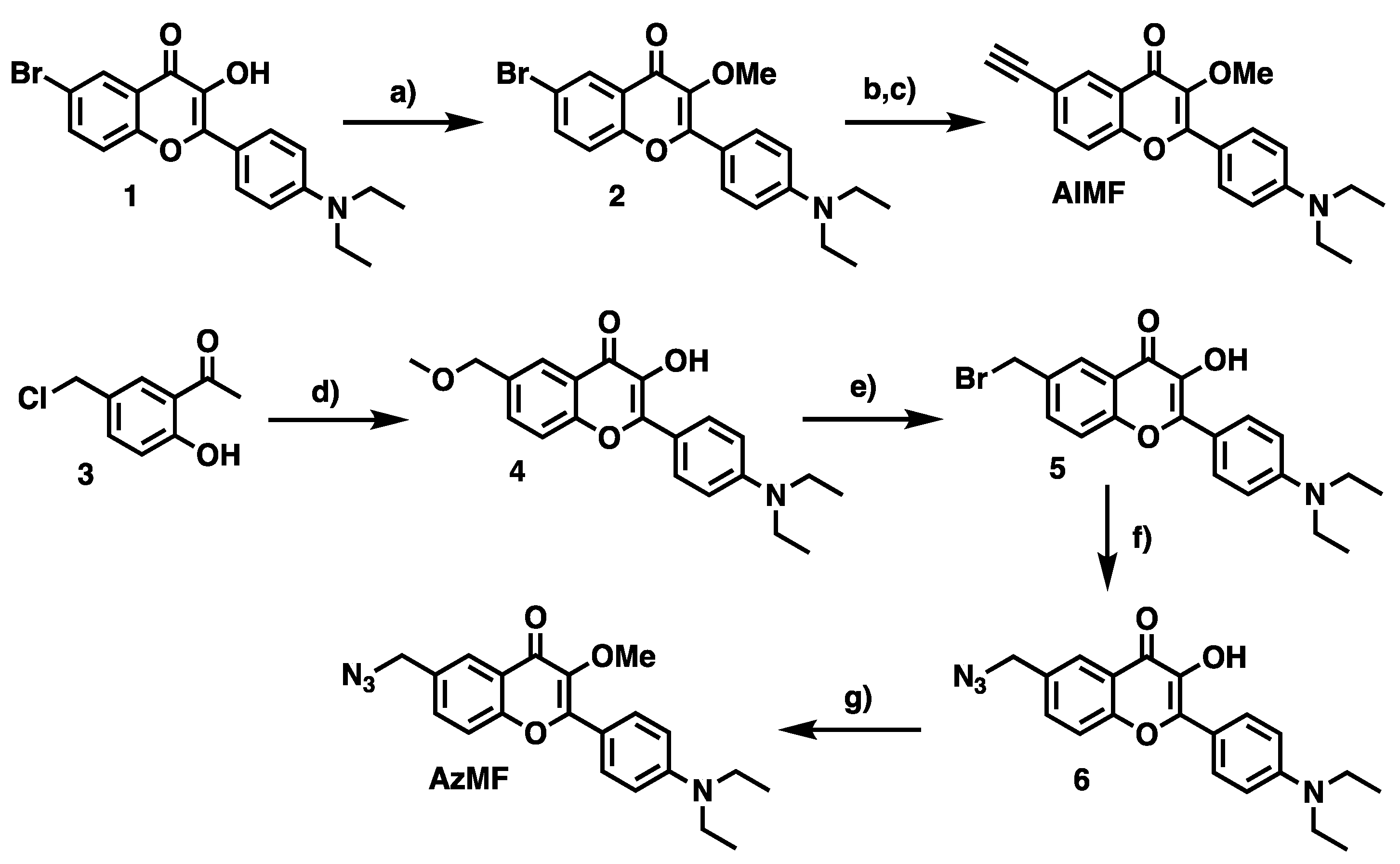
6-Bromo-2-(4-diethylamino)phenyl-3-hydroxy-4H-chromen-4-one (1): 1-(5-Bromo-2-hydroxyphenyl)ethanone (1.7 g, 7.75 mmol, 1 eq.) and 4-(diethylamino)benzaldehyde (1.56 g, 8.52 mmol, 1.1 eq.) were solubilized in 1,2-dichloroethane (10 mL) and morpholine (10 mL). The reaction solution was irradiated by microwave (25 min, 100 °C, 200 W). The volatiles were removed in vacuo to give a reddish brown solid, which was solubilized in ethanol (15 mL). To the stirred mixture cooled in an ice bath, were added sequentially an aq. 5 M NaOH solution (20 mL, 13 eq.) and H2O2 (30% w/w in H2O, 7.65 mL, 10 eq.). After stirring overnight, the mixture was neutralized by an aq. 1 N HCl solution, and the resulting precipitate was filtered and washed with water and cyclohexane sequentially. Compound 1 was obtained as an orange solid. (2.24 g, 77%). Rf = 0.38 (Toluene/Acetone 4:1) or 0.58 (DCM/MeOH, 99.5:0.5). 1H-NMR (CDCl3, 200 MHz): δ 1.15 (t, 3J = 7.0 Hz, 6H, NCH2-CH3), 3.41 (q, 3J = 7.0 Hz, 4H, NCH2-CH3), 6.67 (d, 3J = 9.0 Hz, 2H, Hmeta), 7.35 (d, 3J = 8.8 Hz, 1H, H8), 7.62 (dd, 3J = 8.8 Hz, 4J = 2.4 Hz, 1H, H7), 8.05 (d, 3J = 9.0 Hz, 2H, Hortho), 8.26 (d, 4J = 2.4 Hz, 1H, H5), 9.37 (s, 1H, OH). 13C-NMR (CDCl3, 50 MHz): δ 12.7 (N-CH2-CH3), 44.5 (N-CH2-CH3), 110.9 (Cm), 116.7 (Cp), 119.8 (Ci), 122.3 (C8), 127.6 (C6), 129.5 (C7), 135.5 (Co), 136.9 (C5), 147.3 (C10), 149.1 (C2), 153.7 (C9), 171.0 (C4). HRMS (ESI+): m/z calcd for C19H18NO3BrH+: 388.0543, 390.0522 [M + H]+; found: 388.0550, 390.0529 [M + H]+.
6-Bromo-2-(4-diethylamino)phenyl-3-methoxy-4H-chromen-4-one (2): To a stirred suspension of 1 (110 mg, 0.33 mmol, 1 eq.) in DCM (0.25 M, 6 mL), were sequentially added 18-crown-6 (25 mg, 7 mol%), an aq. KOH solution (25% w/w, 0.9 mL) and dimethyl sulfate (139 µL, 4 eq.). The reaction mixture was stirred 30 min at rt. After addition of H2O (8 mL), the organic layer was extracted with DCM (3×), dried over MgSO4, filtered and the volatiles were removed in vacuo. The residue was purified by flash chromatography on silica gel eluted with DCM/DCM–1% MeOH mixture (90:10 → 10:90, v/v) to provide the desired compound 2 as a yellow powder (101 mg, 0.4 mmol, 88%). Rf = 0.29 (DCM/MeOH 99.5:0.5). 1H-NMR (CDCl3, 400 MHz): δ 1.16 (t, 3J = 7.1 Hz, 6H, NCH2-CH3), 3.38 (d, 3J = 7.1 Hz, 4H, NCH2-CH3), 3.81 (s, 3H, OCH3), 6.67 (d, 3J = 9.3 Hz, 2H, Hmeta), 7.32 (d, 3J = 8.9 Hz, 1H, H8), 7.62 (dd, 3J = 8.9 Hz, 4J = 2.4 Hz, 1H, H7), 8.00 (d, 3J = 9.3 Hz, 2H, Hortho), 8.30 (d, 4J = 2.4 Hz, 1H, H5). 13C-NMR (CDCl3, 101 MHz): δ 12.6 (NCH2-CH3), 44.5 (NCH2-CH3), 56.7 (OCH3), 110.8 (Cmeta), 116.4 (C6), 117.6 (Ci), 119.6 (C8), 127.7 (C10), 128.2 (C5), 130.3 (Cortho), 135.7 (C7), 140.0 (C3), 149.6 (Cpara), 153.8 (C2), 157.0 (C9), 173.2 (C4). HRMS (ESI+): m/z calcd for C20H20BrNO3H+: 402.0699, 404.0679 [M + H]+; found: 402.0698, 404.0679 [M + H]+.
2-(4-(Diethylamino)phenyl)-3-methoxy-6-((trimethylsilyl)ethynyl)-4H-chromen-4-one (AlMF-TMS): To a stirred solution of 2 (50 mg, 0.12 mmol) in dry DMF (3 mL) under argon, were sequentially added TMS-acetylene (53 µL, 0.37 mmol, 3 eq.), triethylamine (175 µL, 1.24 mmol, 10 eq.) and a mixture of CuI (5 mg, 20 mol%)/PdCl2(PPh3)2 (18 mg, 20 mol%). The reaction mixture was warmed to 70 °C overnight. The volatiles were removed in vacuo and the residue was directly engaged in the next step.
2-(4-(Diethylamino)phenyl)-6-ethynyl-3-methoxy-4H-chromen-4-one (AlMF): To a 10-mL vial containing AlMF-TMS (20 mg, 45 µmol, 1 eq.), was added a minimum of MeOH (2 mL) to solubilize the compound. K2CO3 (63 mg, 0.45 mmol, 10 eq.) was then poured, and the resulting solution was stirred overnight at room temperature. The reaction mixture was quenched by acetic acid and reduced in vacuo to give the crude product. The residue was purified by preparative TLC (SiO2) eluted with DCM/MeOH (99:1, v/v) to provide the desired product AlMF as a yellow powder (10 mg, 30 µmol, 64% over 2 steps). Rf = 0.24 (DCM/MeOH 99.5:0.5). 1H-NMR (CDCl3, 400 MHz): δ 1.16 (t, 3J = 7.2 Hz, 6H, NCH2-CH3), 3.04 (s, 1H, HC≡C), 3.38 (d, 3J = 7.2 Hz, 4H, NCH2-CH3), 3.82 (s, 3H, OCH3), 6.67 (d, 3J = 9.2 Hz, 2H, Hmeta), 7.38 (d, 3J = 8.7 Hz, 1H, H8), 7.62 (dd, 3J = 8.7, 4J = 2.0 Hz, 1H, H7), 8.00 (d, 3J = 9.2 Hz, 2H, Hortho), 8.31 (d, 4J = 2.0 Hz, 1H, H5). 13C-NMR (CDCl3, 101 MHz): δ 12.6 (NCH2-CH3), 44.5 (NCH2-CH3), 59.7 (OCH3), 77.8 (HC≡C), 82.3 (HC≡C), 110.8 (Cmeta), 116.5 (C6), 118.0 (C8), 118.4 (Ci), 124.2 (C10), 129.9 (C5), 130.2 (Cortho), 136.0 (C7), 140.0 (C3), 149.5 (Cpara), 154.7 (C2), 156.8 (C9), 173.7 (C4). HRMS (ESI+): m/z calcd for C22H21NO3H+: 348.1594 [M + H]+; found: 348.1600.
1-(5-(Chloromethyl)-2-hydroxyphenyl)ethan-1-one (
3): Adapted from the protocol described in [
34]. To a solution of paraformaldehyde (2.4 g, 79.3 mmol, 1.08 eq.) in conc. HCl solution (45 mL), was added 2-hydroxyacetophenone (8.85 mL, 73.4 mmol, 1 eq.). Next, the reaction mixture was stirred at 35 °C for 5 h. A yellow precipitate was formed which was then filtered and washed with water (3×). The resulting solid was solubilized in DCM, dried over MgSO
4, filtered and the volatiles were removed in vacuo to obtain the product as a yellow powder (11.8 g, 63.9 mmol, 87%). R
f = 0.52 (Cyclohexane/EtOAc 4:1).
1H-NMR (DMSO-d
6, 400 MHz):
δ 2.63 (s, 3H, H9), 4.76 (s, 2H, H8), 6.98 (d,
3J = 8.5 Hz, 1H, H4), 7.59 (dd,
3J = 8.5 Hz,
4J = 2.2 Hz, 1H, H5), 7.95 (d,
4J = 2.2 Hz, 1H, H7).
13C-NMR (DMSO-d
6, 101 MHz):
δ 28.0 (C9), 45.9 (C8), 118.2 (C4), 120.5 (C2), 128.5 (C6), 131.8 (C7), 136.9 (C5), 160.6 (C3), 203.8 (C1).
2-(4-(Diethylamino)phenyl)-3-hydroxy-6-methoxymethyl-4H-chromen-4-one (4): To a stirred solution of 3 (2.5 g, 13.5 mmol, 1 eq.) in methanol (45 mL, 0.3 M) was added NaOH (1.66 g, 14.2 mmol, 1.05 eq.) and 4-(diethylamino)benzaldehyde (2.55 g, 14.2 mmol, 1.05 eq.). The reaction mixture was refluxed for 8 h. After cooling to 4 °C, H2O2 (30% w/w in H2O, 5.5 mL) was added dropwise. Then, cold water (200 mL) was poured, and the reaction mixture was acidified with an aq. 2 N HCl solution to pH 6.5. MeOH was evaporated under reduced pressure. DCM (200 mL) was added, and the organic layer was extracted (3×). The combined organic phases were reduced in vacuo to give the crude compound 4 as a brown oil, which was pure enough to be taken to the next step without further purification (4.66 g, 13.2 mmol, 97%). Rf = 0.44 (Cyclohexane/EtOAc 4:1). 1H-NMR (DMSO-d6, 400 MHz): δ 1.16 (t, 3J = 7.0 Hz, 6H, NCH2-CH3), 3.33 (s, 3H, OCH3), 3.42 (q, 3J = 7.0 Hz, 4H, NCH2-CH3), 4.54 (s, 2H, OCH2Ph), 6.80 (d, 3J = 9.2 Hz, 2H, Hmeta), 7.68 (m, 2H, H7 and H8), 8.01 (d, 4J = 2.0 Hz, 1H, H5), 8.09 (d, 3J = 9.2 Hz, 2H, Hortho). 13C-NMR (CDCl3, 101 MHz): δ 12.0 (NCH2-CH3), 43.3 (NCH2-CH3), 57.2 (OCH3), 72.3 (OCH2), 110.3 (Cmeta), 116.5 (C6), 117.7 (C8), 118.1 (Ci), 122.5 (C5), 124.2 (C10), 128.8 (Cortho), 131.9 (C7), 142.6 (C3), 146.6 (Cpara), 148.0 (C2), 153.1 (C9), 171.3 (C4).
6-(Bromomethyl)-2-(4-(diethylamino)phenyl)-3-hydroxy-4H-chromen-4-one (5): A mixture of 4 (610 mg, 1.7 mmol, 1 eq.) and 47% hydrobromic acid in H2O (5 mL) was refluxed for 3 h. After cooling to room temperature, a saturated aq. solution of sodium carbonate was added slowly to neutralize the reaction until pH 7 was reached. Then, the solid was filtered to directly obtain the desired crude solid 5 as a khaki solid (960 mg, 2.4 mmol, quant.), which was directly committed to the next step without further purification. Rf = 0.24 (Cyclohexane/EtOAc 4:1). 1H-NMR (CDCl3, 400 MHz): δ 1.23 (t, 3J = 7.1 Hz, 6H, NCH2-CH3), 3.45 (q, 3J = 7.1 Hz, 4H, NCH2-CH3), 4.60 (s, 2H, OCH2Ph), 6.77 (d, 3J = 9.1 Hz, 2H, Hmeta), 7.54 (d, 3J = 8.7 Hz, 1H, H8), 7.69 (dd, 3J = 8.7 Hz, 4J = 2.1 Hz, 1H, H7), 8.16 (d, 3J = 9.1 Hz, 2H, Hortho), 8.22 (d, 4J = 2.1 Hz, 1H, H5).
6-(Azidomethyl)-2-(4-(diethylamino)phenyl)-3-hydroxy-4H-chromen-4-one (6): 5 (242 mg, 0.60 mmol, 1 eq.) was solubilized in a mixture of acetone (6 mL) and DMF (1.5 mL) to which was added NaN3 (60 mg, 0.90 mmol, 1.5 eq.). The solution was stirred overnight at room temperature. Then, EtOAc (20 mL) was introduced, and acetone was evaporated under reduced pressure. A saturated aq. solution of NH4Cl was added (10 mL) to quench the reaction. The organic layer was extracted with EtOAc (×2), dried over MgSO4, filtered, and concentrated in vacuo. The crude product was purified by flash chromatography on silica gel eluted with cyclohexane/ethyl acetate (9:1 → 7:3, v/v) to give 6 as an orange powder, which was directly engaged in the next step.
6-Azidomethyl-2-(4-(diethylamino)phenyl)-3-methoxy-4H-chromen-4-one (AzMF): To a stirred solution of 6 (144 mg, 0.40 mmol, 1 eq.) in CH2Cl2 (2.6 mL), were sequentially added 18-crown-6 (30 mg, 7 mol%), an aq. KOH solution (25% w/w, 0.4 mL) and dimethyl sulfate (166 µL, 1.58 mmol, 4 eq.). The resulting mixture was stirred overnight at room temperature. After quenching the reaction by addition of H2O (5 mL), the organic layer was extracted with CH2Cl2 (3×). The combined organic phases were dried over MgSO4, filtered and the volatiles were removed in vacuo. The residue was purified by flash chromatography on silica gel eluted with cyclohexane/ethyl acetate (9:1 → 3:1, v/v) to give the desired compound as a yellow solid (146 mg, 0.38 mmol, 70% over 2 steps). 1H-NMR (CDCl3, 400 MHz): δ 1.17 (t, 3J = 7.1 Hz, 6H, NCH2-CH3), 3.39 (q, 3J = 7.1 Hz, 4H, NCH2-CH3), 3.83 (s, 3H, OCH3), 4.39 (s, 2H, N3CH2Ph), 6.68 (d, 3J = 9.3 Hz, 2H, Hmeta), 7.46 (d, 3J = 8.6 Hz, 1H, H8), 7.54 (dd, 3J = 8.6 Hz, 4J = 2.2 Hz, 1H, H7), 8.02 (d, 3J = 9.3 Hz, 2H, Hortho), 8.22 (d, 4J = 2.2 Hz, 1H, H5). 13C-NMR (CDCl3, 101 MHz): δ 12.6 (NCH2-CH3), 44.5 (NCH2-CH3), 54.2 (N3CH2Ph), 59.7 (OCH3), 110.8 (Cmeta), 116.6 (C6), 118.6 (C8), 124.3 (Ci), 125.3 (C5), 130.2 (Cortho), 131.7 (C10), 132.6 (C7), 140.1 (C3), 149.5 (Cpara), 154.8 (C2), 156.8 (C9), 174.2 (C4). HRMS (ESI+): m/z calcd for C21H22N4O3H+: 379.1765; [M + H]+; found: 379.1777.
6-(Bromomethyl)-2-(4-(diethylamino)phenyl)-3-methoxy-4H-chromen-4-one (7): To a stirred solution of 5 (240 mg, 0.60 mmol, 1 eq.) in DCM (12 mL), were sequentially added 18-crown-6 (79 mg, 0.30 mmol, 0.1 eq.), an aq. KOH solution (25% w/w, 1.7 mL, DCM/KOH 7:1) and dimethyl sulfate (282 µL, 2.98 mmol, 5 eq.). The reaction mixture was stirred for 30 min at room temperature. The organic layer was extracted with DCM (3×), dried over MgSO4, filtered, and concentrated in vacuo. The residue was then purified by flash chromatography on silica gel eluted with cyclohexane/ethyl acetate (9:1 → 7:3, v/v) to give the desired compound 7 as a red oil (200 mg, 0.48 mmol, 80%). Rf = 0.58 (Cyclohexane/EtOAc 3:1). 1H-NMR (CDCl3, 400 MHz): δ 1.23 (t, 3J = 7.1 Hz, 6H, NCH2-CH3), 3.45 (q, 3J = 7.1 Hz, 4H, NCH2-CH3), 3.88 (s, 3H, OCH3), 4.59 (s, 2H, BrCH2Ph), 6.74 (d, 3J = 9.2 Hz, 2H, Hmeta), 7.49 (d, 3J = 8.7 Hz, 1H, H8), 7.67 (dd, 3J = 8.7 Hz, 4J = 2.2 Hz, 1H, H7), 8.08 (d, 3J = 9.2 Hz, 2H, Hortho), 8.24 (d, 4J = 2.2 Hz, 1H, H5). 13C-NMR (CDCl3, 101 MHz): δ 12.6 (NCH2-CH3), 32.4 (BrCH2Ph), 44.5 (NCH2-CH3), 59.7 (OCH3), 110.8 (Cmeta), 116.6 (C6), 118.6 (C8), 124.2 (Ci), 125.8 (C5), 130.2 (Cortho), 133.7 (C7), 134.1 (C10), 140.1 (C3), 149.5 (Cpara), 154.7 (C2), 156.8 (C9), 174.1 (C4). HRMS (ESI+): m/z calcd for C21H22BrNO3H+: 416.0856, 418.0835 [M + H]+; found: 416.0870, 418.0848.
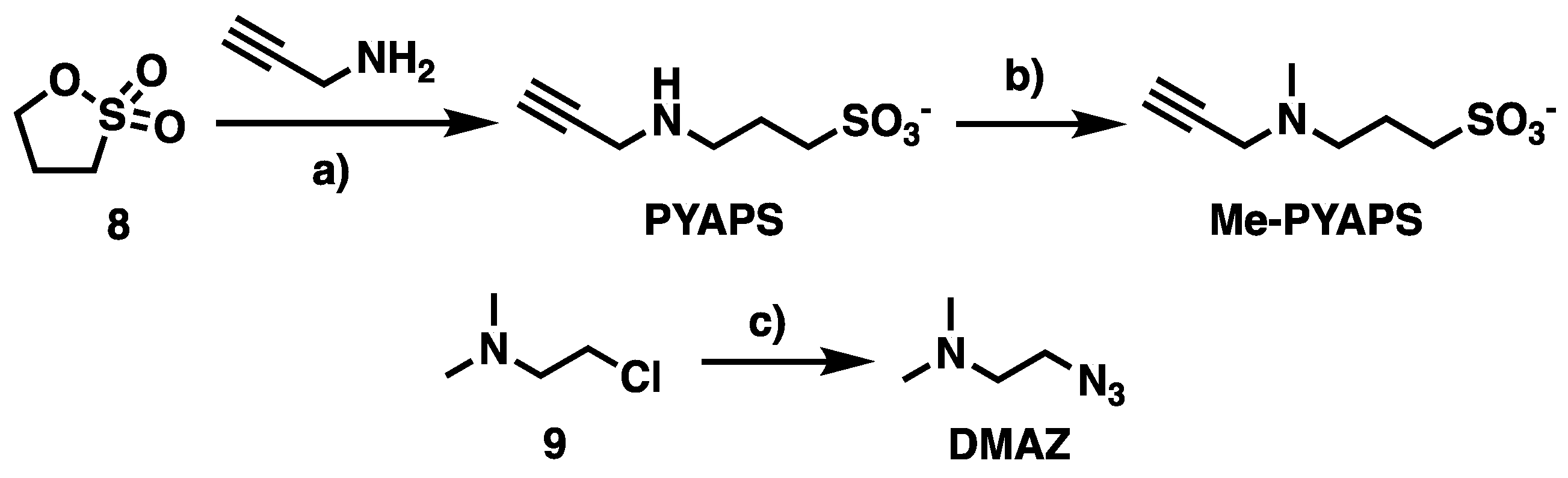
Acid 3-(prop-2-yn-1-ylamino)propane-1-sulfonic (PYAPS): To a solution of propargylamine (116 µL, 1.8 mmol, 1 eq.) in CH3CN (5 mL) was added dropwise 1,3-propane sultone 8 (169 µL, 1.9 mmol, 1.05 eq.). The reaction mixture was stirred at room temperature for 2 d. The resulting precipitate was filtered and washed with CH3CN to afford the desired compound PYAPS as a pinkish solid (230.5 mg, 1.31 mmol, 72%). Rf = 0.25 (DCM/MeOH 4:1). Green staining by ninhydrin. 1H-NMR (DMSO-d6, 400 MHz): δ 1.93 (p, 3J = 6.8 Hz, 2H, H5), 2.60 (t, 3J = 6.8 Hz, 2H, H4), 3.09 (t, 3J = 6.8 Hz, 2H, H6), 3.68 (t, 4J = 2.5 Hz, 1H, H1), 3.90 (d, 4J = 2.5 Hz, 2H, H3). 13C-NMR (DMSO-d6, 101 MHz): δ 21.7 (C5), 35.6 (C3), 46.0 (C4), 48.8 (C6), 75.1 (C2), 79.4 (C1). HRMS (ESI+): m/z calcd for C6H11NO3SH+: 178.0532; [M + H]+; found: 178.0534.
Sodium 3-(methyl(prop-2-yn-1-yl)amino)propane-1-sulfonate (Me-PYAPS): PYAPS (150 mg, 0.85 mmol, 1 eq.), formol (37% formaldehyde in H2O) (900 µL) and formic acid (903 µL) were heated overnight at 70 °C. Then, the solution was concentrated in vacuo until a paste was obtained. NaHCO3 (1 g) was added to the solid until bubbling ceased to provide the sodium sulfonate salt Me-PYAPS as a white solid (375 mg, quant.). Bicarbonate salts contaminate the product due to the excess used. 1H-NMR (DMSO-d6, 400 MHz): δ 1.60–1.73 (m, 2H, H5), 2.15 (s, 3H, H7), 2.32–2.42 (m, 4H, H4 and H6), 3.07 (t, 4J = 2.4 Hz, 1H, H1), 3.25 (d, 4J = 2.4 Hz, 2H, H3). 13C-NMR (DMSO-d6, 101 MHz): δ 22.7 (C5), 40.7 (C3), 44.3 (C6), 48.9 (C7), 53.7 (C4), 75.1 (C2), 81.5 (C1). HRMS (ESI+): m/z calcd for C7H12NO3SH+−: 192.0689; [M + H]+; found: 192.0690.
2-Azido-N,N-dimethylethanamine (DMAZ): 3-Chloro-1,1-dimethylpropylamine 9 (800 mg, 5.6 mmol, 1 eq.) and NaN3 (1.1 g, 16.7 mmol, 3 eq.) were sequentially dissolved in H2O (18.5 mL, 0.3 M). The solution was stirred at 80 °C for 24 h. After cooling to room temperature, the reaction mixture was adjusted to pH 10 by addition of an aq. 0.5 M NaOH solution. The organic layer was then extracted with diethyl ether (3×). The combined extracts were dried over MgSO4, filtered, and concentrated under vacuum to yield the product DMAZ as a yellowish liquid (970 mg, 9.03 mmol, quant.). 1H-NMR (CDCl3, 400 MHz): δ 2.24 (s, 6H, H3 and H3′), 2.47 (t, 3J = 6.2 Hz, 2H, H2), 3.31 (t, 3J = 6.2 Hz, 2H, H1). 13C-NMR (CDCl3, 101 MHz): δ 45.5 (C3 and C3′), 49.1 (C1), 58.0 (C2). HRMS (ESI+): m/z calcd for C4H10N4H+−: 115.0978; [M − H]+; found: 115.0982.
Potassium N(3-(((2-(4-(diethylamino)phenyl)-3-methoxy-4H-chromen-6-yl)methyl), N(prop-2-yn-1-yl)amino)propane-1-sulfonate (AlMF−): To a solution of 7 (22 mg, 50 µmol, 1 eq.) in DMF (0.5 mL), were added PYAPS (19 mg, 0.11 mmol, 2 eq.), H2O (0.4 mL), and K2CO3 (19 mg, 0.13 mmol, 2.5 eq.). The solution was stirred at room temperature for 24 h. Then, DMF and water were evaporated in vacuo and the crude (solubilized with DCM/MeOH) was purified by flash chromatography on silica gel eluted with i-PrOH/NH3/H2O (12:2:1, v/v) to afford the desired product AlMF− as an orange solid (26 mg, 50 µmol, 95%). 1H-NMR (MeOD, 400 MHz): δ 1.22 (t, 3J = 7.0 Hz, 6H, NCH2-CH3), 1.97–2.08 (m, 2H, H6′), 2.73 (t, 3J = 6.9 Hz, 2H, H5′), 2.90 (m, 2H, H7′), 3.49 (q, 3J = 7.0 Hz, 4H, NCH2-CH3), 3.78 (m, 2H, H2′), 3.79 (s, 3H, OCH3), 3.94 (d, 3J = 2.6 Hz, 1H, H4′), 4.84 (s, 2H, H1′), 6.80 (d, 3J = 7.0 Hz, 2H, Hmeta), 7.60 (d, 3J = 8.6 Hz, 1H, H8), 7.77 (dd, 3J = 8.6 Hz, 2J = 2.1 Hz 1H, H7), 8.07 (s, 1H, H5), 8.08 (m, 3J = 7.0 Hz, 2H, Hortho). HRMS (ESI+): m/z calcd for C27H32N2O6SH+: 513.2054; [M − H]+; found: 513.2064.
3-(((2-(4-(diethylamino)phenyl)-3-methoxy-4-oxo-4H-chromen-6-yl)methyl)(methyl)(prop-2-yn-1-yl)ammonio)propane-1-sulfonate (AlMF+−): To a solution of 7 (80 mg, 0.19 mmol, 1 eq.) in DMF (4.5 mL), were added Me-PYAPS and water (2 mL). The reaction mixture was stirred at room temperature for 24 h before the volatiles were removed under vacuum. The residue (diluted with DCM/MeOH) was purified by flash chromatography on silica gel eluted with i-PrOH/NH3/H2O (12:2:1, v/v) to give the desired product AlMF+− as a yellow oil (36 mg, 68 µmol, 36%). 1H-NMR (MeOD, 400 MHz): δ 1.23 (t, 3J = 7.1 Hz, 6H, NCH2-CH3), 2.07–2.17 (m, 2H, H6′), 2.86 (s, 3H, H8′), 2.88 (m, 2H, H5′), 3.07–3.14 (m, 2H, H7′), 3.50 (q, 3J = 7.1 Hz, 4H, NCH2-CH3), 3.68–3.69 (m, 5H, H2′ and OCH3), 4.30 (m, 1H, H4′), 4.79 (s, 2H, H1′), 6.81 (d, 3J = 9.0 Hz, 2H, Hmeta), 7.72 (d, 3J = 8.6 Hz, 1H, H8), 7.96 (dd, 3J = 8.6 Hz, 2J = 2.0 Hz 1H, H7), 8.07 (d, 3J = 9.0 Hz, 2H, Hortho), 8.35 (s, 1H, H5). 13C-NMR (MeOD, 101 MHz): δ 11.5, 21.0, 44.0, 44.7, 53.8, 58.7, 60.9, 64.4, 71.1, 77.6, 82.6, 110.7, 115.4, 119.2, 123.5, 123.9, 130.0, 130.2, 137.1, 139.5, 150.1, 156.0, 158.0, 173.9. HRMS (ESI+): m/z calcd for C28H34N2O6SH+: 527.2210; [M + H]+; found: 527.2215.
2-Azidoethyl-N-((2-(4-(diethylamino)phenyl)-3-methoxy-4-oxo-4H-chromen-6-yl)methyl)-N,N-dimethylethanammonium bromide (AzMF+): To a solution of 7 (12 mg, 29 µmol, 1 eq.) in anhydrous THF (0.4 mL, 80 mM), was added DMAZ (16 mg, 0.144 mmol, 5 eq.). The reaction mixture was stirred at room temperature for 6 h. The formed precipitate was centrifuged, and the upper layer was discarded. The compound was washed twice more after suspension in dry THF, centrifugation and removal of the supernatant. The recovered solid was dried under reduced pressure to provide AzMF+ as a yellow powder (9.6 mg, 18 µmol, 63%). 1H-NMR (MeOD, 400 MHz): δ 1.24 (t, 3J = 7.1 Hz, 6H, NCH2-CH3), 3.16 (s, 6H, H2′and H3′), 3.52 (q, 3J = 7.1 Hz, 4H, NCH2-CH3), 3.62 (t, 3J = 5.4 Hz, 2H, H5′), 3.83 (s, 3H, OCH3), 4.08 (t, 3J = 5.4 Hz, 2H, H4′), 4.76 (s, 2H, H1′), 6.84 (d, 3J = 9.3 Hz, 2H, Hmeta), 7.82 (d, 3J = 8.6 Hz, 1H, H8), 7.93 (d, 3J = 8.6 Hz, 1H, H7), 8.13 (d, 3J = 9.3 Hz, 2H, Hortho), 8.35 (s, 1H, H5). 13C-NMR (MeOD, 101 MHz): δ 11.5 (NCH2-CH3), 44.0 (NCH2-CH3), 44.6 (C2′, C3′), 49.5 (N3CH2), 54.2 (N+CH2Ph), 58.7 (OCH3), 62.6 (CH2N+), 67.6 (N+CH2Ph), 110.8 (Cmeta), 115.4 (C6), 119.1 (C8), 123.8 (Ci), 123.9 (C10), 130.2 (C5), 130.2 (Cortho), 137.2 (C7), 137.2 (C3), 139.6 (Cpara), 150.2 (C2), 156.1 (C9), 174.1 (C4). HRMS (ESI+): m/z calcd for C25H32N5O3+: 450.2500 [M − H]+; found: 450.2500.
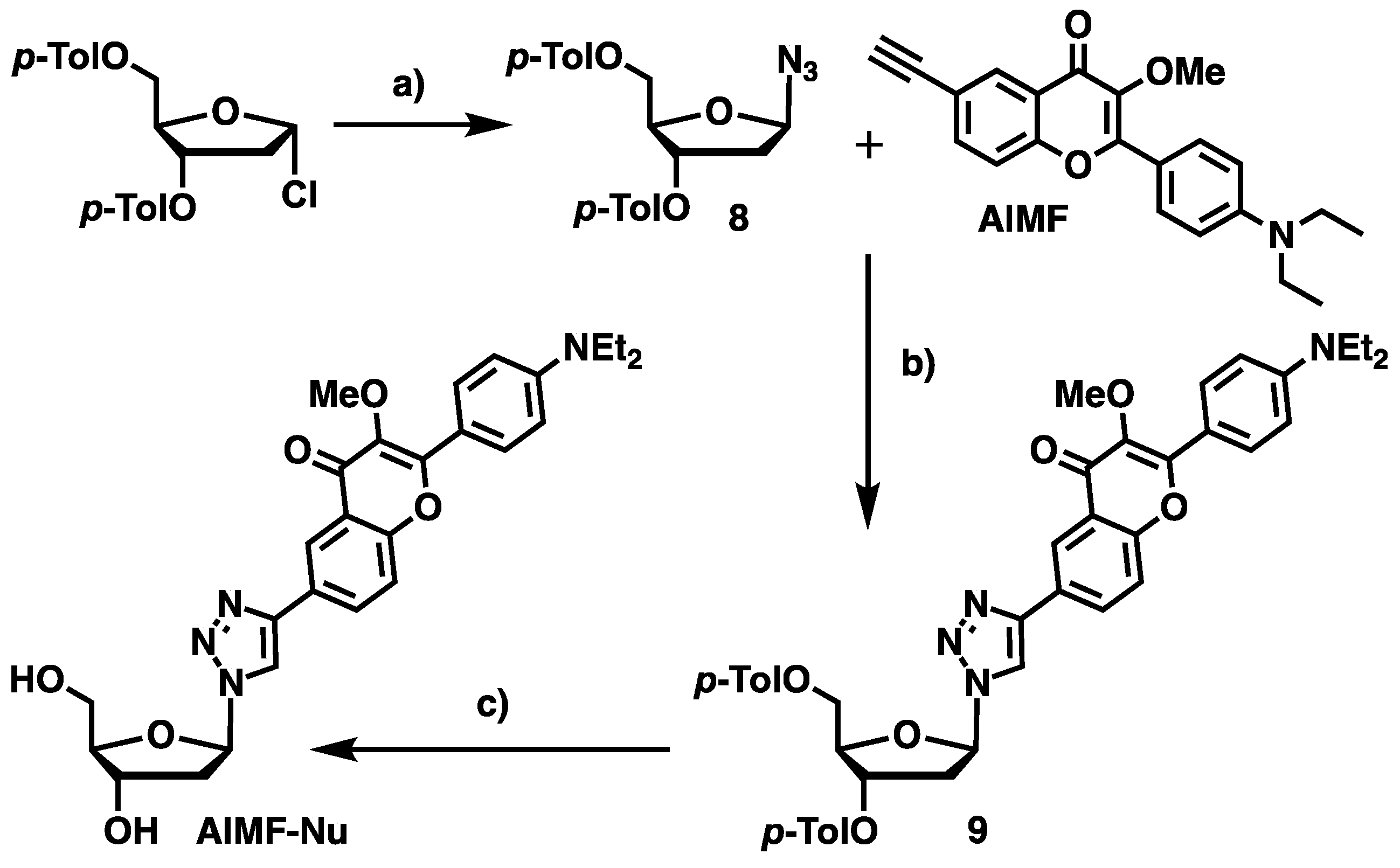
1-Azido-1,2-dideoxy-3,5-di-O-p-toluoyl-β-d-ribofuranose (
8): Adapted from the procedure described in [
35,
36]. To a stirred solution of 1α-chloro-3,5-di-toluoyl-2-deoxy-
l-ribose (1.4 g, 3.64 mmol, 1 eq.) in acetone (20 mL) at 0 °C, was slowly added NaN
3 (1.22 g, 18.2 mmol, 5.0 eq.). The reaction mixture was kept under vigorous stirring for 5 h. After completion, as evidenced by TLC monitoring, the reaction mixture was quenched with a saturated aq. NaHCO
3 solution (15 mL), and the organic layer was extracted with CH
2Cl
2 (3×). The combined extracts were dried over MgSO
4, filtered, and evaporated under reduced pressure. The crude oil obtained is a mixture of both anomers (
β/
α 9:1), which was subjected to flash chromatography on silica gel eluted with PE/Et
2O (19:1 → 4:1,
v/
v) to furnish the pure
β anomer of
8 as a white crystalline compound (1.0 g, 80%). C
21H
23O
5N
3 (384.13). R
f = 0.67 (Toluene/Acetone 9:1).
1H-NMR (CD
2Cl
2, 400 MHz):
δ 2.43 (s, 3H, C
H3-Tol), 2.44 (s, 3H, C
H3-Tol), 2.46–2.49 (m, 2H, H2′), 4.55–4.67 (m, 3H, H4′, H5′), 5.64 (ddd,
3J = 7.9 Hz,
3J = 4.4Hz,
4J = 2.2 Hz, 1H, H3′), 5.77 (t,
3J = 5.1 Hz, 1H, H1′), 7.28–7.31 (m, 4H, H
m-Tol), 7.98 (d,
3J = 8.2 Hz, 2H, H
o-Tol), 8.05 (d,
3J = 8.2 Hz, 2H, H
o-Tol).
13C-NMR (CDCl
3, 50 MHz):
δ 22.1 (CH
3), 22.1 (CH
3), 39.3 (C2′), 65.0 (C5′), 75.7 (C4′), 83.5 (C3′), 93.0 (C1′), 127.5 (C
i), 127.9 (C
i), 129.9 (C
m), 129.9 (C
m), 130.4 (C
o), 130.4 (C
o), 144.7 (C
p), 145.1 (C
p), 166.5 (CO), 166.8 (CO).
1,2-Dideoxy-1β-(4-(2-(4-(diethylamino)phenyl)-3-methoxy-4H-chromen-6-yl)-1H-1,2,3-triazol-1-yl)-3,5-di-O-p-toluoyl-β-d-ribofuranose (9): To a stirred solution of AlMF (10 mg, 30 µmol, 1 eq.) in DCE (2.5 mL) inside a 4-mL vial, were sequentially added 8 (18 mg, 50 µmol, 1.8 eq.), DIPEA (61 µL, 0.35 mmol, 12 eq.), acetic acid (10 µL, 0.17 mmol, 6 eq.) and CuI (16 mg, 80 µmol, 2.8 eq.). The reaction was heated at 40 °C for 1 h under an argon atmosphere to give a homogeneous blue solution. The mixture was concentrated under reduced pressure and the residue was purified by preparative TLC (SiO2) eluted with DCM/MeOH (99:1, v/v) to give the desired product 9 as a yellow powder (21 mg, 30 µmol, 98%). 1H-NMR (CDCl3, 400 MHz): δ 1.14 (t, 3J = 7.0 Hz, 6H, NCH2-CH3), 2.28 and 2.37 (2s, 6H, H12 and H14), 2.84 (ddd, 2J = 14.2 Hz, 3J = 6.1 Hz, 3J = 3.3 Hz, 1H, H2′), 3.16–3.25 (m, 1H, H2′), 3.3–3.44 (m, 4H, NCH2-CH3), 3.84 (s, 3H, OCH3), 4.49–4.54 (m, 1H, H5′), 4.58–4.66 (m, 2H, H5′, H4′), 5.71–5.77 (m, 1H, H3′), 6.47 (t, 3J = 6.3 Hz, 1H, H1′), 6.69 (d, 3J = 9.2 Hz, 2H, Hm), 7.12 (d, 3J = 8.0 Hz, 2H, Hm′/Hm′′), 7.21 (d, 3J = 8.2 Hz, 2H, Hm′/Hm′′), 7.49 (d, 3J = 8.7 Hz, 1H, H8), 7.80 (d, 3J = 8.0 Hz, 2H, Ho′/Ho′′), 7.90 (d, 3J = 8.2 Hz, 2H, Ho′/Ho′′), 8.04 (m, 3H, Hα, Ho), 8.15 (dd, 3J = 8.7 Hz, 2J = 2.2 Hz, 1H, H7), 8.36 (d, 2J = 2.2 Hz, 1H, H5). 13C-NMR (CDCl3, 101 MHz): δ 12.6 (NCH2-CH3), 15.3 (C12/C14), 21.7 (C12/C14), 38.2 (C2′), 44.5 (NCH2-CH3), 59.7 (OCH3), 63.8 (C5′), 74.8 (C3′), 83.8 (C4′), 89.0 (C1′), 110.9 (Cm), 116.7 (Ci), 118.4 (C8), 118.9 (Cα), 122.3 (C5), 124.4 (C6), 126.4 (Ci′/Ci′′), 126.6 (Ci′/Ci′′), 126.8 (C10), 129.3 (Cm′/Cm′′), 129.7 (Co′/Co′′), 129.8 (Co′/Co′′), 130.2 (Co), 130.5 (C7), 140.3 (C3), 144.1 (Cp′/Cp′′), 144.5 (Cp′/Cp′′), 147.0 (Cβ), 149.5 (Cp), 154.8 (C9), 156.7 (C2), 165.9 (C11/C13), 166.2 (C11/C13), 172.7 (C4).
1,2-Dideoxy-1-(4-(2-(4-(diethylamino)phenyl)-3-methoxy-4-oxo-4H-chromen-6-yl)-1H-1,2,3-triazol-1-yl)-β-d-ribofuranose (AlMF-Nu): In a 4-mL vial, 9 (21 mg, 30 µmol, 1 eq.) was solubilized in MeOH (0.6 mL) and few drops of DCM. Then, K2CO3 (20 mg, 0.14 mmol, 5 eq.) was added and the reaction mixture was stirred overnight at 35 °C. The mixture was then quenched by a minimum of acetic acid and the solvents were removed in vacuo. The crude was purified by preparative RP-TLC (C18) eluted with H2O/acetonitrile (50:50, v/v) to give the desired product AlMF-Nu as a yellow solid (8 mg, 20 µmol, 56%). Rf = 0.11 (DCM/MeOH, 99.8:0.2). 1H-NMR (CDCl3, 400 MHz): δ 1.13 (t, 3J = 7.0 Hz, 6H, NCH2-CH3), 2.47 (ddd, 3J = 13.6 Hz, 3J = 6.0 Hz, 3J = 4.7 Hz, 1H, H2′), 2.70–2.79 (m, 1H, H2′), 3.35–3.44 (m, 4H, NCH2-CH3), 3.58 (dd, 3J = 12.0 Hz, 3J = 5.0 Hz, 1H, H5′), 3.68 (dd, 3J = 12.0 Hz, 3J = 3.9 Hz, 1H, H5′), 3.72 (s, 3H, OCH3), 3.97 (m, 1H, H4′), 4.50 (m, 1H, H3′), 6.38 (t, 3J = 6.0 Hz, 1H, H1′), 6.71 (d, 3J = 9.3 Hz, 2H, Hmeta), 7.62 (d, 3J = 8.8 Hz, 1H, H8), 8.00 (d, 3J = 9.2 Hz, 2H, Hortho), 8.12 (dd, 3J = 8.8 Hz, 3J = 2.1 Hz, 1H, H7), 8.44 (d, 3J = 2.1 Hz, 1H, H5), 8.56 (s, 1H, Hα). 13C-NMR (CDCl3, 101 MHz): δ 11.5 (NCH2-CH3), 40.4 (C2′), 44.0 (NCH2-CH3), 58.7 (OCH3), 61.9 (C5′), 70.8 (C3′), 88.4 (C4′), 89.0 (C1′), 110,7 (Cmeta), 115.8 (Ci), 118.5 (C8), 119.9 (Cα), 121.3 (C5), 123.9 (C10), 127.3 (C6), 130.1 (Cortho), 130.6 (C7), 139.5 (C3), 146.2 (Cβ), 150.0 (Cpara), 154.8 (C9), 158.0 (C2), 174.6 (C4). HRMS (ESI+): m/z calcd for C27H30N4O6H+: 507.2238 [M + H]+; found: 507.2240.


 ,
, 












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





