3.1. General Information
Unless stated otherwise, all commercially available reagents and solvents were used in the form in which they were supplied without any further purification. The stated yields are based on isolated material. All reactions were performed under an argon atmosphere using Schlenk techniques. Thin layer chromatography was performed on silica gel 60 F254 aluminum-backed plates fabricated by Merck. Flash column chromatography was performed on silica gel 60 (40–63 µm) produced by Merck. NMR spectra were recorded on a Bruker Ascend TM 400 (Bruker, Billerica, MA, USA) or Bruker AVI600 spectrometer (Bruker, Billerica, MA, USA) at 600 MHz or 400 MHz, respectively, for
1H NMR and at 150 MHz or 100 MHz, respectively, for
13C NMR. Coupling constants (
J) are reported in hertz and chemical shifts are reported in parts per million (δ) relative to the central residual protium solvent resonance in
1H NMR (CDCl
3 = δ 7.26, DMSO-
d6 = δ 2.50 and MeOD-
d4 = δ 3.31) and the central carbon solvent resonance in
13C NMR (CDCl
3 = δ 77.00 ppm, DMSO-
d6 = δ 39.43 and MeOD-
d4 = δ 49.00).
19F NMR experiments were run in CDCl
3 with signals calibrated against CF
3CH
2OH. Mass spectra were recorded at 70 eV on Waters Prospec Q spectrometer (Waters Corporation, Milford, MA, USA) using EI, ES or CI as the methods of ionization. High resolution mass spectra were recorded on Waters Prospec Q spectrometer using EI or ES as the methods of ionization. Optical rotations were measured using a 1 mL cell with a 1.0 dm path length on a Perkin Elmer 341 polarimeter (PerkinElmer Inc., Waltham, MA, USA). IR, MS,
1H and
13C NMR spectra as well as UV and HPLC chromatograms are found in the
Supplementary Material.
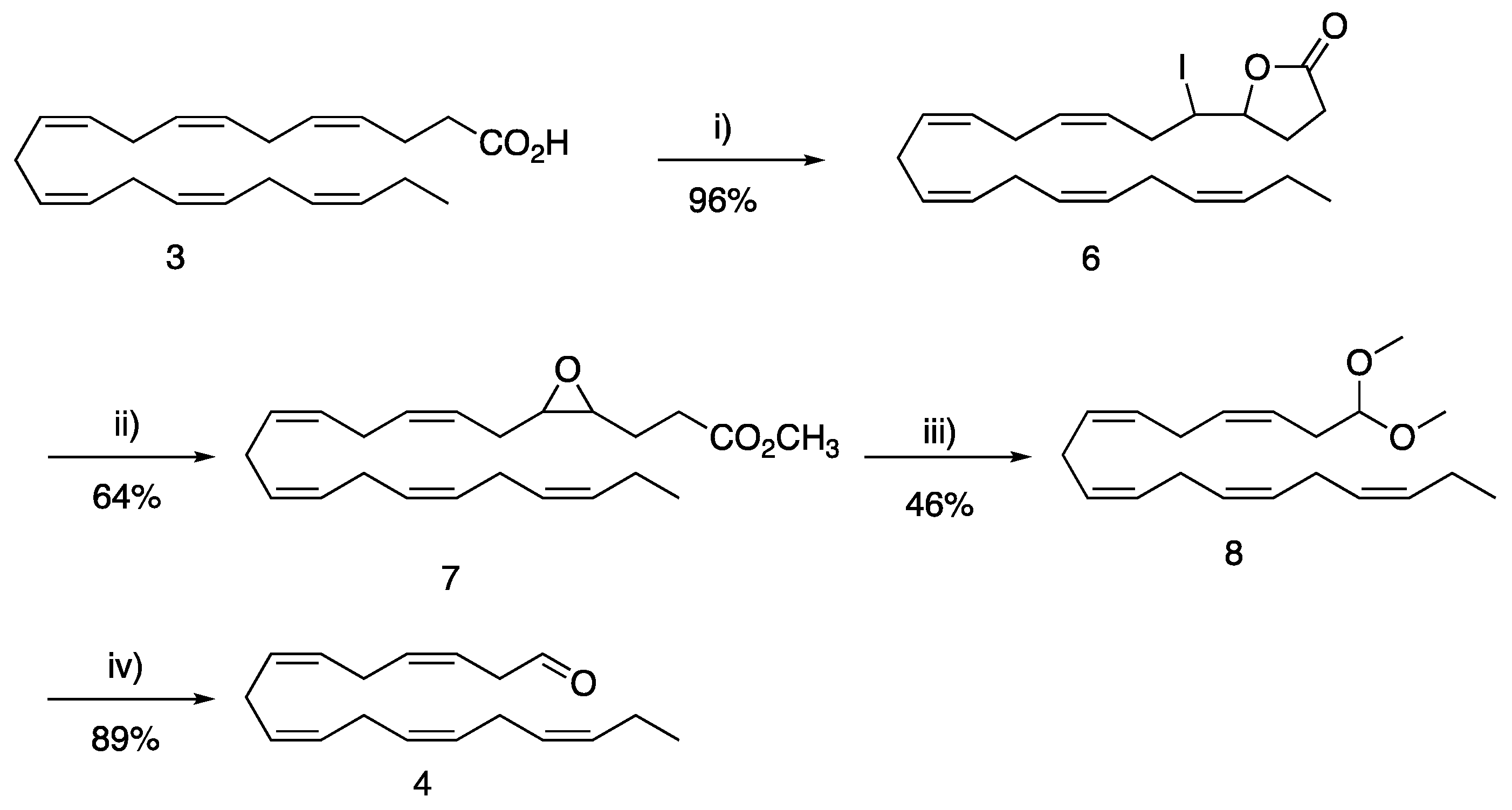
3.1.1. 5-((3Z,6Z,9Z,12Z,15Z)-1-Iodooctadeca-3,6,9,12,15-pentaenyl)dihydro-2(3H)-furanone (6)
Iodolactone
6 was prepared according to a literature procedure by Ulven and co-workers [
35]. 2,6-Lutidine (7.1 mL, 60.9 mmol, 2 eq.) was added dropwise to a solution of
3 (10.0 g, 30.4 mmol, 1 eq.) dissolved in CH
2Cl
2 (100 mL). The mixture was cooled to 0 °C and I
2 (15.453 g, 60.9 mmol, 2 eq.) was added. The reaction continued overnight (15 h) at 0 °C. The reaction was quenched by adding saturated aqueous Na
2S
2O
3 (60 mL). Ethyl acetate was used for extraction (2 × 60 mL) and the combined organic layers were washed with saturated aqueous NaH
2PO
4 (50 mL) and brine, and dried using Na
2SO
4. The solvent was removed in vacuo and the product was passed through a short silica plug (hexane/EtOAc 1:1) to afford iodolactone
6 (13.279 g) in a 96% yield. All spectroscopic and physical data were in agreement with those reported in the literature [
35].
Rf = 0.28 (hexanes/EtOAc 3:1 KMnO
4 stain);
1H NMR (400 MHz, CDCl
3) δ 5.58 (m. 1H), 5.46–5.29 (m, 9H), 4.28 (dt,
J = 7.7, 3.0 Hz, 1H), 4.13 (dt,
J = 7.4, 3.0 Hz, 1H), 2.90–2.69 (m, 11H), 2.63–2.52 (m, 1H), 2.42 (m, 1H), 2.09 (m, 3H), 0.98 (t,
J = 7.5 Hz, 3H).
13C NMR (101 MHz, CDCl
3) δ 176.2, 132.0, 131.6, 128.8, 128.6, 128.4, 127.9, 127.9, 127.4, 127.0, 126.7, 80.7, 37.7, 34.6, 28.5, 27.3, 25.9, 25.7, 25.7, 25.6, 20.6, 14.3.
3.1.2. Methyl 3-(3-((2Z,5Z,8Z,11Z,14Z)-heptadeca-2,5,8,11,14-pentaen-1-yl)oxiran-2-yl) Propanoate (7)
Iodolactone
6 (8.482 g, 18.66 mmol, 1 eq.) was dissolved in MeOH (60 mL) and K
2CO
3 (3.096 g, 22.40 mmol, 1.2 eq.) was added. The reaction mixture was stirred at ambient temperature for 3 h. Water (50 mL) was added and the aqueous phase was extracted using hexane (3 × 50 mL). The combined organic layers were dried (MgSO
4) and the solvent removed in vacuo. Epoxide
7 was obtained as a light brown oil (6.023 g) in a 90% yield. All spectroscopic and physical data were in agreement with those reported in the literature [
36].
Rf = 0.45 (hexanes/EtOAc 3:1 KMnO
4 stain);
1H NMR (400 MHz, CDCl
3) δ 5.55–5.21 (m, 10H), 3.65 (s, 3H), 3.00–2.88 (m, 2H), 2.85–2.75 (m, 8H), 2.55–2.32 (m, 3H), 2.25–2.18 (m, 1H), 2.09–1.98 (m, 2H), 1.90–1.82 (m, 1H), 1.80–1.70 (m, 1H), 0.93 (t,
J = 7.5 Hz, 3H).
13C NMR (101 MHz, CDCl
3) δ 173.1, 132.0, 130.6, 128.5, 128.4, 128.3, 127.9, 127.8, 127.7, 127.0, 124.2, 56.5, 55.9, 51.7, 31.0, 26.2, 25.8, 25.6, 25.5, 23.3, 20.5, 14.3.
3.1.3. (3Z,6Z,9Z,12Z,15Z)-1,1-Dimethoxyoctadeca-3,6,9,12,15-pentaene (8)
Epoxide
7 (6.023 g, 16.80 mmol, 1 eq.) was dissolved in MeOH (120 mL) and periodic acid (4.595 g, 20.16 mmol, 1.2 eq.) was added. The reaction mixture was stirred for 6 h. Water (100 mL) was added and the aqueous phase was extracted (3 × 100 mL) using hexane. The combined organic layers were washed with brine and dried (MgSO
4). The solvent was removed in vacuo. The crude product was purified by column chromatography on silica (hexane/EtOAc 95:5) to obtain dimetylacetal
8 as a colorless oil (2.086 g) in a 41% yield. All spectroscopic and physical data were in agreement with those reported in the literature [
35].
Rf = 0.46 (hexanes/EtOAc 9:1 KMnO
4 stain).
1H NMR (400 MHz, CDCl
3) δ 5.54–5.24 (m, 10H), 4.37 (t,
J = 5.8 Hz, 1H), 3.32 (s, 6H), 2.89–2.74 (m, 8H), 2.39 (ddd,
J = 7.2, 55.9, 1.5 Hz, 2H), 2.06 (dd,
J = 7.3, 1.3 Hz, 2H), 0.96 (t,
J = 7.6 Hz, 3H).
13C NMR (101 MHz, CDCl
3) δ 132.1, 130.3, 128.6, 128.3, 128.1, 128.0, 127.9, 127.1, 124.0, 104.1, 53.0, 31.1, 25.9, 25.7 (2C), 25.6, 20.6, 14.3.
3.1.4. (3Z,6Z,9Z,12Z,15Z)-Octadeca-3,6,9,12,15-pentaenal (4)
Acetal
8 (1.010 g, 3.32 mmol, 1 eq.) was dissolved in 1,4-dioxane (14 mL) and an 80% (
w/
w) aqueous solution of formic acid (16 mL) was added. The reaction mixture was stirred for 1.5 h at ambient temperature. The reaction was quenched by the addition of water (50 mL). The aqueous layer was extracted (3 × 30 mL) with hexane. The combined organic layers were washed with an aqueous NaHCO
3 solution (50 mL), then with brine (50 mL) and dried (MgSO
4). The solvent was removed in vacuo and aldehyde
4 (0.771 g) was obtained as a colorless oil in a 90% yield. All spectroscopic and physical data were in agreement with those reported in the literature [
35].
Rf = 0.40 (hexanes/EtOAc 9:1 KMnO
4 stain);
1H NMR (400 MHz, CDCl
3) δ 9.66 (t,
J = 2.0 Hz, 1H), 5.42–5.30 (m, 8H), 3.21 (dt,
J = 7.3, 1.9 Hz, 2H), 2.86–2.80 (m, 8H), 2.11–2.00 (m, 2H), 0.96 (t,
J = 7.6 Hz, 3H).
13C NMR (101 MHz, CDCl
3) δ 199.3, 133.2, 132.1, 128.9, 128.7, 128.5, 127.9 (2C), 127.2, 127.1, 118.8, 42.6, 26.1, 25.7 (2C), 25.6, 20.7, 14.4.
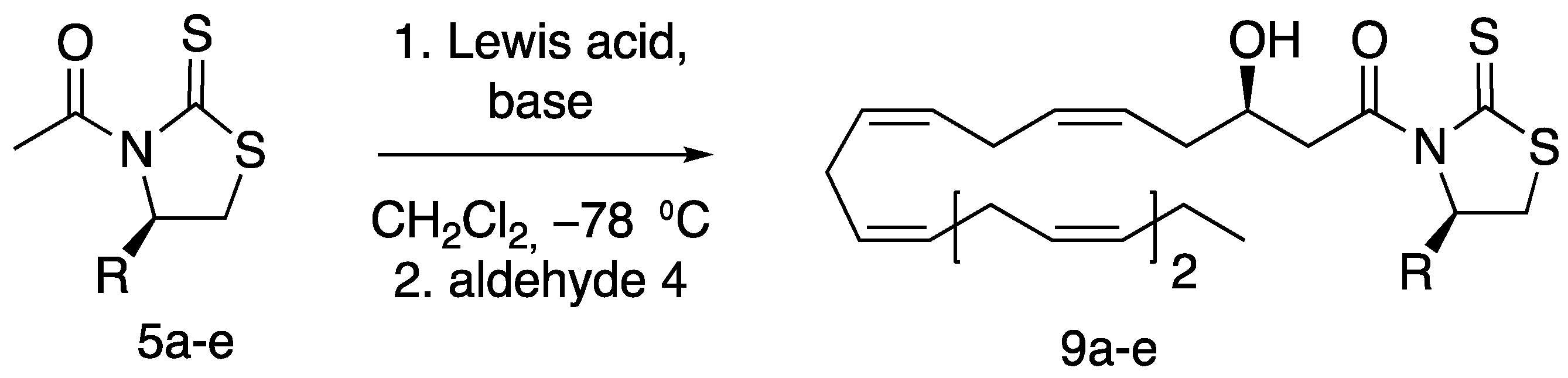
3.1.5. (R)-1-(4-Isopropyl-2-thioxothiazolidin-3-yl) Ethan-1-one (5b)
(
R)-valinol (1.010 g, 9.79 mmol, 1 eq.) was dissolved in EtOH (5 mL) and CS
2 (2.35 mL, 39.1 mmol, 4 eq.) was added. A 2.25 M solution of KOH (17.40 mL, 39.1 mmol, 4 eq.) in 1:1 EtOH/H
2O was added dropwise at room temperature over 20 min. The reaction mixture was stirred and heated at reflux for 72 h. After cooling, the mixture was acidified by slowly adding 0.5 M HCl (20 mL). The slightly acidic mixture was extracted with CH
2Cl
2 (3 × 20 mL) and the combined organic layers were concentrated in vacuo. The crude product was purified by column chromatography on silica (hexane/EtOAc 4:1) to afford
5b as a yellow oil (1.40 g) in a 88% yield. All spectroscopic and physical data were in agreement with those reported in the literature [
37].
Rf = 0.33 (hexanes/EtOAc 7:3 KMnO
4 stain);
1H NMR (400 MHz, CDCl
3) δ 8.19 (bs, 1H), 4.15–3.96 (m,1H), 3.49 (dd,
J = 11.1, 8.2 Hz, 1H), 3.30 (dd,
J = 11.1, 8.3 Hz, 1H), 2.04–1.89 (m, 1H), 1.02 (d,
J = 6.8 Hz, 3H), 0.99 (d,
J = 6.8 Hz, 3H).
13C NMR (101 MHz, CDCl
3) δ 201.1, 70.1, 36.0, 32.1, 18.9, 18.3. The obtained thioxothiazolidin (1.40 g, 9.3 mmol, 1. eq.) was dissolved in dry THF (18 mL) and cooled to 0 °C. A 60% dispersion of sodium hydride in mineral oil (0.409 g, 10.2 mmol, 1.1 eq.) was dissolved in dry THF (18 mL) and the thioxothiazolidin solution was added dropwise. The mixture was stirred for 10 min at 0 °C, followed by the addition of AcCl (0.73 mL, 10.2 mmol, 1.1 eq.) and the mixture was stirred for another 10 min at the same temperature. The heterogenous solution was allowed to reach ambient temperature and stirred for 1 h. The reaction was quenched by the addition of an aqueous 5% HCl solution (15 mL) and the aqueous layer was extracted with EtOAc (3 × 20 mL). The combined organic layers were dried (Na
2SO
4), filtrated and the solvent was removed in vacuo. The crude product was purified by column chromatography (hexane/EtOAc 9:1) to afford the chiral auxiliary
5b (1.70 g) as a bright yellow oil in a 90% yield. All spectroscopic and physical data were in agreement with those reported in the literature [
37].
Rf = 0.23 (hexanes/EtOAc 9:1 KMnO
4 stain);
1H NMR (400 MHz, CDCl
3) δ 5.14 (ddd,
J = 7.7, 6.1, 1.2 Hz, 1H), 3.50 (dd,
J = 11.5, 8.1 Hz, 1H), 3.01 (dd,
J = 11.5, 1.2 Hz, 1H), 2.76 (s, 3H), 2.41–2.31 (m, 1H), 1.05 (d,
J = 6.8 Hz, 3H), 0.96 (d,
J = 7.0 Hz, 3H).
13C NMR (101 MHz, CDCl
3) δ 203.3, 170.8, 71.4, 30.9, 30.5, 27.0, 19.2, 17.8. The auxiliaries
5a and
5c-e were produced by the same method described above for
5b.
3.1.6. (R)-1-(4-Methyl-2-thioxothiazolidin-3-yl) Ethan-1-one (5a)
Rf = 0.22 (hexanes/EtOAc 7:3 KMnO4 stain); 1H NMR (400 MHz, CDCl3) δ 5.34 (tt, J = 6.4, 0.9 Hz, 1H), 3.64 (dd, J = 11.2, 7.3 Hz, 1H), 2.79 (dd, J = 11.2, 0.9 Hz, 1H), 2.76 (s, 3H) 1.51 (d, J = 6.4 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 201.3, 170.7, 63.2, 35.4, 27.1, 18.1. Exact mass for C6H9NOS2 [M]Na+: 198.0018.
3.1.7. (R)-1-(4-Isobutyl-2-thioxothiazolidin-3-yl) Ethan-1-one (5c)
Rf = 0.46 (hexanes/EtOAc 7:3 KMnO4 stain); 1H NMR (400 MHz, CDCl3) δ 5.29 (dddd, J = 10.8, 7.3, 3.6, 0.8 Hz, 1H), 3.56 (ddd, J = 11.2, 7.2, 1.1 Hz, 1H), 3.07–2.86 (m, 1H), 2.76 (s, 3H), 1.91 (ddd, J = 13.1, 10.2, 4.2 Hz, 1H), 1.72–1.61 (m, 1H), 1.56 (dddd, J = 13.3, 9.7, 3.6, 1.1 Hz, 1H), 1.01 (dd, J = 6.4, 3.2 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 201.91, 170.54, 65.75, 39.61, 33.01, 27.06, 25.45, 23.56, 21.35. Exact mass for C9H15NOS2 [M]Na+: 240.0487.
3.1.8. (R)-1-(4-Benzyl-2-thioxothiazolidin-3-yl) Ethan-1-one (5d)
Rf = 0.44 (hexanes/EtOAc 3:1 KMnO4 stain); 1H NMR (400 MHz, CDCl3) δ 7.32 (ddd, J = 20.2, 7.3, 1.5 Hz, 5H), 5.46–5.25 (m, 1H), 3.40 (ddd, J = 11.6, 7.3, 1.1 Hz, 1H), 3.23 (dd, J = 13.2, 3.9 Hz, 1H), 3.05 (dd, J = 13.2, 10.5 Hz, 1H), 2.90 (dd, J = 11.5, 0.7 Hz, 1H), 2.81 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 201.6, 170.7, 136.5, 129.5 (2C), 128.9 (2C), 127.2, 68.2, 36.7, 31.8, 27.1. Exact mass for C12H13NOS2 [M]Na+: 274.0331.
3.1.9. (R)-1-(4-Phenyl-2-thioxothiazolidin-3-yl) Ethan-1-one (5e)
Rf = 0.20 (hexanes/EtOAc 7:3 KMnO4 stain); 1H NMR (400 MHz, CDCl3) δ 7.45–7.33 (m, 5H), 6.26 (dd, J = 8.2, 1.3 Hz, 1H), 3.95 (dd, J = 11.2, 8.2 Hz, 1H), 3.09 (dd, J = 11.2, 1.5 Hz, 1H), 2.82 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 202.6, 170.6, 139.1, 129.0 (2C), 128.5, 125.4 (2C), 69.4, 36.6, 27.2. Exact mass for C11H11NOS2 [M]Na+: 260.0174.
3.1.10. (R,5Z,8Z,11Z,14Z,17Z)-3-Hydroxy-1-((R)-4-isopropyl-2-thioxothiazolidin-3-yl) Icosa-5,8,11,14,17-pentaen-1-one (9b)
The aldol product
9b was prepared according to a literature procedure by Tungen and co-workers [
31]. (
R)-4-isopropyl-1,3-thiazolidine-2-thione (
5b) (1.213 g, 5.97 mmol mmol, 2 eq.) was dissolved in CH
2Cl
2 (60 mL) and 1 M TiCl
4 in CH
2Cl
2 (6.56 mL, 6.56 mmol mmol, 2.2 eq.) was added at −78 °C. After 5 min, DIPEA (1.25 mL, 7.16 mmol, 2.4 eq.) was added. The reaction mixture was stirred at −78 °C for 1 h. Aldehyde
4 (0.771 g, 2.98 mmol, 1 eq.) in CH
2Cl
2 (16 mL) was added dropwise over 20 min, and the reaction mixture was stirred for 4 h at −78 °C. The reaction was quenched by adding aqueous saturated NH
4Cl (60 mL) and the mixture was allowed to reach ambient temperature. The aqueous phase was extracted using CH
2Cl
2 (3 × 60 mL). The combined organic layers were dried (Na
2SO
4) and the solvent removed in vacuo. The crude product was purified by column chromatography on silica (hexane/EtOAc 9:1) yielding aldol product
9b (630 mg, 46%) as a yellow oil. In addition, its diastereomer (85 mg, 6%) was also obtained.
Rf = 0.25 (hexanes/EtOAc 7:3 KMnO
4 stain);
= −214.3° (CHCl
3, c 1.08);
1H NMR (400 MHz, CDCl
3) δ 5.53–5.24 (m, 10H), 5.21–5.10 (m, 1H), 4.18 (ddt,
J = 9.0, 6.5, 3.2 Hz, 1H), 3.64 (dd,
J = 17.7, 2.6 Hz, 1H), 3.51 Hz (dd,
J = 11.5 Hz, 7.9 Hz, 1H), 3.16 (dd,
J = 17.7, 9.4 Hz, 1H), 3.02 (dd,
J = 11.5 Hz, 1.0 Hz, 1H), 2.87–2.77 (m, 9H), 2.40–2.25 (m, 3H), 2.14–1.99 (m, 2H), 1.05 (d,
J = 6.7 Hz, 3H), 0.96 (t,
J = 7.5 Hz, 6H).
13C NMR (101 MHz, CDCl
3) δ 203.0, 173.0, 132.1, 131.2, 128.6, 128.5, 128.4, 128.1, 128.0 (2C), 127.1, 125.1, 71.4, 67.9, 45.0, 34.2, 30.9, 30.7, 25.9, 25.8, 25.7, 25.6, 20.7, 19.2, 17.9, 14.4. Exact mass for C
26H
39NO
2S
2 [M]Na
+: 484.2314.
3.1.11. 9b Diastereomer (Minor Product)
Rf = 0.37 (hexanes/EtOAc 7:3 KMnO4 stain); = −197.0° (CHCl3, c 0.46); 1H NMR (400 MHz, CDCl3) δ 5.66–5.25 (m, 10H), 5.17 (ddd, J = 7.8, 6.3, 1.2 Hz, 1H), 4.11–4.01 (m, 1H), 3.57–3.34 (m, 3H), 3.24 (bs, 1H), 3.03 (dd, J = 11.5, 1.2 Hz, 1H), 2.95–2.69 (m, 8H), 2.45–2.26 (m, 3H), 2.06 (tdd, J = 7.6, 7.0, 1.5 Hz, 2H), 1.05 (d, J = 6.8 Hz, 3H), 0.96 (t, J = 7.5 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 203.1, 173.6, 132.1, 131.1, 128.7, 128.5, 128.4, 128.1, 128.0 (2C), 127.1, 125.1, 71.4, 68.3, 44.7, 34.4, 30.9, 30.7, 25.9, 25.8, 25.7 (2C), 20.7, 19.2, 17.9, 14.4. Exact mass for C26H39NO2S2 [M]Na+: 484.2314.
3.1.12. (S)-1-((R,5Z,8Z,11Z,14Z,17Z)-3-((Tert-butyldimethylsilyl)oxy)icosa-5,8,11,14,17-pentaenoyl)-5-isopropylpyrrolidin-2-one (10)
The aldol product 9b (0.085 g, 0.18 mmol) was dissolved in CH2Cl2 (9.5 mL). The solution was cooled on a dry ice/ethanol bath. After 10 min, 2,6-lutidine (0.064 mL, 0.059 g, 0.55 mmol) was added. The reaction mixture was stirred for an additional 10 min, and TBSOTf (0.063 mL, 0.073 g, 0.28 mmol) was added dropwise. The reaction was stirred at this temperature for two hours before it was quenched with a saturated aqueous solution of NH4Cl (5 mL). The phases were separated. The water phase was extracted with CH2Cl2 (3 × 10 mL). The combined organic phases were dried (Na2SO4), filtered and concentrated in vacuo. The crude oil was purified by column chromatography on silica (EtOAc/hexane 5:95) to give compound 10 (0.077 g, 0.13 mmol, 72%) as a yellow oil. Rf: 0.70 (EtOAc:Heksan 3:7 KMnO4 stain). [α]: −126.5° (CHCl3, c 0.68) IR (film): 3014, 2964, 1700 cm−1. UV: λmax 262, 310 nm. 1H NMR (400 MHz, CDCl3): δ 5.53–5.27 (m, 11H), 5.03 (ddd, J = 7.6, 6.3, 1.0 Hz, 1H), 4.39–4.31 (m, 1H), 3.58–3.42 (m, 2H), 3.14 (dd, J = 17.1, 3.7 Hz, 1H), 3.02 (dd, J = 11.4, 1.0 Hz, 1H), 2.88–2.77 (m, 8H), 2.42–2.26 (m, 3H), 2.14–2.02 (m, 2H), 1.06 (d, J = 6.7 Hz, 3H), 0.99–0.94 (m, 6H), 0.89–0.84 (m, 8H), 0.09 (s, 3H), 0.04 (s, 3H). 13C NMR (100 MHz, CDCl3): δ 202.8, 172.0, 132.1, 130.6, 128.7, 128.4, 128.2, 128.0, 127.2, 125.4, 71.7, 69.2, 45.3, 35.7, 31.1, 30.9, 26.0, 25.8, 25.8, 25.7, 20.7, 19.3, 18.1, 18.0, 14.4, −4.3, −4.6.
3.1.13. Ethyl (R,5Z,8Z,11Z,14Z,17Z)-3-((Tert-butyldimethylsilyl)oxy)icosa-5,8,11,14,17-penta-enoate (11)
Silyl ether 10 (0.041 g, 0.072 mmol) was dissolved in absolute EtOH (1.45 mL) and cooled to 0 °C. K2CO3 (0.015 g, 0.11 mmol) was added to the solution, and the reaction mixture was left stirring for 3 h, then slowly allowed to reach room temperature, and stirred for an additional 23 h. The reaction was stopped by adding saturated aqueous NH4Cl (10 mL). The product was extracted with CH2Cl2 (3 × 10 mL). The combined organic phase was washed with 1 M KOH (5 mL), saturated aqueous NaCl (10 mL) and dried over Na2SO4. Filtration and concentration in vacuo gave a crude oil, which was purified by column chromatography on silica (EtOAc/hexane 1:39) to give ethyl ester 11 (0.026 g, 0.056 mmol, 78%) as a colorless oil. Rf: 0.53 (EtOAc:hexane 1:9). [α]: −22.6° (CHCl3, c 0.39). IR (film): 3014, 2931, 2858, 1739 cm−1. UV: λmax 246 nm. 1H NMR (400 MHz, CDCl3): δ 5.57–5.18 (m, 10H), 4.28–4.01 (m, 3H), 2.89–2.73 (m, 8H), 2.42 (dd, J = 6.3, 2.5 Hz, 2H), 2.35–2.23 (m, 2H), 2.16–2.00 (m, 2H), 1.25 (t, J = 7.2 Hz, 3H), 0.97 (t, J = 7.5 Hz, 3H), 0.86 (s, 9H), 0.07 (s, 3H), 0.04 (s, 3H). 13C NMR (100 MHz, CDCl3): δ 171.96, 132.18, 130.43, 128.72, 128.44, 128.42, 128.20, 128.16, 128.01, 127.16, 125.41, 69.47, 60.46, 42.47, 35.62, 25.95, 25.90, 25.80, 25.77, 25.69, 20.71, 18.13, 14.43, 14.35, −4.31, −4.79.
3.1.14. Synthesis of 3R-HEPE (2)
At 0 °C, TBAF in THF (1 M, 0.26 mL, 0.26 mmol) was added to a stirred solution of ethyl ester 11 (0.024 g, 0.052 mmol) in THF (1 mL). The reaction was stirred at this temperature for seven hours before it was quenched with phosphate buffer (pH = 7.29, 1 mL). Saturated aqueous NaCl (5 mL) and EtOAc (5 mL) was added, followed by the separation of the phases. The aqueous phase was extracted with EtOAc (2 × 5 mL). The combined organic phases were dried (Na2SO4), filtered and concentrated in vacuo. The crude oil was purified by column chromatography on silica (EtOAc/hexane 3:7) to give the ethyl ester of 3R-HEPE (2) (0.010 g, 0.030 mmol, 58%) as a colorless oil. Rf: 0.49 (EtOAc:Heksan 3:7) [α]: −19.4° (CHCl3, c 1.03). IR (film): 3507, 3014, 2964, 1734 cm−1. UV: λmax 245 nm. 1H NMR (400 MHz, CDCl3): δ 5.70–5.15 (m, 10H), 4.17 (q, J = 7.1 Hz, 2H), 4.12–4.01 (m, 1H), 2.94 (d, J = 2.8 Hz, 1H), 2.90–2.68 (m, 8H), 2.52 (dd, J = 16.4, 3.4 Hz, 1H), 2.42 (dd, J = 16.4, 8.9 Hz, 1H), 2.38–2.21 (m, 2H), 2.07 (pd, J = 7.5, 1.3 Hz, 2H), 1.27 (t, J = 7.2 Hz, 3H), 0.97 (t, J = 7.5 Hz, 3H). 13C NMR (100 MHz, CDCl3): δ 172.98, 132.19, 131.21, 128.74, 128.56, 128.47, 128.14, 127.99, 127.96, 127.15, 125.04, 68.00, 60.86, 40.78, 34.48, 25.93, 25.80, 25.78, 25.69, 20.70, 14.41, 14.32. The obtained ethyl ester (0.036 g, 0.10 mmol) was dissolved in THF/EtOH/H2O 2:2:1 (11 mL), followed by the addition of LiOH·H2O (0.153 g, 3.64 mmol) 0 °C. The reaction mixture was allowed to reach room temperature and stirred for 1.5 h. The solvent was evaporated in vacuo. EtOAc (10 mL) and NaH2PO4 (5 mL) were added. The phases were separated. The water phase was extracted with EtOAc (2 × 10 mL). The combined organic phases were dried over Na2SO4, filtered and concentrated in vacuo. This gave acid 3R-HEPE (2) (0.030 g, 0.095 mmol, 95%) as a colorless oil. Rf: 0.81 (MeOH:CH2Cl2 1:3). [α]: −10.5° (CHCl3, c 0.86). IR (film): 3014, 2964, 2931, 1712 cm−1. UV: λmax 253 nm (ε 561). 1H NMR (400 MHz, CDCl3): δ 5.62–5.51 (m, 1H), 5.50–5.26 (m, 9H), 4.16–4.02 (m, 1H), 2.93–2.73 (m, 8H), 2.59 (dd, J = 16.5, 3.4 Hz, 1H), 2.49 (dd, J = 16.6, 8.9 Hz, 1H), 2.42–2.24 (m, 2H), 2.07 (pd, J = 7.3, 1.3 Hz, 2H), 0.97 (t, J = 7.5 Hz, 3H). 13C NMR (100 MHz, CDCl3): δ 177.69, 132.19, 131.63, 128.74, 128.65, 128.50, 128.09, 127.97, 127.82, 127.14, 124.65, 67.89, 40.61, 34.46, 25.93, 25.79, 25.77, 25.68, 20.69, 14.40.
3.1.15. 3R-HEPE (2) from Aldol Product 9b
The aldol product 9b (100 mg, 0.216 mmol, 1 eq.) was dissolved in a solution of THF/MeOH/H2O (2:2:1) (30 mL) and the solution was cooled to 0 °C. LiOH·H2O (0.318 g, 7.56 mmol, 35 eq.) was added and the reaction mixture was stirred for 2 h, allowed to reach ambient temperature during this time. The solvent was removed in vacuo and EtOAc (30 mL) was added. The solution was acidified with aqueous saturated NaH2PO4 (20 mL) and the aqueous layer was extracted with EtOAc (2 × 30 mL). The combined organic layers were dried (Na2SO4) and the solvent was removed in vacuo. The crude product was purified by column chromatography on silica (CH2Cl2/MeOH/HCO2H 95:5:0.0125) and 2 (60.0 mg) was obtained as a light-yellow oil in 87% yield. Rf = 0.20 (DCM/MeOH 95:5 KMnO4 stain); = −10.5° (CHCl3, c 1.08). The spectral data were similar as those obtained above.
3.1.16. (R)-2-Hydroxy-1,2,2-triphenylethyl (S,5Z,8Z,11Z,14Z,17Z)-3-hydroxyicosa-5,8,11,14,17-pentaenoate (13)
The aldol product
13 was prepared according to a literature procedure by Braun and co-workers [
27]. Auxiliary
12 (81.5 mg, 0.245 mmol, 1 eq.) was added to dry THF (1.28 mL) under argon atmosphere. The slurry was cooled to −78 °C and the 2.5 M LDA solution (0.27 mL, 0.27 mmol, 1.1 eq.) was added dropwise over 10 min. The mixture was slowly warmed to −10 °C over 2 h and stirred further for 40 min at this temperature. The solution was cooled to −78 °C and
4 (85 mg, 0.33 mmol, 1.35 eq.) in THF (0.25 mL) was added dropwise over 30 min. The mixture was stirred for 2 h at −78 °C. The reaction was quenched by a dropwise addition of saturated ammonium chloride solution (1.5 mL). The solution was warmed to −5 °C and diluted with water (1.5 mL). The mixture was extracted with EtOAc (2 × 3 mL). The combined organic layers were dried (Na
2SO
4), filtered and concentrated in vacuo. The crude oil was purified by column chromatography on silica gel (EtOAc/Et
2O/Toluene/hexane 10:15:10:65), affording the aldol product
13 (81.3 mg) in a 70% yield.
Rf = 0.25 (hexane/EtOAc 8:2 KMnO
4 stain);
1H NMR (400 MHz, CDCl
3) δ 7.63–7.54 (m, 2H), 7.38 (dd,
J = 8.4, 6.8 Hz, 2H), 7.33–7.28 (m, 1H), 7.22–7.11 (m, 8H), 7.10–7.01 (m, 2H), 6.74 (s, 1H), 5.66–5.45 (m, 1H), 5.36 (m, 9H), 3.96–3.85 (m, 1H), 2.75–2.85 (m, 8H), 2.54–2.31 (m, 2H), 2.25–2.13 (m, 2H), 2.13–2.04 (m, 2H), 0.98 (t,
J = 7.5 Hz, 3H).
13C NMR (101 MHz, CDCl
3) δ 171.3, 144.5, 142.5, 135.4, 132.1, 131.1, 128.6, 128.4 (3C), 128.1, 128.0, 127.9 (2C), 127.8, 127.6, 127.5, 127.2, 127.0, 126.2 (2C), 124.7, 80.3, 79.0, 67.8, 41.1, 34.2, 25.8, 25.7, 25.6, 20.6, 14.3. Exact mass for C
40H
46O
4 [M]Na
+: 613.3288.
3.1.17. Methyl (S,5Z,8Z,11Z,14Z,17Z)-3-Hydroxyicosa-5,8,11,14,17-pentaenoate (S-14)
To a solution of 13 (16.3 mg, 0.027 mmol, 1 eq.) in MeOH (0.5 mL) was added K2HPO4 (0.6 mg, 0.0027 mmol, 0.1 eq.). The mixture was heated at reflux for 20 h and then concentrated in vacuo. The crude product was purified by column chromatography on silica gel (EtOAc/hexanes 0:100 → 20:80), affording 14 (7.3 mg) in a 80% yield. Rf = 0.28 (hexanes/EtOAc 8:2 KMnO4 stain); = +9.3° (CHCl3, c 1.08); 1H NMR (400 MHz, CDCl3) δ 5.60–5.50 (m, 1H), 5.49–5.29 (m, 9H), 4.20–3.94 (m, 1H), 3.72 (s, 3H), 2.84 (m, 8H), 2.55 (dd, J = 16.4, 3.4 Hz, 1H), 2.50–2.42 (m, 1H), 2.40–2.22 (m, 2H), 2.20–1.90 (m, 2H), 0.99 (t, J = 7.5 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 173.0, 132.2, 131.2, 128.7, 128.6, 128.5, 128.1, 128.0 (2C), 127.2, 125.0, 68.0, 60.9, 40.8, 34.5, 25.9, 25.8 (2C), 25.7, 20.7, 14.4. Exact mass for C21H32NaO3 [M]Na+: 355.2244.
3.1.18. 3S-HEPE (2)
Methyl ester S-14 (11.1 mg, 0.033 mmol, 1 eq.) was dissolved in a solution of THF/MeOH/H2O (2:2:1) (5 mL) and the solution was cooled to 0 °C. LiOH·H2O (49.1 mg, 1.17 mmol, 35 eq.) was added and the reaction mixture was stirred for 2 h, allowed to reach ambient temperature during this time. The solvent was removed in vacuo and EtOAc (5 mL) was added. The solution was acidified with aqueous saturated NaH2PO4 (5 mL) and the aqueous layer was extracted with EtOAc (2 × 5 mL). The combined organic layers were dried (Na2SO4) and the solvent was removed in vacuo. The crude product was purified by column chromatography on silica (CH2Cl2/MeOH/HCO2H 95:5:0.0125) and (S)-2 (10.0 mg) was obtained as a light-yellow oil in a 94% yield. Rf = 0.20 (DCM/MeOH 95:5 KMnO4 stain); = +11.1 (CHCl3, c 0.90); 1H NMR (400 MHz, CDCl3) δ 5.62–5.51 (m, 1H), 5.50–5.26 (m, 9H), 4.16–4.02 (m, 1H), 2.93–2.73 (m, 8H), 2.59 (dd, J = 16.5, 3.4 Hz, 1H), 2.49 (dd, J = 16.6, 8.9 Hz, 1H), 2.42–2.24 (m, 2H), 2.07 (dd, J = 7.3 Hz, 1.3 Hz, 2H), 0.97 (t, J = 7.5 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 177.69, 132.19, 131.63, 128.74, 128.65, 128.50, 128.09, 127.97, 127.82, 127.14, 124.65, 67.89, 40.61, 34.46, 25.93, 25.79, 25.77, 25.68, 20.69, 14.40. Exact mass for C20H30NaO3 [M]Na+: 341.2087
3.1.19. Methyl (S,5Z,8Z,11Z,14Z,17Z)-3-Hydroxyicosa-5,8,11,14,17-pentaenoate (R-14)
For the comparison of both enantiomers in Mosher analysis, (
R)-14 was prepared as follows [
38]. Trimethylsilyl diazomethane (2.0 M in hexanes, 1 mL, 2 mmol) was added dropwise to a solution of the 3R-HEPE (2) in methanol (3 mL) at 0 °C until a yellow color persisted in the reaction. The reaction was quenched by the addition of acetic acid (2 drops) and concentrated. The crude residue was submitted to the next reaction without further purification.
1H NMR (300 MHz, CDCl
3) δ (ppm): 5.88–5.75 (m, 1H), 5.18–5.11 (m, 2H), 3.97 (major, ddd,
J = 6.9, 5.7, 3.9 Hz, 0.8H), 3.81–3.73 (minor, 0.2H), 3.72 (s, 3H), 2.58 (dddd,
J = 6.9, 6.9, 6.9, 4.2 Hz, 1H), 2.32–3.19 (m, 2H), 1.22 (d,
J = 6.9 Hz, 3H).


 ,
, 






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}