
Adsorption Characteristics of Gas Molecules Adsorbed on Graphene Doped with Mn: A First Principle Study

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Computational Details and Methods
3. Results and Discussion
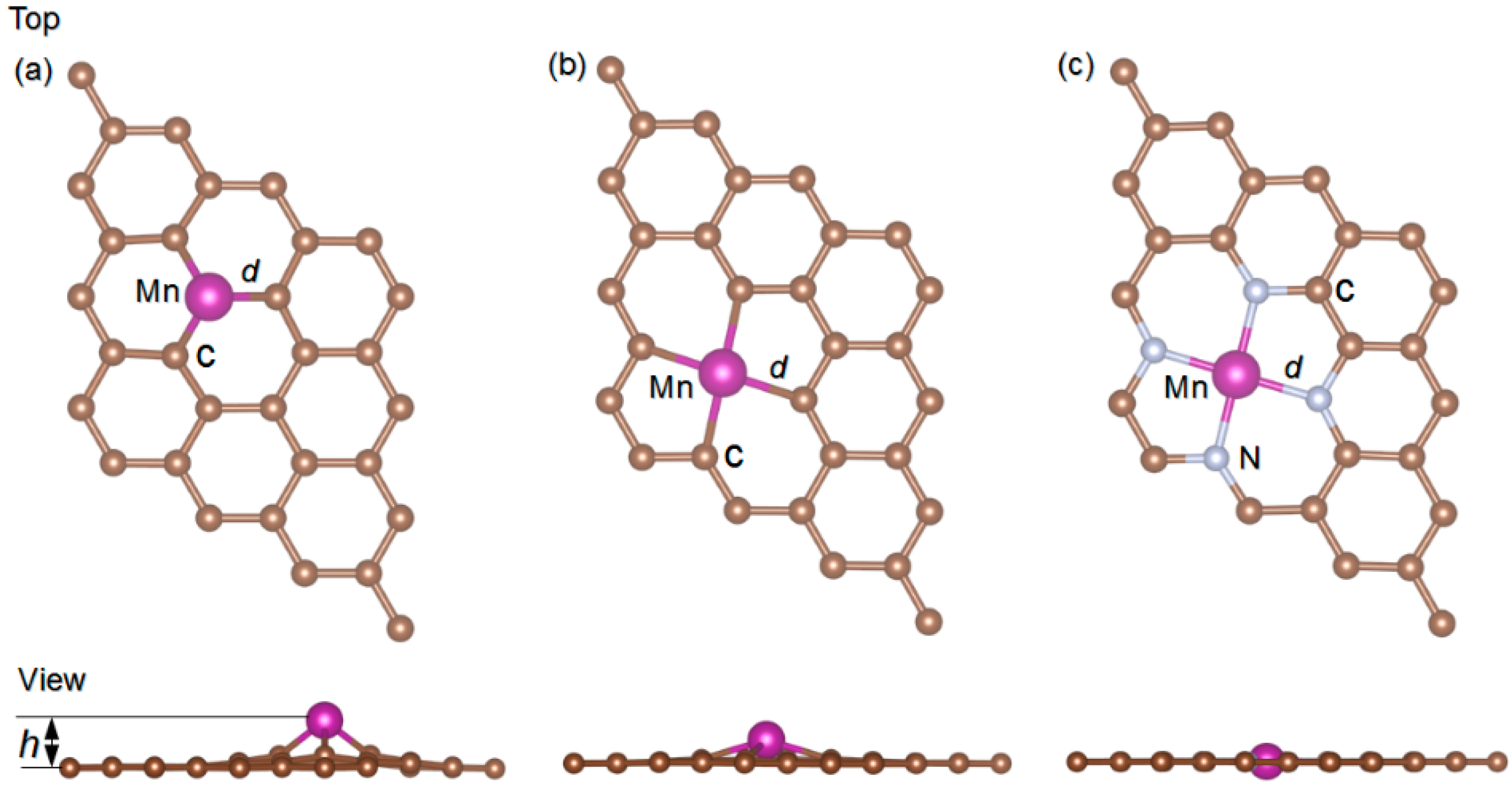
3.1. Model of MnX-GN
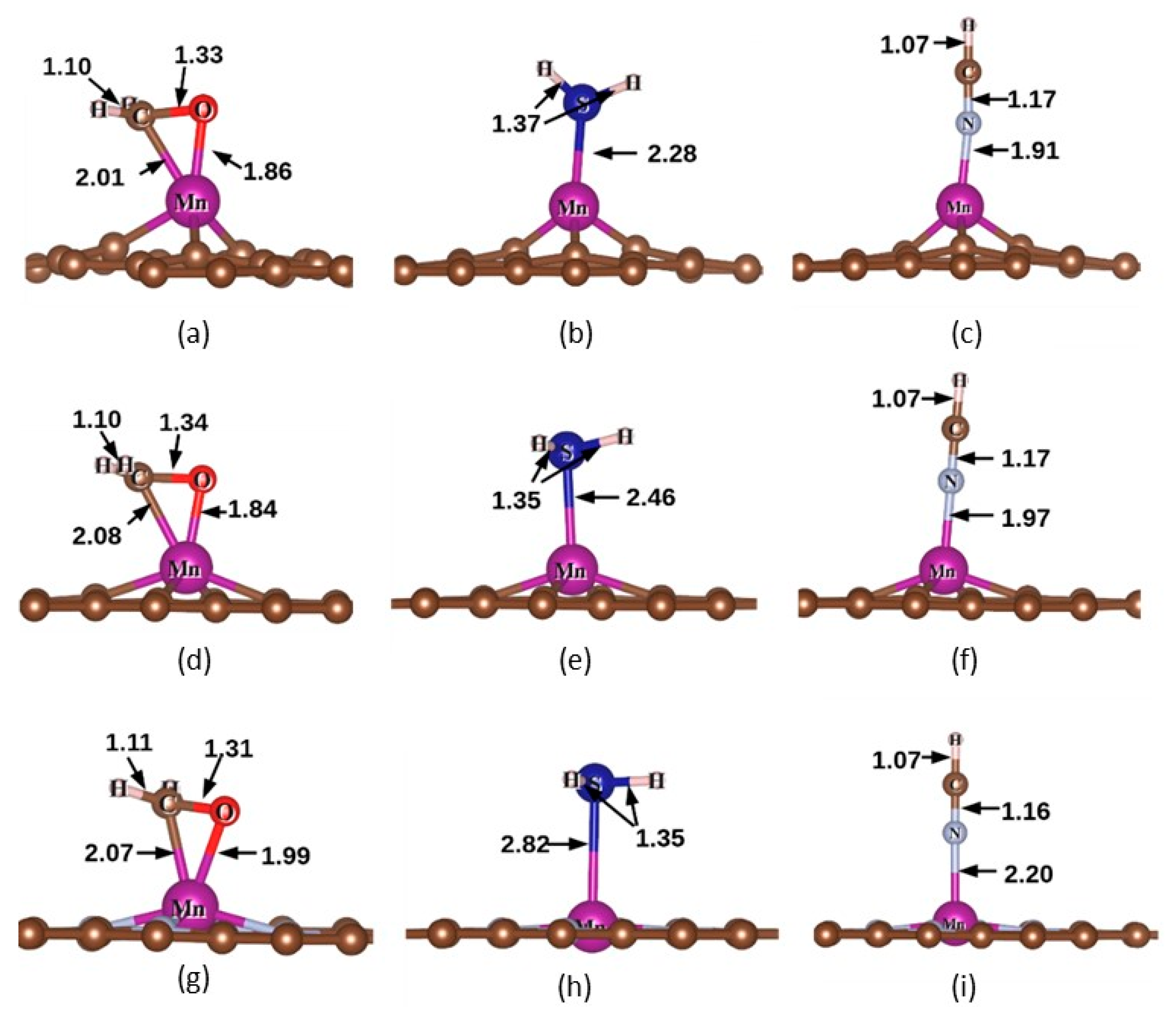
3.2. Adsorption Structure, Adsorption Energy
3.3. Electronic Structure and Magnetic Properties
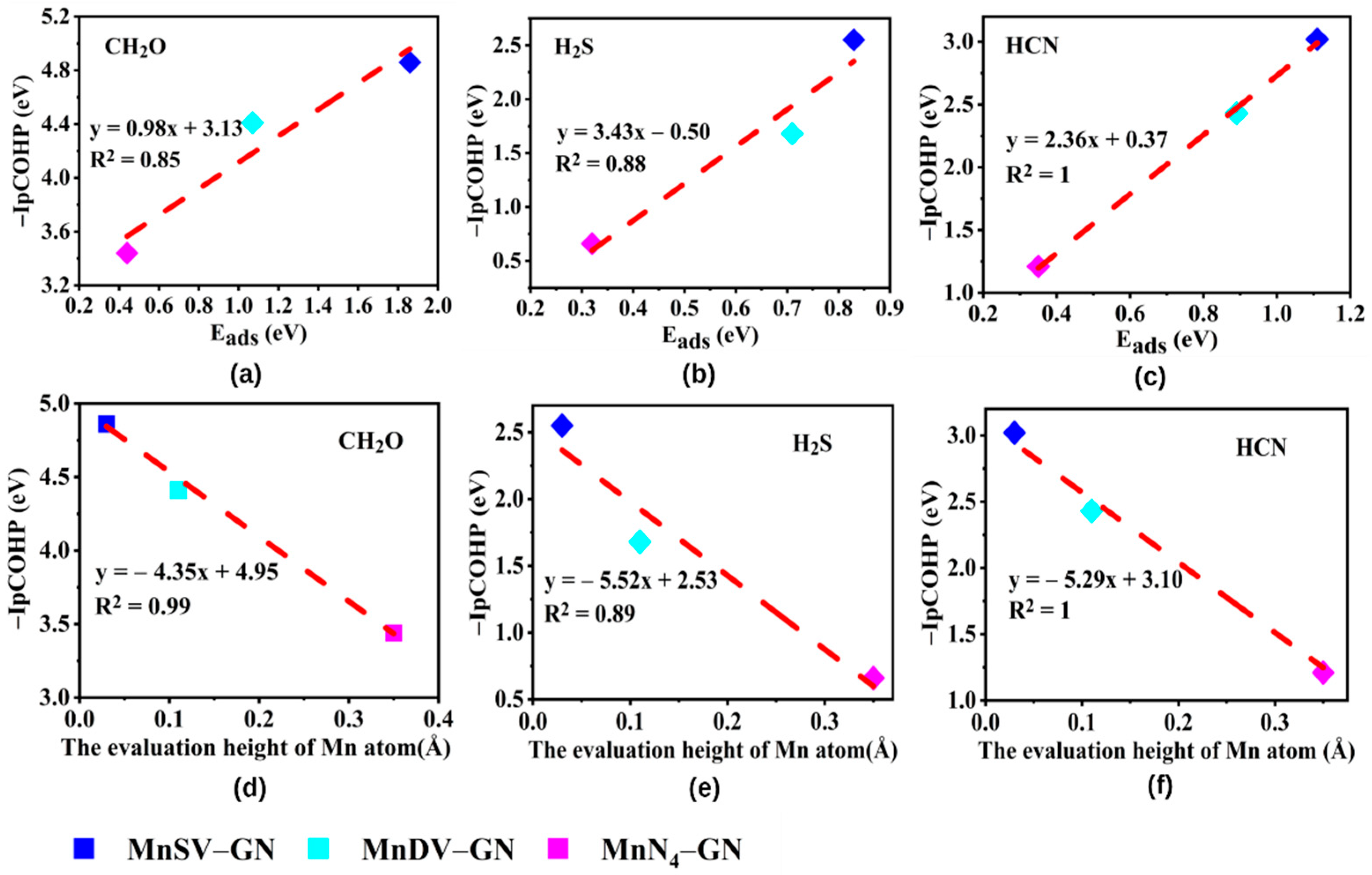
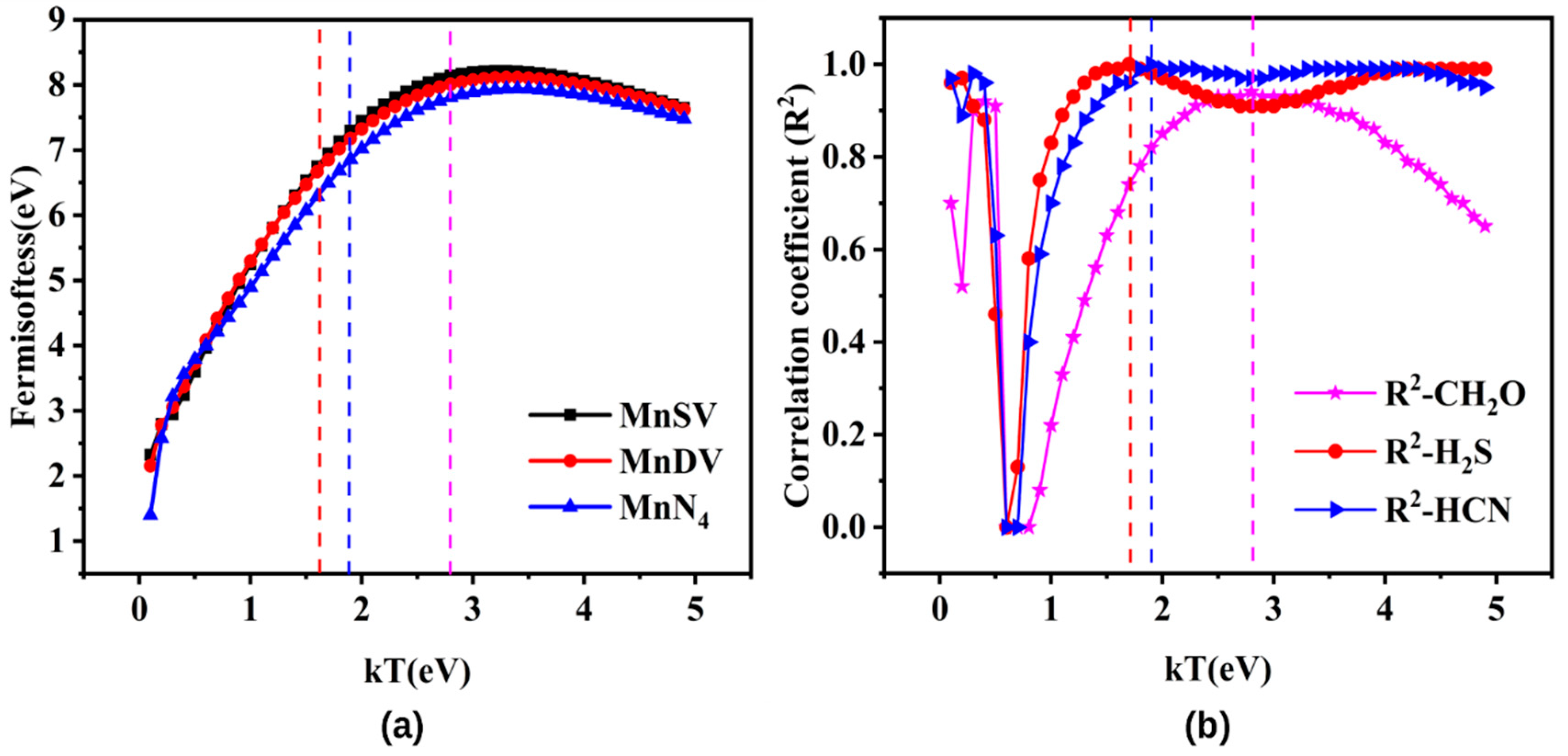
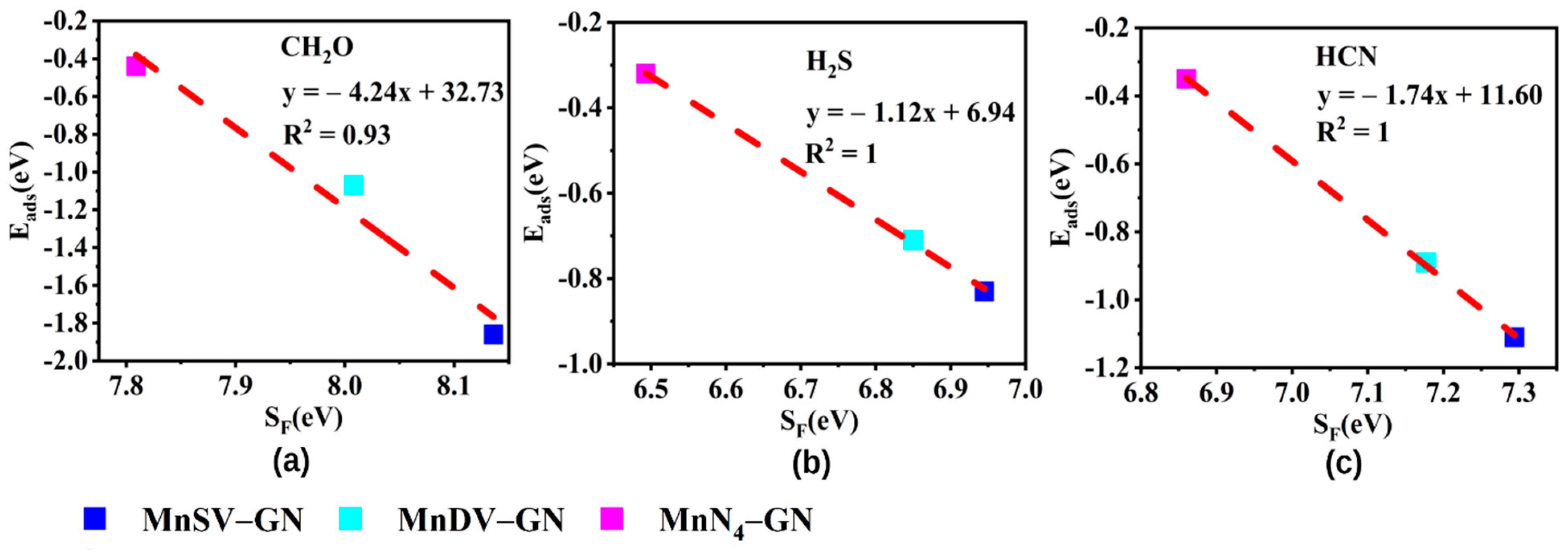
3.4. Electronic Structure–Reactivity Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Faye, O.; Raj, A.; Mittal, V.; Beye, A.C. H2S Adsorption on Graphene in the Presence of Sulfur: A Density Functional Theory Study. Comp. Mater. Sci. 2016, 117, 110–119. [Google Scholar] [CrossRef]
- Ashori, E.; Nazari, F.; Illas, F. Adsorption of H2S on Carbonaceous Materials of Different Dimensionality. Int. J. Hydrog. Energy 2014, 39, 6610–6619. [Google Scholar] [CrossRef]
- Salthammer, T. Formaldehyde in the Ambient Atmosphere: From an Indoor Pollutant to an Outdoor Pollutant. Angew. Chem. Int. Ed. 2013, 52, 3320–3327. [Google Scholar] [CrossRef] [PubMed]
- Noorizadeh, S.; Shakerzadeh, E. Formaldehyde Adsorption on Pristine, Al-doped and Mono-vacancy Defected Boron Nitride Nanosheets: A First Principles Study. Comp. Mater. Sci. 2012, 56, 122–130. [Google Scholar] [CrossRef]
- Rastegar, S.F.; Peyghan, A.A.; Hadipour, N.L. Response of Si- and Al-Doped Graphenes toward HCN: A Computational Study. Appl. Surf. Sci. 2013, 265, 412–417. [Google Scholar] [CrossRef]
- Peyghan, A.A.; Hadipour, N.L.; Bagheri, Z. Effects of Al Doping and Double-Antisite Defect on the Adsorption of HCN on a BC2N Nanotube: Density Functional Theory Studies. J. Phys. Chem. C 2013, 117, 2427–2432. [Google Scholar] [CrossRef]
- Wang, L.Z.; Mullen, K. Transparent, Conductive Graphene Electrodes for Dye-sensitized Solar Cells. Nano Lett. 2008, 8, 323–327. [Google Scholar] [CrossRef]
- Tombros, C.Z.N.; Popinciuc, M.; Jonkman, H.T.; Wees, B.J.V. Electronic Spin Transport and Spin Precession in Single Graphene Layers at Room Temperature. Nature 2007, 448, 571–574. [Google Scholar] [CrossRef] [Green Version]
- Schedin, F.; Geim, A.K.; Morozov, S.V.; Hill, E.W.; Blake, P.; Katsnelson, M.I.; Novoselov, K.S. Detection of Individual Gas Molecules Adsorbed on Graphene. Nat. Mater. 2007, 6, 652–655. [Google Scholar] [CrossRef]
- Stankovich, S.; Dikin, D.A.; Dommett, G.H.; Kohlhaas, K.M.; Zimney, E.J.; Stach, E.A.; Piner, R.D.; Nguyen, S.T.; Ruoff, R.S. Graphene-Based Composite Materials. Nature 2006, 442, 282–286. [Google Scholar] [CrossRef]
- Chhowalla, M.; Jena, D.; Zhang, H. Two-Dimensional Semiconductors for Transistors. Nat. Rev. Mater. 2016, 1, 16052. [Google Scholar] [CrossRef]
- Novoselov, K.S.; Geim, A.K.; Morozov, S.V.; Jiang, D.; Zhang, Y.; Dubonos, S.V.; Grigorieva, I.V.; Firsov, A.A. Electric Field Effect in Atomically Thin Carbon Films. Science 2004, 306, 666–669. [Google Scholar] [CrossRef] [Green Version]
- He, C.Z.; Wang, R.; Yang, H.Y.; Li, S.; Fu, L. The Growth Pattern of Ptn (n = 1–6) Clusters on Pentagonal B2C Monolayer Support: A Computational Study. Appl. Surf. Sci. 2020, 507, 145076. [Google Scholar] [CrossRef]
- He, C.Z.; Wang, R.; Xiang, D.; Li, X.Y.; Fu, L.; Jian, Z.Y.; Huo, J.R.; Li, S. Charge-regulated CO2 Capture Capacity of Metal Atom Embedded Graphyne: A First-Principles Study. Appl. Surf. Sci. 2020, 509, 145392. [Google Scholar] [CrossRef]
- Wang, J.; He, C.Z.; Huo, J.R.; Fu, L.; Zhao, C.X. A Theoretical Evaluation of Possible N2 Reduction Mechanism on Mo2B2. Adv. Theory Simul. 2021, 4, 2100003. [Google Scholar] [CrossRef]
- Zhou, D.W.; Li, C.P.; Yin, F.R.; Tang, X.; Pu, C.Y.; He, C.Z. Two-dimensional 1T-PS2 as a Promising Anode Material for Sodium-ion Batteries with Ultra-high Capacity, Low Average Voltage and Appropriate Mobility. Chin. Chem. Lett. 2020, 31, 2325–2329. [Google Scholar] [CrossRef]
- Pu, C.Y.; Yu, J.H.; Fu, L.; Wang, J.; Yang, H.Y.; Zhou, D.W.; He, C.Z. Two-dimensional MgSiP2 with anisotropic electronic properties and good performances for Na-ion batteries. Chin. Chem. Lett. 2021, 32, 1081–1085. [Google Scholar] [CrossRef]
- Fu, X.; Yang, H.Y.; Fu, L.; He, C.Z.; Huo, J.R.; Guo, J.Y.; Li, L.M. Prediction of Semiconducting SiP2 Monolayer with Negative Possion’s Ratio, Ultrahigh Carrier Mobility and CO2 Capture Ability. Chin. Chem. Lett. 2021, 32, 1089–1094. [Google Scholar] [CrossRef]
- Fu, L.; Yan, L.B.; Lin, L.; Xie, K.; Zhu, L.H.; He, C.Z.; Zhang, Z.Y. Fe-embedded Au (111) Monolayer as An Electrocatalyst for N2 Reduction Reaction: A First-principles Investigation. J. Alloys Compd. 2021, 875, 159907. [Google Scholar] [CrossRef]
- Wang, R.; He, C.Z.; Chen, W.X.; Zhao, C.X.; Huo, J.R. Rich B Active Centers in Penta-B2C as High-performance Photocatalyst for Nitrogen Reduction. Chin. Chem. Lett. 2021, 32, 3821–3824. [Google Scholar] [CrossRef]
- Yang, H.Y.; He, C.Z.; Fu, L.; Huo, J.R.; Zhao, C.X.; Li, X.Y.; Song, Y. Capture and Separation of CO2 on BC3 Nanosheets: A DFT Study. Chin. Chem. Lett. 2021, 32, 3202–3206. [Google Scholar] [CrossRef]
- Sun, R.S.; He, C.Z.; Fu, L.; Huo, J.R.; Zhao, C.X.; Li, X.Y.; Song, Y.; Wang, S.M. Defect Engineering for high-selection-performance of NO reduction to NH3 over CeO2(111) Surface: A DFT Study. Chin. Chem. Lett. 2022, 33, 527–532. [Google Scholar] [CrossRef]
- Fu, L.; Wang, R.; Zhao, C.X.; Huo, J.R.; He, C.Z.; Kim, K.H.; Zhang, W. Construction of Cr-embedded Graphyne Electrocatalyst for Highly Selective Reduction of CO2 to CH4: A DFT Study. Chem. Engin. J. 2021, 414, 128857. [Google Scholar] [CrossRef]
- Ma, D.W.; Wang, Q.G.; Yang, G.; He, C.Z.; Ma, B.Y.; Lu, Z.S. Graphyne as a Promising Substrate for the Noble-Metal Single-atom Catalysts. Carbon 2015, 95, 756–765. [Google Scholar] [CrossRef]
- Deng, D.H.; Bao, X.H. A Graphene Composite Material with Single Cobalt Active Sites: A Highly Efficient Counter Electrode for Dye-Sensitized Solar Cells. Angew. Chem. Int. Ed. 2016, 55, 6708–6712. [Google Scholar]
- Lu, Y.H.; Zhang, C.; Feng, Y.P. Metal-embedded Graphene: A Possible Catalyst with High Activity. J. Phys. Chem. C 2009, 113, 20156–20160. [Google Scholar] [CrossRef]
- Gao, Z.; Li, A.; Liu, X.; Ma, C.; Li, X.; Yang, W.; Ding, X. Density Functional Study of the Adsorption of NO on Ni (n = 1, 2, 3 and 4) Clusters Doped Functionalized Graphene Support. Appl. Surf. Sci. 2019, 481, 940–950. [Google Scholar] [CrossRef]
- Gao, Z.; Xu, S.; Li, L.; Yan, G.; Yang, W.; Wu, C.; Gates, I.D. On the Adsorption of Elemental Mercury on Single-atom TM (TM = V, Cr, Mn, Co) Decorated Graphene Substrates. Appl. Surf. Sci. 2020, 516, 146037. [Google Scholar] [CrossRef]
- Shi, L.B.; Wang, Y.P.; Dong, H.K. First-Principle Study of Structural, Electronic, Vibrational and Magnetic Properties of HCN Adsorbed Graphene Doped With Cr, Mn and Fe. Appl. Surf. Sci. 2015, 329, 330–336. [Google Scholar] [CrossRef]
- Sharma, S.; Verma, A.S.A. Theoretical Study of H2S Adsorption on Graphene Doped with B, Al and Ga. Phys. B 2013, 427, 12–16. [Google Scholar] [CrossRef]
- Gao, Z.; Yang, W.; Ding, X.; Lv, G.; Yan, W. Support Effects in Single Atom Iron Catalysts on Adsorption Characteristics of Toxic Gases (NO2, NH3, SO3 and H2S). Appl. Surf. Sci. 2018, 436, 585–595. [Google Scholar] [CrossRef]
- Zhang, Y.H.; Han, L.F.; Xiao, Y.H.; Jia, D.Z.; Guo, Z.H.; Li, F. Understanding Dopant and Defect Effect on H2S Sensing Performances of Graphene: A First-Principles Study. Comp. Mater. Sci. 2013, 69, 222–228. [Google Scholar] [CrossRef]
- Zhang, H.P.; Luo, X.G.; Song, H.T.; Lin, X.Y.; Lu, X.; Tang, Y. DFT Study of Adsorption and Dissociation Behavior of H2S on Fe-doped Graphene. Appl. Surf. Sci. 2014, 317, 511–516. [Google Scholar] [CrossRef]
- Impeng, S.; Junkaew, A.; Maitarad, P.; Kungwan, N.; Zhang, D.; Shi, L.; Namuangruk, S. A MnN4 Moiety Embedded Graphene as a Magnetic Gas Sensor for CO Detection: A First Principle Study. Appl. Surf. Sci. 2019, 473, 820–827. [Google Scholar] [CrossRef]
- Chi, M.; Zhao, Y.P. Adsorption of Formaldehyde Molecule on the Intrinsic and Al-Doped Graphene: A First Principle Study. Comp. Mater. Sci. 2009, 46, 1085–1090. [Google Scholar] [CrossRef]
- Robertson, A.W.; Montanari, B.; He, K.; Kim, J.; Allen, C.S.; Wu, Y.A.; Olivier, J.; Neethling, J.; Harrison, N.; Kirkland, A.I.; et al. Dynamics of Single Fe Atoms in Graphene Vacancies. Nano Lett. 2013, 13, 1468–1475. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.C.; Teng, P.Y.; Yeh, C.H.; Koshino, M.; Chiu, P.W.; Suenaga, K. Structural and Chemical Dynamics of Pyridinic-Nitrogen Defects in Graphene. Nano Lett. 2015, 15, 7408–7413. [Google Scholar] [CrossRef]
- Fujimoto, Y.; Saito, S. Formation, Stabilities, and Electronic Properties of Nitrogen Defects in Graphene. Phys. Rev. B 2011, 84, 245446. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.Y.; Liu, K.K.; Chen, W.G.; Tang, Y.N. Structural, Electronic and Catalytic Performances of Single-Atom Fe Stabilized by Divacancy-Nitrogen-Doped Graphene. RSC Adv. 2017, 7, 7920–7928. [Google Scholar] [CrossRef] [Green Version]
- Lu, Z.S.; He, C.Z.; Wang, T.X.; Yang, L.; Yang, Z.X.; Ma, D.W. Novel Catalytic Activity for Oxygen Reduction Reaction on MnN4 Embedded Graphene: A Dispersion-Corrected Density Functional Theory Study. Carbon 2015, 84, 500–508. [Google Scholar] [CrossRef]
- Banhart, J.K.F.; Krasheninnikov, A.V. Structural Defects in Graphene. ACS Nano 2011, 5, 26–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dewapriya, M.A.N. Effects of Free Edges and Vacancy Defects on the Mechanical Properties of Graphene. Nanotechnology 2014, 12, 908–912. [Google Scholar]
- Kresse, G.; Furthmuller, J.J.B. Efficient Iterative Schemes for Ab Initio Total-Energy Calculations Using a Plane-Wave Basis Set. Phys. Rev. B 1996, 54, 11169. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kresse, G.; Joubert, D. From Ultrasoft Pseudopotentials to the Projector Augmented-Wave Method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Xie, T.; Wang, P.; Tian, C.; Zhao, G.; Jia, J.; He, C.; Zhao, C.; Wu, H.; Michel, C. The Investigation of Adsorption Behavior of Gas Molecules on FeN3-Doped Graphene. J. Sens. 2022, 2022, 9306741. [Google Scholar] [CrossRef]
- Xie, T.; Wang, P.; Tian, C.; Zhao, G.; Jia, J.; Zhao, C.; Wu, H. The Adsorption Behavior of Gas Molecules on Co/N Co-Doped Graphene. Molecules 2021, 26, 7700. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [Green Version]
- Santos, E.J.G.; Ayuela, A.; Sanchez-Portal, D. First-principles Study of Substitutional Metal Impurities in Graphene: Structural, Electronic and Magnetic Properties. New J. Phys. 2010, 12, 053012. [Google Scholar] [CrossRef] [Green Version]
- Kattel, S.; Atanassov, P.; Kiefer, B. Stability, Electronic and Magnetic Properties of In-plane Defects in Graphene: A First-Principles Study. J. Phys. Chem. C 2012, 116, 8161–8166. [Google Scholar] [CrossRef]
- Sun, S.; Zhang, G.; Gauquelin, N.; Chen, N. Single-Atom Catalysis Using Pt/Graphene Achieved through Atomic Layer Deposition. Sci. Rep. 2013, 3, 1775. [Google Scholar] [CrossRef] [Green Version]
- Momma, K.; Izumi, F. Three-Dimensional Visualization of Crystal, Volumetric and Morphology Data. J. Appl. Crystallogr. 2011, 44, 1272–1276. [Google Scholar] [CrossRef]
- Dronskowski, R. Crystal Orbital Hamilton Populations (COHP), Energy-Resolved Visualization of Chemical Bonding in Solids Based on Density-Functional Calculations. J. Phys. Chem. 1993, 97, 8617–8624. [Google Scholar] [CrossRef]
- Deringer, V.L.; Tchougreeff, A.L.; Dronskowski, R. Crystal Orbital Hamilton Population (COHP) Analysis as Projected from Plane-Wave Basis Sets. J. Phys. Chem. A 2011, 115, 5461–5466. [Google Scholar] [CrossRef] [PubMed]
- Maintz, S.; Deringer, V.L.; Tchougreeff, A.L.; Dronskowski, R. LOBSTER: A Tool to Extract Chemical Bonding from Plane-Wave Based DFT. J. Comput. Chem. 2016, 37, 1030–1035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henkelman, G.; Arnaldsson, A.; Jonsson, H. A Fast and Robust Algorithm for Bader Decomposition of Charge Density. Comput. Mater. Sci. 2006, 36, 354–360. [Google Scholar] [CrossRef]
- Krasheninnikov, A.V.; Lehtinen, P.O.; Foster, A.S.; Pyykko, P.; Nieminen, R.M. Embedding Transition-Metal Atoms in Graphene: Structure, Bonding, and Magnetism. Phys. Rev. Lett. 2009, 102, 126807. [Google Scholar] [CrossRef] [Green Version]
- Woolley, J.C. Introduction to Solid State Physics: C. Kittel: 2nd Edition. Chapman and Hall, 1956. 617 pp., 96s. J. Mech. Phys. Solids 1957, 6, 83. [Google Scholar] [CrossRef] [Green Version]
- Feng, C.; Yang, D.G.; Zhang, G.Q. First-Principles Investigation of the Adsorption Behaviors of CH2O on BN, AlN, GaN, InN, BP and P Monolayers. Materials 2019, 12, 676. [Google Scholar] [CrossRef] [Green Version]
- Gao, X.; Wang, J.X.; Xu, L.N.; Zeng, W. Performance of Intrinsic and Modified Graphene for the Adsorption of H2S and CH4: A DFT Study. Nanomaterials 2020, 10, 299. [Google Scholar] [CrossRef] [Green Version]
- Tang, Y.; Chen, W.; Li, C.; Pan, L.; Dai, X.; Ma, D. Adsorption Behavior of Co Anchored on Graphene Sheets toward NO, SO2, NH3, CO and HCN Molecules. Appl. Surf. Sci. 2015, 342, 191–199. [Google Scholar] [CrossRef]
- Fukui, K. Role of Frontier Orbitals in Chemical Reactions. Science 1982, 218, 747–754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, B.; Xiao, L.; Lu, J.; Zhuang, L. Spatially Resolved Quantification of the Surface Reactivity of Solid Catalysts. Angew. Chem. Int. Ed. 2016, 55, 6239–6243. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Gao, Z.; Ding, X.; Lv, G.; Yan, W. The Adsorption Characteristics of Mercury Species on Single Atom Iron Catalysts with Different Graphene-Based Substrates. Appl. Surf. Sci. 2018, 455, 940–951. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xie, T.; Wang, P.; Tian, C.; Zhao, G.; Jia, J.; He, C.; Zhao, C.; Wu, H. Adsorption Characteristics of Gas Molecules Adsorbed on Graphene Doped with Mn: A First Principle Study. Molecules 2022, 27, 2315. https://doi.org/10.3390/molecules27072315
Xie T, Wang P, Tian C, Zhao G, Jia J, He C, Zhao C, Wu H. Adsorption Characteristics of Gas Molecules Adsorbed on Graphene Doped with Mn: A First Principle Study. Molecules. 2022; 27(7):2315. https://doi.org/10.3390/molecules27072315
Chicago/Turabian StyleXie, Tingyue, Ping Wang, Cuifeng Tian, Guozheng Zhao, Jianfeng Jia, Chaozheng He, Chenxu Zhao, and Haishun Wu. 2022. "Adsorption Characteristics of Gas Molecules Adsorbed on Graphene Doped with Mn: A First Principle Study" Molecules 27, no. 7: 2315. https://doi.org/10.3390/molecules27072315