
Introducing a Chemically Intuitive Core-Substituent Fingerprint Designed to Explore Structural Requirements for Effective Similarity Searching and Machine Learning



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
2.1. Fingerprint Design Principles
2.2. Molecular Fragments
2.2.1. Fragment Categories
2.2.2. Core Structure Fragmentation
2.2.3. Ring and Substituent Fragments
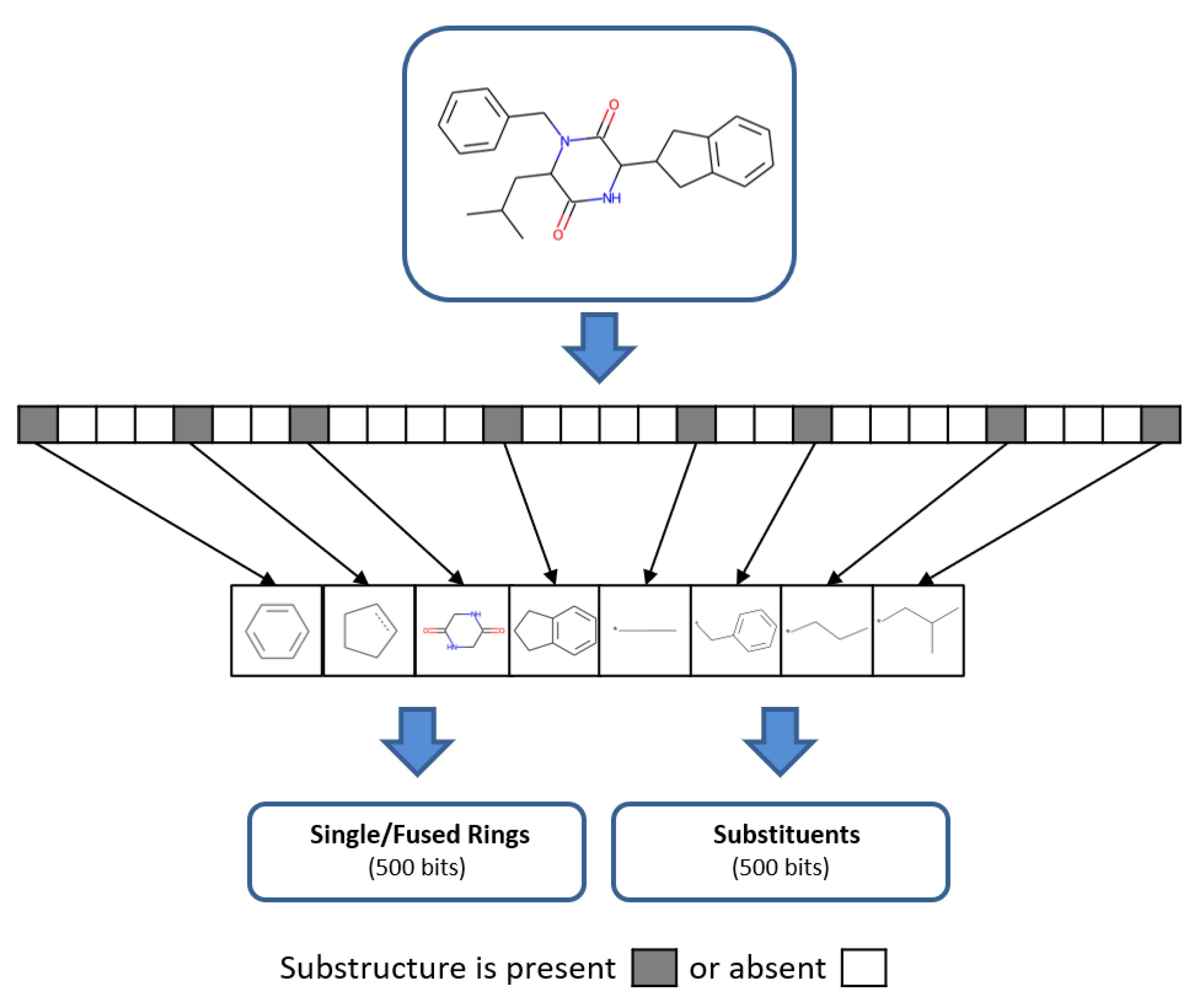
2.3. Fingerprint Assembly and Feature Mapping
2.4. Compound Activity Classes
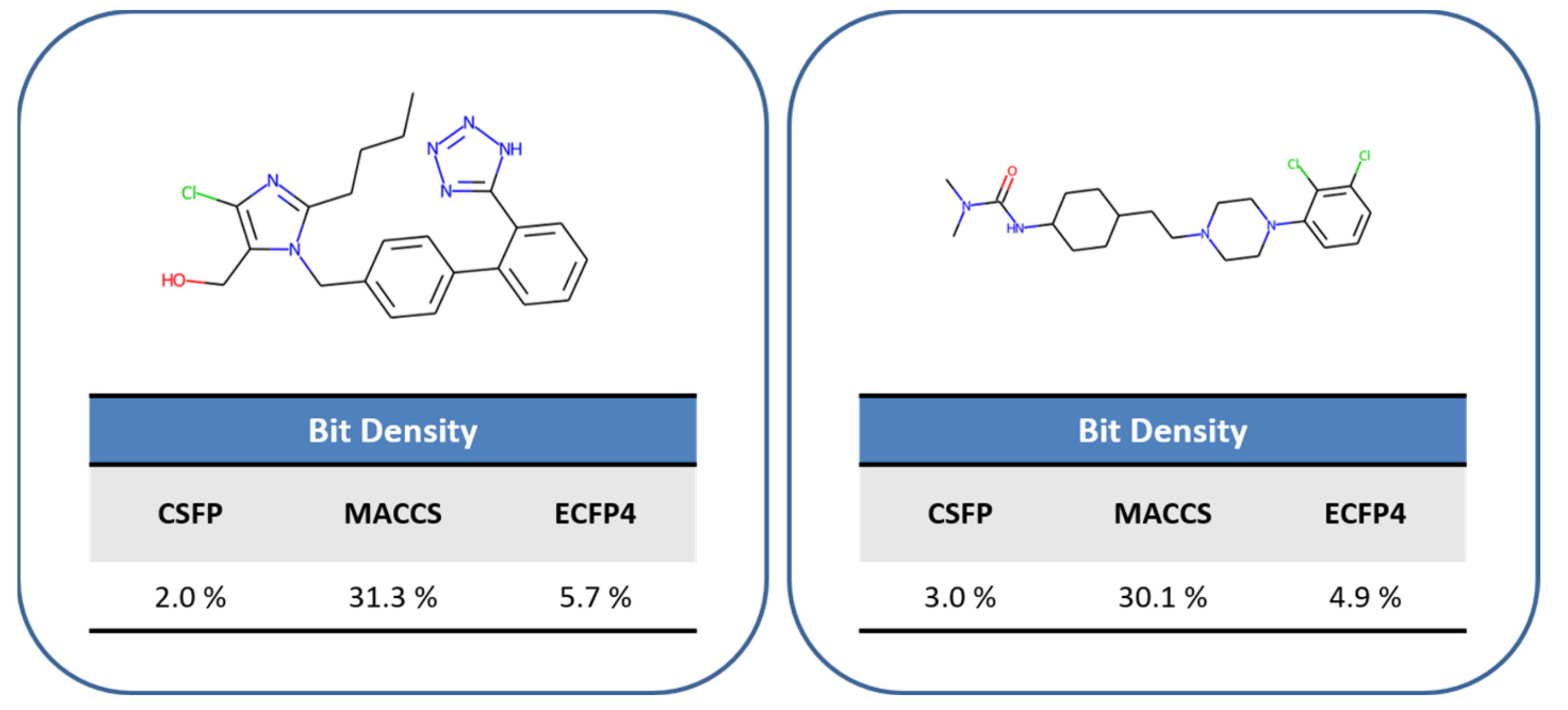
2.5. Feature Distribution
2.6. Performance Evaluation
2.6.1. Similarity Searching
2.6.2. Compound Classification
3. Materials and Methods
3.1. Compound Activity Classes
3.2. Core Generation and Fragmentation
3.3. Molecular Representations
3.4. Similarity Searching
3.5. Machine Learning
3.5.1. Random Forest
3.5.2. Support Vector Machine
3.5.3. Model Building and Hyperparameter Optimization
3.5.4. Predictions
3.6. Performance Measures
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Willett, P. Searching Techniques for Databases of Two- and Three-Dimensional Chemical Structures. J. Med. Chem. 2005, 48, 4183–4199. [Google Scholar] [CrossRef] [PubMed]
- Willett, P. Similarity-based virtual screening using 2D fingerprints. Drug Discovery Today 2006, 11, 1046–1053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stumpfe, D.; Bajorath, J. Similarity Searching. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2011, 1, 260–282. [Google Scholar] [CrossRef]
- Vogt, M.; Stumpfe, D.; Geppert, H.; Bajorath, J. Scaffold Hopping Using Two-Dimensional Fingerprints: True Potential, Black Magic, or a Hopeless Endeavor? Guidelines for Virtual Screening. J. Med. Chem. 2010, 53, 5707–5715. [Google Scholar] [CrossRef]
- Maggiora, G.; Vogt, M.; Stumpfe, D.; Bajorath, J. Molecular Similarity in Medicinal Chemistry. J. Med. Chem. 2014, 57, 3186–3204. [Google Scholar] [CrossRef]
- Cereto-Massagué, A.; Ojeda, M.J.; Valls, C.; Mulero, M.; Garcia-Vallvé, S.; Pujadas, G. Molecular Fingerprint Similarity Search in Virtual Screening. Methods 2015, 71, 58–63. [Google Scholar] [CrossRef]
- Muegge, I.; Mukherjee, P. An Overview of Molecular Fingerprint Similarity Search in Virtual Screening. Expert Opin. Drug Discov. 2016, 11, 137–148. [Google Scholar] [CrossRef]
- McGregor, M.J.; Muskal, S.M. Pharmacophore Fingerprinting. 1. Application to QSAR and Focused Library Design. J. Chem. Inf. Comput. Sci. 1999, 39, 569–574. [Google Scholar] [CrossRef]
- Matter, H.; Potter, T. Comparing 3D Pharmacophore Triplets and 2D Fingerprints for Selecting Diverse Compound Subsets. J. Chem. Inf. Comput. Sci. 1999, 39, 1211–1225. [Google Scholar] [CrossRef] [Green Version]
- Mason, J.S.; Morize, I.; Menard, P.R.; Cheney, D.L.; Hulme, C.; Labaudiniere, R.F. New 4-Point Pharmacophore Method for Molecular Similarity and Diversity Applications: Overview of the Method and Applications Including a Novel Approach to the Design of Combinatorial Libraries Containing Privileged Substructures. J. Med. Chem. 1995, 38, 144−150. [Google Scholar] [CrossRef]
- Singh, J.; Deng, Z.; Narale, G.; Chuaqui, C. Structural Interaction Fingerprints: A New approach to Organizing, Mining, Analyzing, and Designing Protein–Small Molecule Complexes. Chem. Biol. Drug Des. 2006, 67, 5–12. [Google Scholar] [CrossRef]
- Brewerton, S.C. The Use of Protein-Ligand Interaction Fingerprints in Docking. Curr. Opin. Drug Discov. Develop. 2008, 11, 356–364. [Google Scholar]
- Bonachéra, F.; Parent, B.; Barbosa, F.; Froloff, N.; Horvath, D. Fuzzy Tricentric Pharmacophore Fingerprints. 1. Topological Fuzzy Pharmacophore Triplets and Adapted Molecular Similarity Scoring Schemes. J. Chem. Inf. Model. 2006, 46, 2457–2477. [Google Scholar] [CrossRef] [Green Version]
- Chemical Computing Group. TGD and TGT Fingerprints. In Molecular Operating Environment (MOE); Chemical Computing Group Inc.: Montreal, QC, Canada, 2013. [Google Scholar]
- Xue, L.; Godden, J.W.; Stahura, F.L.; Bajorath, J. Design and Evaluation of a Molecular Fingerprint Involving the Transformation of Property Descriptor Values into a Binary Classification Scheme. J. Chem. Inf. Comput. Sci. 2003, 43, 1151−1157. [Google Scholar] [CrossRef]
- Xue, L.; Godden, J.W.; Bajorath, J. Evaluation of Descriptors and Mini-Fingerprints for the Identification of Molecules with Similar Activity. J. Chem. Inf. Comput. Sci. 2000, 40, 1227–1234. [Google Scholar] [CrossRef]
- MDL information Systems. MACCS (Molecular ACCess System) Structural Keys; MDL information Systems: San Leandro, CA, USA, 2002. [Google Scholar]
- Durant, J.; Leland, B.; Henry, D.; Nourse, J. Reoptimization of MDL Keys for Use in Drug Discovery. J. Chem. Inf. Comput. Sci. 2002, 42, 1273–1280. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Geppert, H.; Bajorath, J. Random Reduction in Fingerprint Bit Density Improves Compound recall in Search Calculations Using Complex Reference Molecules. Chem. Biol. Drug Des. 2008, 71, 511–517. [Google Scholar] [CrossRef]
- Barnard, J.M.; Downs, G.M. Chemical Fragment Generation and Clustering Software. J. Chem. Inf. Comput. Sci. 1997, 37, 141–142. [Google Scholar] [CrossRef]
- Bolton, E.E.; Wang, Y.; Thiessen, P.A.; Bryant, S.H. PubChem: Integrated Platform of Small Molecules and Biological Activities. Ann. Rep. Comput. Chem. 2008, 4, 217–241. [Google Scholar]
- Carhart, R.E.; Smith, D.H.; Venkataraghavan, R. Atom Pairs as Molecular Features in Structure-Activity Studies: Definition and Application. J. Chem. Inf. Comput. Sci. 1985, 25, 64–73. [Google Scholar] [CrossRef]
- Ahmed, H.E.; Vogt, M.; Bajorath, J. Design and Evaluation of Bonded Atom Pair Descriptors. J. Chem. Inf. Model. 2010, 50, 487–499. [Google Scholar] [CrossRef]
- Awale, M.; Reymond, J.L. Atom Pair 2D-Fingerprints Perceive 3D-Molecular Shape and Pharmacophores for Very Fast Virtual Screening of ZINC and GDB-17. J. Chem. Inf. Model. 2014, 54, 1892–1907. [Google Scholar] [CrossRef]
- Daylight Fingerprints; Daylight Chemical Information Systems, Inc.: Mission Viejo, CA, USA, 2015.
- Morgan, H.L. The Generation of a Unique Machine Description for Chemical Structures—A Technique Developed at Chemical Abstracts Service. J. Chem. Doc. 1965, 5, 107–112. [Google Scholar] [CrossRef]
- Bender, A.; Mussa, H.Y.; Glen, R.C.; Reiling, S. Similarity Searching of Chemical Databases Using Atom Environment descriptors (MOLPRINT 2D): Evaluation of Performance. J. Chem. Inf. Comput. Sci. 2004, 44, 1708–1718. [Google Scholar] [CrossRef]
- Glen, R.C.; Bender, A.; Arnby, C.H.; Carlsson, L.; Boyer, S.; Smith, J. Circular Fingerprints: Flexible Molecular Descriptors with Applications from Physical Chemistry to ADME. IDrugs 2006, 9, 199–204. [Google Scholar]
- Rogers, D.; Hahn, M. Extended-Connectivity Fingerprints, J. Chem. Inf. Model. 2010, 50, 742–754. [Google Scholar] [CrossRef]
- Hu, Y.; Lounkine, E.; Batista, J.; Bajorath, J. RelACCS-FP: A Structural Minimalist Approach to Fingerprint Design. Chem. Biol. Drug Des. 2008, 72, 341–349. [Google Scholar] [CrossRef]
- Takeuchi, K.; Kunimoto, R.; Bajorath, J. R-Group Replacement Database for Medicinal Chemistry. Future Sci. OA 2021, 7, 742. [Google Scholar] [CrossRef]
- Takeuchi, K.; Kunimoto, R.; Bajorath, J. Global Assessment of Substituents on the Basis of Analogue Series. J. Med. Chem. 2020, 63, 15013–15020. [Google Scholar] [CrossRef]
- Bento, A.P.; Gaulton, A.; Hersey, A.; Bellis, L.J.; Chambers, J.; Davies, M.; Krüger, F.A.; Light, Y.; Mak, L.; McGlinchey, S.; et al. The ChEMBL Bioactivity Database: An Update. Nucleic Acids Res. 2014, 42, D1083–D1090. [Google Scholar] [CrossRef] [Green Version]
- Heikamp, K.; Bajorath, J. Large-Scale Similarity Search Profiling of ChEMBL Compound Data Sets. J. Chem. Inf. Model. 2011, 51, 1831–1839. [Google Scholar] [CrossRef] [PubMed]
- RDKit: Cheminformatics and Machine Learning Software. 2013. Available online: http://www.rdkit.org (accessed on 1 July 2021).
- Bruns, R.F.; Watson, I.A. Rules for Identifying Potentially Reactive or Promiscuous Compounds. J. Med. Chem. 2012, 55, 9763–9772. [Google Scholar] [CrossRef] [PubMed]
- Irwin, J.J.; Duan, D.; Torosyan, H.; Doak, A.K.; Ziebart, K.T.; Sterling, T.; Tumanian, G.; Shoichet, B.K. An Aggregation Advisor for Ligand Discovery. J. Med. Chem. 2015, 58, 7076–7087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weininger, D. SMILES, a Chemical Language and Information System: 1: Introduction to Methodology and Encoding Rules. J. Chem. Inf. Comput. Sci. 1988, 28, 31–36. [Google Scholar] [CrossRef]
- Naveja, J.J.; Vogt, M.; Stumpfe, D.; Medina-Franco, J.L.; Bajorath, J. Systematic Extraction of Analogue Series from Large Compound Collections Using a New Computational Compound-Core Relationship Method. ACS Omega 2019, 4, 1027–1032. [Google Scholar] [CrossRef] [Green Version]
- Hert, J.; Willett, P.; Wilton, D.J.; Acklin, P.; Azzaoui, K.; Jacoby, E.; Schuffenhauer, A. Comparison of Fingerprint-Based Methods for Virtual Screening Using Multiple Bioactive Reference Structures. J. Chem. Inf. Comput. Sci. 2004, 44, 1177–1185. [Google Scholar] [CrossRef]
- Willett, P.; Barnard, J.M.; Downs, G.M. Chemical Similarity Searching. J. Chem. Inf. Comput. Sci. 1998, 38, 983–996. [Google Scholar] [CrossRef] [Green Version]
- Breiman, L. Random Forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef] [Green Version]
- Vapnik, V.N. The Nature of Statistical Learning Theory, 2nd ed.; Springer: New York, NY, USA, 2000. [Google Scholar]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-Learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Ralaivola, L.; Swamidass, S.J.; Saigo, H.; Baldi, P. Graph Kernels for Chemical Informatics. Neur. Netw. 2005, 18, 1093–1110. [Google Scholar] [CrossRef]
- Brodersen, K.H.; Ong, C.S.; Stephan, K.E.; Buhmann, J.M. The Balanced Accuracy and Its Posterior Distribution. In Proceedings of the 20th International Conference on Pattern Recognition (ICPR), Istanbul, Turkey, 23–26 August 2010; pp. 3121–3124. [Google Scholar]
- Matthews, B.W. Comparison of the Predicted and Observed Secondary Structure of T4 Phage Lysozyme. BBA—Protein Struct. 1975, 405, 442–451. [Google Scholar] [CrossRef]
- Van Rijsbergen, C.J. Information Retrieval, 2nd ed.; Butterworth-Heinemann: Oxford, UK, 1979. [Google Scholar]
- Bradley, A.P. The Use of the Area under the ROC Curve in the Evaluation of Machine Learning Algorithms. Pattern Recognit. 1997, 30, 1145–1159. [Google Scholar] [CrossRef] [Green Version]
- Conover, W.J. On Methods of Handling Ties in the Wilcoxon Signed-Rank Test. J. Am. Stat. Assoc. 1973, 68, 985–988. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Janela, T.; Takeuchi, K.; Bajorath, J. Introducing a Chemically Intuitive Core-Substituent Fingerprint Designed to Explore Structural Requirements for Effective Similarity Searching and Machine Learning. Molecules 2022, 27, 2331. https://doi.org/10.3390/molecules27072331
Janela T, Takeuchi K, Bajorath J. Introducing a Chemically Intuitive Core-Substituent Fingerprint Designed to Explore Structural Requirements for Effective Similarity Searching and Machine Learning. Molecules. 2022; 27(7):2331. https://doi.org/10.3390/molecules27072331
Chicago/Turabian StyleJanela, Tiago, Kosuke Takeuchi, and Jürgen Bajorath. 2022. "Introducing a Chemically Intuitive Core-Substituent Fingerprint Designed to Explore Structural Requirements for Effective Similarity Searching and Machine Learning" Molecules 27, no. 7: 2331. https://doi.org/10.3390/molecules27072331
APA StyleJanela, T., Takeuchi, K., & Bajorath, J. (2022). Introducing a Chemically Intuitive Core-Substituent Fingerprint Designed to Explore Structural Requirements for Effective Similarity Searching and Machine Learning. Molecules, 27(7), 2331. https://doi.org/10.3390/molecules27072331