
Molecular Dynamics and MM-PBSA Analysis of the SARS-CoV-2 Gamma Variant in Complex with the hACE-2 Receptor




Abstract
:1. Introduction
2. Materials and Methods
2.1. Molecular Dynamics
2.2. Structure and Trajectory Analysis
2.2.1. Statistical Analysis
2.2.2. Molecular Mechanics Poison-Boltzmann Surface Area (MM-PBSA)
2.2.3. Entropy Calculations
3. Results and Discussion
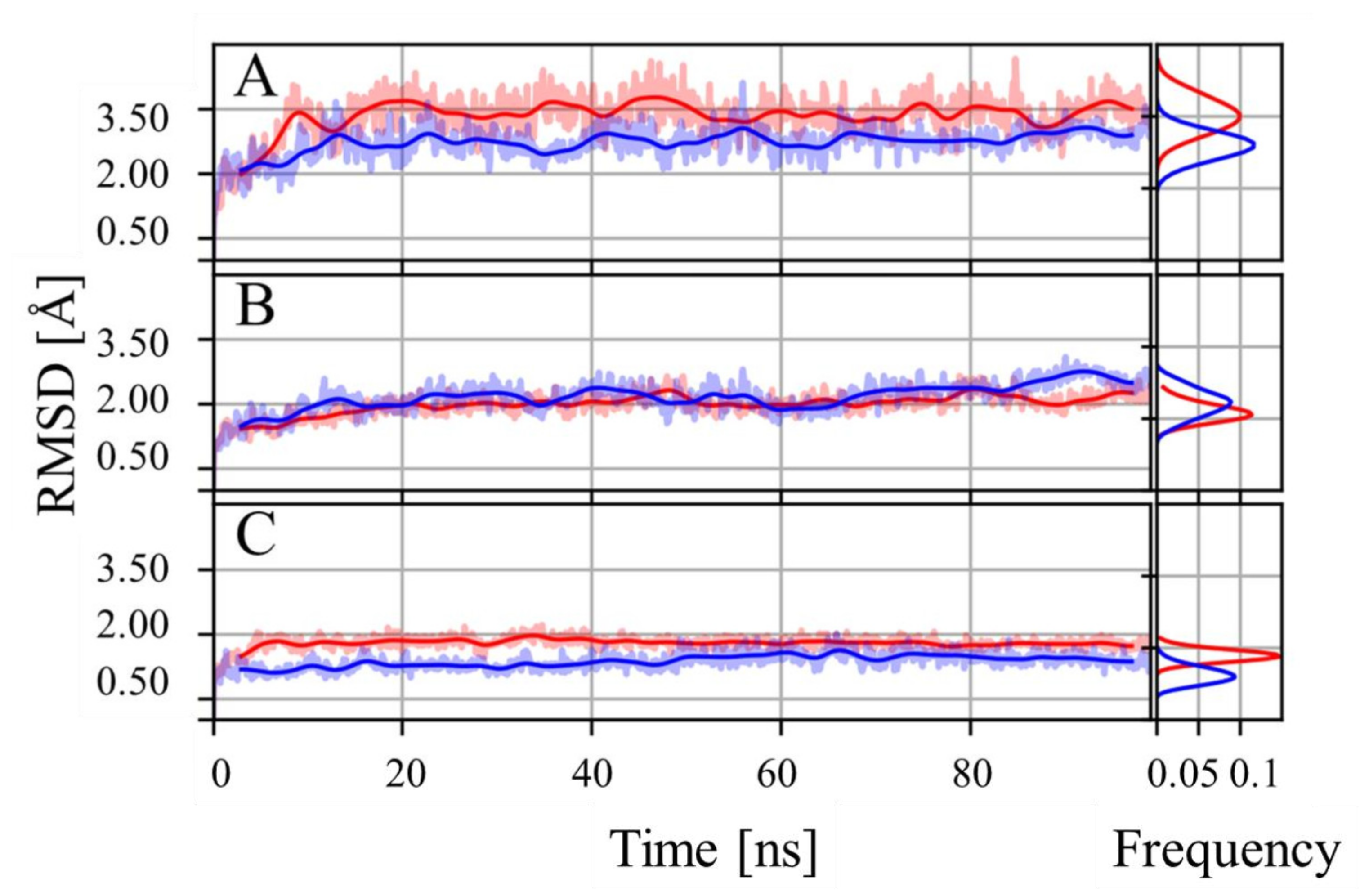
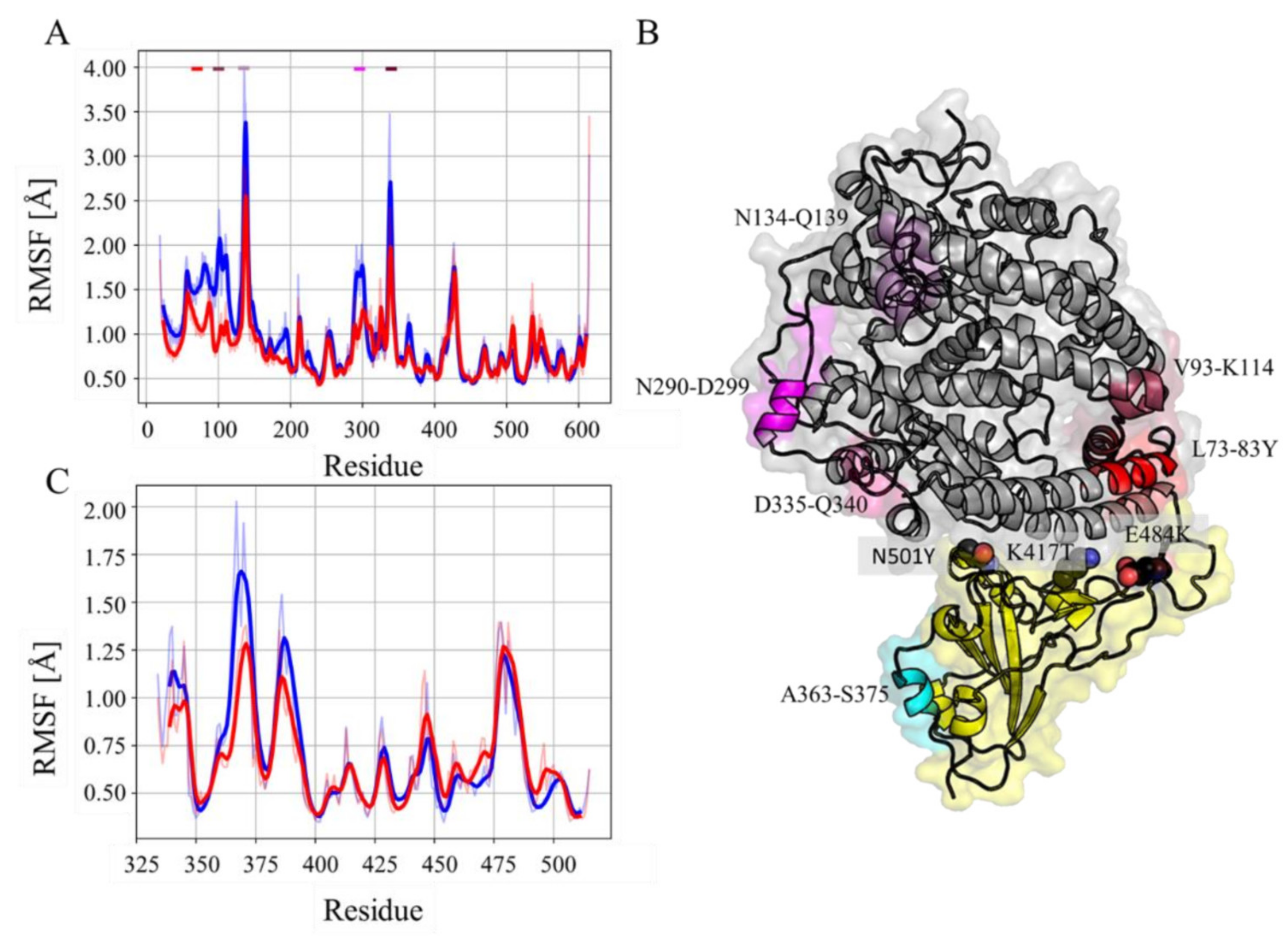
3.1. Molecular Dynamics Analysis
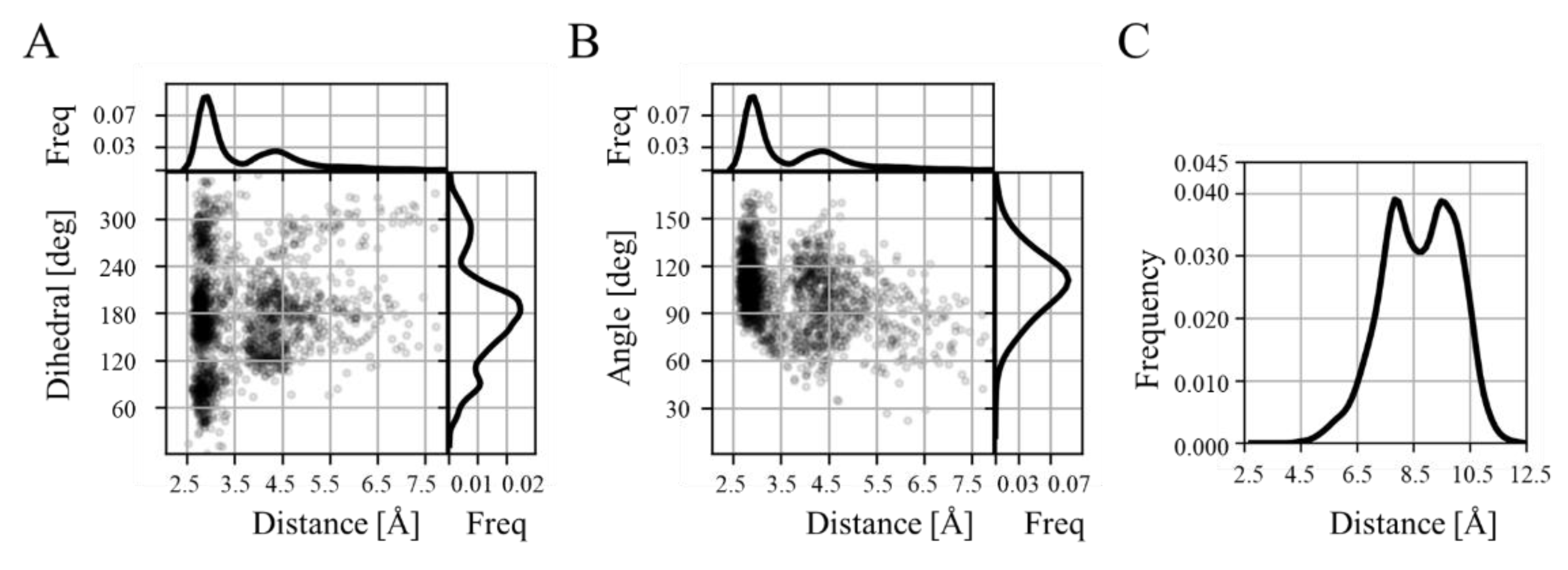
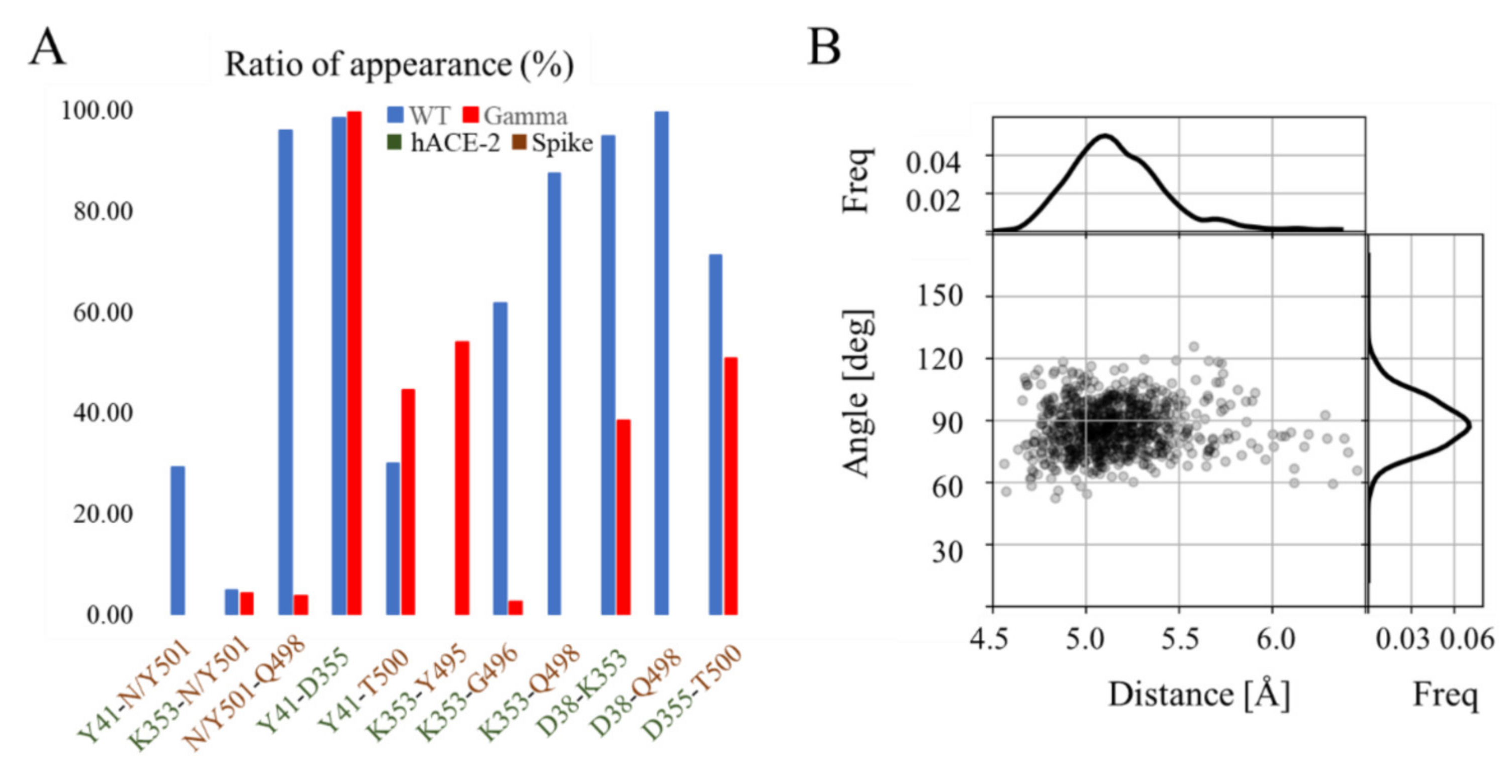
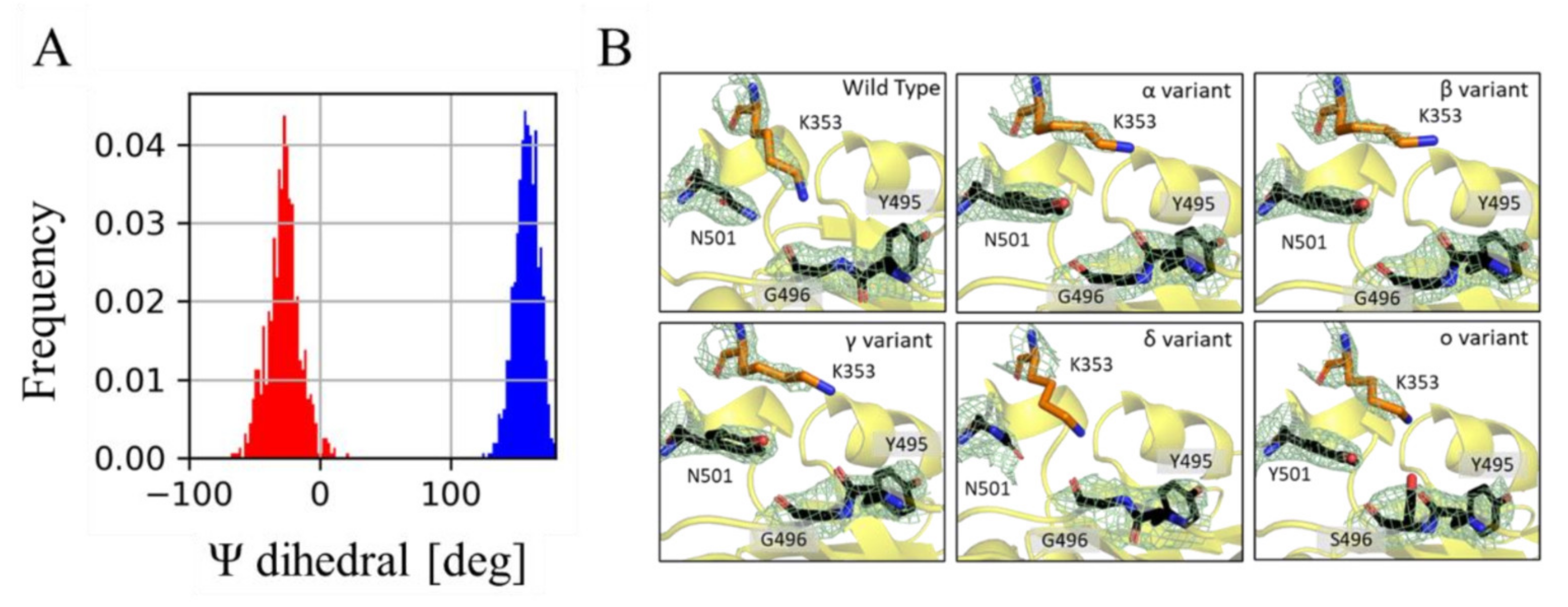
3.2. Effect of Mutations on the hACE-RBD Interaction
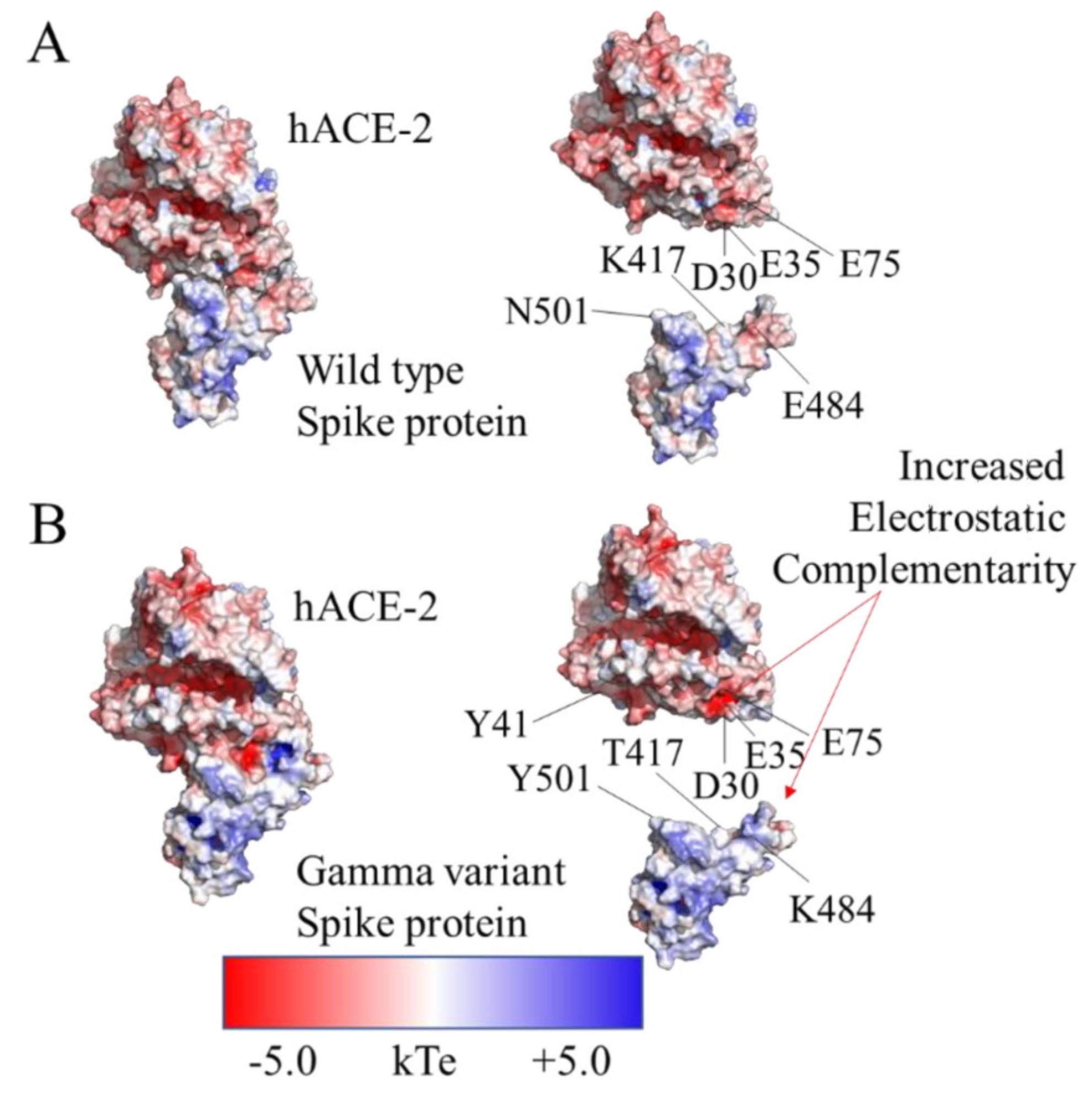
3.3. Effect of Mutations on Binding Affinity
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
Appendix A
References
- Coronavirus Tracker: The Latest Figures as Countries Fight the COVID-19 Resurgence | Free to Read | Financial Times. Available online: https://www.ft.com/content/a2901ce8-5eb7-4633-b89c-cbdf5b386938 (accessed on 15 August 2021).
- Plante, J.A.; Mitchell, B.M.; Plante, K.S.; Debbink, K.; Weaver, S.C.; Menachery, V.D. The Variant Gambit: COVID’s Next Move. Cell Host Microbe 2021, 29, 508–515. [Google Scholar] [CrossRef]
- Sternberg, A.; Naujokat, C. Structural Features of Coronavirus SARS-CoV-2 Spike Protein: Targets for Vaccination. Life Sci. 2020, 257, 118056. [Google Scholar] [CrossRef]
- Guruprasad, L. Human SARS CoV-2 Spike Protein Mutations. Proteins 2021, 89, 569–576. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.Y.; Smith, D.M. SARS-CoV-2 Variants of Concern. Yonsei Med. J. 2021, 62, 961. [Google Scholar] [CrossRef] [PubMed]
- Davies, N.G.; Abbott, S.; Barnard, R.C.; Jarvis, C.I.; Kucharski, A.J.; Munday, J.D.; Pearson, C.A.B.; Russell, T.W.; Tully, D.C.; Washburne, A.D.; et al. Estimated Transmissibility and Impact of SARS-CoV-2 Lineage B.1.1.7 in England. Science 2021, 372, eabg3055. [Google Scholar] [CrossRef] [PubMed]
- Hussain, M.; Shabbir, S.; Amnaullah, A.; Raza, F.; Imdad, M.J.; Zahid, S. Immunoinformatic Analysis of Structural and Epitope Variations in Spike and Orf8 Proteins of SARS-CoV-2/B.1.1.7. J. Med. Virol. 2021, 93, 4461–4468. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Liu, L.; Iketani, S.; Luo, Y.; Guo, Y.; Wang, M.; Yu, J.; Zhang, B.; Kwong, P.D.; Graham, B.S.; et al. Increased Resistance of SARS-CoV-2 Variants B.1.351 and B.1.1.7 to Antibody Neutralization. bioRxiv 2021. [Google Scholar] [CrossRef]
- Supasa, P.; Zhou, D.; Dejnirattisai, W.; Liu, C.; Mentzer, A.J.; Ginn, H.M.; Zhao, Y.; Duyvesteyn, H.M.E.; Nutalai, R.; Tuekprakhon, A.; et al. Reduced Neutralization of SARS-CoV-2 B.1.1.7 Variant by Convalescent and Vaccine Sera. Cell 2021, 184, 2201. [Google Scholar] [CrossRef]
- Tegally, H.; Wilkinson, E.; Giovanetti, M.; Iranzadeh, A.; Fonseca, V.; Giandhari, J.; Doolabh, D.; Pillay, S.; San, E.J.; Msomi, N.; et al. Emergence and Rapid Spread of a New Severe Acute Respiratory Syndrome-Related Coronavirus 2 (SARS-CoV-2) Lineage with Multiple Spike Mutations in South Africa. medRxiv 2020. [Google Scholar] [CrossRef]
- Zhou, D.; Dejnirattisai, W.; Supasa, P.; Liu, C.; Mentzer, A.J.; Ginn, H.M.; Zhao, Y.; Duyvesteyn, H.M.E.; Tuekprakhon, A.; Nutalai, R.; et al. Evidence of Escape of SARS-CoV-2 Variant B.1.351 from Natural and Vaccine-Induced Sera. Cell 2021, 184, 2348. [Google Scholar] [CrossRef] [PubMed]
- Taylor, P.C.; Adams, A.C.; Hufford, M.M.; de la Torre, I.; Winthrop, K.; Gottlieb, R.L. Neutralizing Monoclonal Antibodies for Treatment of COVID-19. Nat. Rev. Immunol. 2021, 21, 382–393. [Google Scholar] [CrossRef] [PubMed]
- Barnes, C.O.; Jette, C.A.; Abernathy, M.E.; Dam, K.M.A.; Esswein, S.R.; Gristick, H.B.; Malyutin, A.G.; Sharaf, N.G.; Huey-Tubman, K.E.; Lee, Y.E.; et al. SARS-CoV-2 Neutralizing Antibody Structures Inform Therapeutic Strategies. Nature 2020, 588, 682–687. [Google Scholar] [CrossRef] [PubMed]
- Faria, N.R.; Mellan, T.A.; Whittaker, C.; Claro, I.M.; Candido, D.d.S.; Mishra, S.; Crispim, M.A.E.; Sales, F.C.; Hawryluk, I.; McCrone, J.T.; et al. Genomics and Epidemiology of a Novel SARS-CoV-2 Lineage in Manaus, Brazil. medRxiv 2021. [Google Scholar] [CrossRef]
- Sabino, E.C.; Buss, L.F.; Carvalho, M.P.S.; Prete, C.A.; Crispim, M.A.E.; Fraiji, N.A.; Pereira, R.H.M.; Parag, K.V.; da Silva Peixoto, P.; Kraemer, M.U.G.; et al. Resurgence of COVID-19 in Manaus, Brazil, despite High Seroprevalence. Lancet 2021, 397, 452–455. [Google Scholar] [CrossRef]
- Tracking SARS-CoV-2 Variants. Available online: https://www.who.int/en/activities/tracking-SARS-CoV-2-variants/ (accessed on 15 August 2021).
- Rees-Spear, C.; Muir, L.; Griffith, S.A.; Heaney, J.; Aldon, Y.; Snitselaar, J.L.; Thomas, P.; Graham, C.; Seow, J.; Lee, N.; et al. The Effect of Spike Mutations on SARS-CoV-2 Neutralization. Cell Rep. 2021, 34, 108890. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Wang, M.; Yu, J.; Cerutti, G.; Nair, M.S.; Huang, Y.; Kwong, P.D.; Shapiro, L.; Ho, D.D. Increased Resistance of SARS-CoV-2 Variant P.1 to Antibody Neutralization. bioRxiv 2021. [Google Scholar] [CrossRef]
- Dejnirattisai, W.; Zhou, D.; Supasa, P.; Liu, C.; Mentzer, A.J.; Ginn, H.M.; Zhao, Y.; Duyvesteyn, H.M.E.; Tuekprakhon, A.; Nutalai, R.; et al. Antibody Evasion by the P.1 Strain of SARS-CoV-2. Cell 2021, 184, 2939–2954.e9. [Google Scholar] [CrossRef]
- Zahradník, J.; Marciano, S.; Shemesh, M.; Zoler, E.; Chiaravalli, J.; Meyer, B.; Rudich, Y.; Dym, O.; Elad, N.; Schreiber, G. SARS-CoV-2 RBD in Vitro Evolution Follows Contagious Mutation Spread, yet Generates an Able Infection Inhibitor. bioRxiv 2021. [Google Scholar] [CrossRef]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; et al. Structure of the SARS-CoV-2 Spike Receptor-Binding Domain Bound to the ACE2 Receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef] [Green Version]
- Berman, H.M.; Kleywegt, G.J.; Nakamura, H.; Markley, J.L. The Protein Data Bank Archive as an Open Data Resource. J. Comput.-Aided Mol. Des. 2014, 28, 1009–1014. [Google Scholar] [CrossRef] [Green Version]
- DeLano, W. Pymol: An Open-Source Molecular Graphics Tool. CCP4 Newsl. Protein Crystallogr. 2002, 40, 82–92. [Google Scholar]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. GROMACS: Fast, Flexible, and Free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef] [PubMed]
- Lindorff-Larsen, K.; Piana, S.; Palmo, K.; Maragakis, P.; Klepeis, J.L.; Dror, R.O.; Shaw, D.E. Improved Side-Chain Torsion Potentials for the Amber Ff99SB Protein Force Field. Proteins 2010, 78, 1950–1958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Kumari, R.; Kumar, R.; Lynn, A. G-Mmpbsa—A GROMACS Tool for High-Throughput MM-PBSA Calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef] [PubMed]
- Baker, N.A.; Sept, D.; Joseph, S.; Holst, M.J.; McCammon, J.A. Electrostatics of Nanosystems: Application to Microtubules and the Ribosome. Proc. Natl. Acad. Sci. USA 2001, 98, 10037–10041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheng, Y.J.; Yin, Y.W.; Ma, Y.Q.; Ding, H.M. Improving the Performance of MM/PBSA in Protein-Protein Interactions via the Screening Electrostatic Energy. J. Chem. Inf. Model. 2021, 61, 2454–2462. [Google Scholar] [CrossRef] [PubMed]
- Duan, L.; Liu, X.; Zhang, J.Z.H. Interaction Entropy: A New Paradigm for Highly Efficient and Reliable Computation of Protein-Ligand Binding Free Energy. J. Am. Chem. Soc. 2016, 138, 5722–5728. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Yan, Y.N.; Yang, M.; Zhang, J.Z.H. Interaction Entropy for Protein-Protein Binding. J. Chem. Phys. 2017, 146, 124124. [Google Scholar] [CrossRef]
- Huang, K.; Luo, S.; Cong, Y.; Zhong, S.; Zhang, J.Z.H.; Duan, L. An Accurate Free Energy Estimator: Based on MM/PBSA Combined with Interaction Entropy for Protein–Ligand Binding Affinity. Nanoscale 2020, 12, 10737–10750. [Google Scholar] [CrossRef]
- Duan, L.; Feng, G.; Wang, X.; Wang, L.; Zhang, Q. Effect of Electrostatic Polarization and Bridging Water on CDK2–Ligand Binding Affinities Calculated Using a Highly Efficient Interaction Entropy Method. Phys. Chem. Chem. Phys. 2017, 19, 10140–10152. [Google Scholar] [CrossRef]
- Han, Y.; Wang, Z.; Wei, Z.; Schapiro, I.; Li, J. Binding Affinity and Mechanisms of SARS-CoV-2 Variants. Comput. Struct. Biotechnol. J. 2021, 19, 4184. [Google Scholar] [CrossRef] [PubMed]
- Donald, J.E.; Kulp, D.W.; DeGrado, W.F. Salt Bridges: Geometrically Specific, Designable Interactions. Proteins 2011, 79, 898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, F.; Tong, B.; Sun, L.; Shi, S.; Zheng, B.; Wang, Z.; Dong, X.; Zheng, P. Mutation N501Y in RBD of Spike Protein Strengthens the Inter-Action between COVID-19 and Its Receptor ACE2. bioRxiv 2021, 19. [Google Scholar] [CrossRef]
- Koehler, M.; Ray, A.; Moreira, R.A.; Juniku, B.; Poma, A.B.; Alsteens, D. Molecular Insights into Receptor Binding Energetics and Neutralization of SARS-CoV-2 Variants. Nat. Commun. 2021, 12, 6977. [Google Scholar] [CrossRef] [PubMed]
- Socher, E.; Conrad, M.; Heger, L.; Paulsen, F.; Sticht, H.; Zunke, F.; Arnold, P. Computational Decomposition Reveals Reshaping of the SARS-CoV-2–ACE2 Interface among Viral Variants Expressing the N501Y Mutation. J. Cell. Biochem. 2021, 122, 1863–1872. [Google Scholar] [CrossRef]
- Kim, S.; Liu, Y.; Lei, Z.; Dicker, J.; Cao, Y.; Zhang, X.F.; Im, W. Differential Interactions between Human ACE2 and Spike RBD of SARS-CoV-2 Variants of Concern. J. Chem. Theory Comput. 2021, 17, 7972–7979. [Google Scholar] [CrossRef] [PubMed]
- Tian, F.; Tong, B.; Sun, L.; Shi, S.; Zheng, B.; Wang, Z.; Dong, X.; Zheng, P. N501y Mutation of Spike Protein in Sars-Cov-2 Strengthens Its Binding to Receptor Ace2. Elife 2021, 10, e69091. [Google Scholar] [CrossRef]
- Han, P.; Su, C.; Zhang, Y.; Bai, C.; Zheng, A.; Qiao, C.; Wang, Q.; Niu, S.; Chen, Q.; Zhang, Y.; et al. Molecular Insights into Receptor Binding of Recent Emerging SARS-CoV-2 Variants. Nat. Commun. 2021, 12, 6103. [Google Scholar] [CrossRef] [PubMed]
- Han, P.; Li, L.; Liu, S.; Wang, Q.; Zhang, D.; Xu, Z.; Han, P.; Li, X.; Peng, Q.; Su, C.; et al. Receptor Binding and Complex Structures of Human ACE2 to Spike RBD from Omicron and Delta SARS-CoV-2. Cell 2022, 185, 630–640.e10. [Google Scholar] [CrossRef] [PubMed]
- Cong, Y.; Feng, Y.; Ni, H.; Zhi, F.; Miao, Y.; Fang, B.; Zhang, L.; Zhang, J.Z.H. Anchor-Locker Binding Mechanism of the Coronavirus Spike Protein to Human ACE2: Insights from Computational Analysis. J. Chem. Inf. Model. 2021, 61, 3529–3542. [Google Scholar] [CrossRef] [PubMed]
- Forouzesh, N.; Mishra, N. An Effective MM/GBSA Protocol for Absolute Binding Free Energy Calculations: A Case Study on SARS-CoV-2 Spike Protein and the Human ACE2 Receptor. Molecules 2021, 26, 2383. [Google Scholar] [CrossRef] [PubMed]
- Shah, M.; Ahmad, B.; Choi, S.; Woo, H.G. Mutations in the SARS-CoV-2 Spike RBD Are Responsible for Stronger ACE2 Binding and Poor Anti-SARS-CoV MAbs Cross-Neutralization. Comput. Struct. Biotechnol. J. 2020, 18, 3402–3414. [Google Scholar] [CrossRef] [PubMed]
- Miton, C.M.; Buda, K.; Tokuriki, N. Epistasis and Intramolecular Networks in Protein Evolution. Curr. Opin. Struct. Biol. 2021, 69, 160–168. [Google Scholar] [CrossRef]
- Greaney, A.J.; Starr, T.N.; Gilchuk, P.; Zost, S.J.; Binshtein, E.; Loes, A.N.; Hilton, S.K.; Huddleston, J.; Eguia, R.; Crawford, K.H.D.; et al. Complete Mapping of Mutations to the SARS-CoV-2 Spike Receptor-Binding Domain That Escape Antibody Recognition. Cell Host Microbe 2021, 29, 44. [Google Scholar] [CrossRef] [PubMed]
- Jones, B.E.; Brown-Augsburger, P.L.; Corbett, K.S.; Westendorf, K.; Davies, J.; Cujec, T.P.; Wiethoff, C.M.; Blackbourne, J.L.; Heinz, B.A.; Foster, D.; et al. The Neutralizing Antibody, LY-CoV555, Protects against SARS-CoV-2 Infection in Nonhuman Primates. Sci. Transl. Med. 2021, 13, 1906. [Google Scholar] [CrossRef] [PubMed]
- Starr, T.N.; Greaney, A.J.; Dingens, A.S.; Bloom, J.D. Complete Map of SARS-CoV-2 RBD Mutations That Escape the Monoclonal Antibody LY-CoV555 and Its Cocktail with LY-CoV016. Cell Rep. Med. 2021, 2, 100255. [Google Scholar] [CrossRef] [PubMed]
- Luchsinger, L.L.; Hillyer, C.D. Vaccine Efficacy Probable against COVID-19 Variants. Science 2021, 371, 1116. [Google Scholar] [CrossRef]
- Fiolet, T.; Kherabi, Y.; MacDonald, C.-J.; Ghosn, J.; Peiffer-Smadja, N. Comparing COVID-19 Vaccines for Their Characteristics, Efficacy and Effectiveness against SARS-CoV-2 and Variants of Concern: A Narrative Review. Clin. Microbiol. Infect. 2021, 28, 202–221. [Google Scholar] [CrossRef] [PubMed]
- Ciotti, M.; Ciccozzi, M.; Pieri, M.; Bernardini, S. The COVID-19 Pandemic: Viral Variants and Vaccine Efficacy. Crit. Rev. Clin. Lab. Sci. 2021, 59, 66–75. [Google Scholar] [CrossRef]
- Shu, Y.; McCauley, J. GISAID: Global Initiative on Sharing All Influenza Data—From Vision to Reality. Eurosurveillance 2017, 22, 30494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elbe, S.; Buckland-Merrett, G. Data, Disease and Diplomacy: GISAID’s Innovative Contribution to Global Health. Glob. Chall. 2017, 1, 33–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Wild Type [kcal/mol] | Gamma Variant [kcal/mol] | |
|---|---|---|
| ∆Eelec | −102.84 ± 13.36 | −70.17 ± 8.50 |
| ∆Gnon pol | −11.92 ± 0.79 | −10.99 ± 0.85 |
| ∆Gpol | 159.86 ± 19.70 | 114.52 ± 21.11 |
| ∆EVdW | −100.98 ± 6.16 | −96.73 ± 5.59 |
| ∆H | −55.88 ± 22.15 | −63.37 ± 20.59 |
| −T∆S | 29.45 ± 0.56 | 26.27 ± 1.47 |
| ∆G | −26.43 ± 24.68 | −37.10 ± 23.50 |
| ∆Gexp | −10.40 | −11.39 |
| WT | Gamma | ∆∆H | ||||
|---|---|---|---|---|---|---|
| Residue | ∆H | Mean Std | Residue | ∆H | Mean Std | |
| S19 | 3.60 | 0.47 | S19 | 0.86 | 0.11 | −2.74 |
| D30 | 5.55 | 0.68 | D30 | 1.33 | 0.16 | −4.23 |
| K31 | 3.89 | 0.50 | K31 | 0.93 | 0.12 | −2.96 |
| H34 | −2.93 | 0.12 | H34 | −0.70 | 0.03 | 2.23 |
| Y41 | −1.81 | 0.08 | Y41 | −0.43 | 0.02 | 1.38 |
| E329 | −1.79 | 0.08 | E329 | −0.43 | 0.02 | 1.36 |
| K353 | 5.17 | 0.20 | K353 | 1.24 | 0.05 | −3.93 |
| D405 | 2.43 | 0.16 | D405 | 0.58 | 0.04 | −1.85 |
| K417 | 4.02 | 0.63 | T417 | 0.96 | 0.15 | −3.06 |
| L455 | −2.90 | 0.04 | L455 | −0.69 | 0.01 | 2.21 |
| F456 | −1.96 | 0.06 | F456 | −0.47 | 0.01 | 1.49 |
| E484 | 2.35 | 0.24 | K484 | 0.56 | 0.06 | −1.79 |
| F486 | −2.55 | 0.11 | F486 | −0.61 | 0.03 | 1.94 |
| Q498 | −2.29 | 0.09 | Q498 | −0.55 | 0.02 | 1.74 |
| T500 | −1.66 | 0.10 | T500 | −0.40 | 0.02 | 1.27 |
| N501 | −1.54 | 0.11 | Y501 | −0.37 | 0.03 | 1.17 |
| Y505 | −3.63 | 0.09 | Y505 | −0.87 | 0.02 | 2.76 |
| WT | Gamma | −T∆∆S | ||||
|---|---|---|---|---|---|---|
| Residue | −T∆S | Mean Std | Residue | −T∆S | Mean Std | |
| S19 | −0.86 | 0.16 | S19 | 2.31 | 1.44 | 3.17 |
| D30 | −6.67 | 0.71 | D30 | −0.43 | 0.22 | 6.24 |
| K31 | 0.01 | 0.97 | K31 | −2.74 | 0.89 | −2.75 |
| H34 | 1.63 | 0.79 | H34 | −0.24 | 0.63 | −1.87 |
| E35 | 0.70 | 0.25 | E35 | −2.29 | 1.37 | −3.00 |
| D38 | 0.49 | 0.64 | D38 | −1.80 | 0.97 | −2.29 |
| K353 | 0.49 | 0.34 | K353 | 0.72 | 0.98 | 0.23 |
| K417 | −5.56 | 2.85 | T417 | −0.18 | 0.08 | 5.39 |
| E484 | −0.57 | 0.27 | K484 | −2.10 | 0.61 | −1.53 |
| Q493 | 0.08 | 0.12 | Q493 | −1.67 | 1.24 | −1.75 |
| N501 | 0.05 | 0.09 | Y501 | −0.59 | 0.31 | −0.64 |
| −T∆S | −T∆∆S | ∆H | ∆∆H | ∆G | ∆∆G | ||||
|---|---|---|---|---|---|---|---|---|---|
| WT | Gamma | WT | Gamma | WT | Gamma | ||||
| D30 | −6.67 | −0.43 | 6.24 | 5.55 | 1.33 | −4.23 | −1.12 | 0.89 | 2.01 |
| K31 | 0.01 | −2.74 | −2.75 | 3.89 | 0.93 | −2.96 | 3.90 | −1.81 | −5.71 |
| E35 | 0.70 | −2.29 | −3.00 | 0.61 | 0.15 | −0.46 | 1.31 | −2.15 | −3.46 |
| D38 | 0.49 | −1.80 | −2.29 | −0.05 | −0.01 | 0.04 | 0.44 | −1.81 | −2.25 |
| E75 | −0.21 | −1.43 | −1.22 | 1.02 | 0.24 | −0.78 | 0.82 | −1.19 | −2.00 |
| E329 | 0.17 | 0.90 | 0.73 | −1.79 | −0.43 | 1.36 | −1.62 | 0.47 | 2.09 |
| K353 | 0.49 | 0.72 | 0.23 | 5.17 | 1.24 | −3.93 | 5.66 | 1.95 | −3.71 |
| D405 | 1.04 | 0.21 | −0.83 | 2.43 | 0.58 | −1.85 | 3.47 | 0.79 | −2.68 |
| K417T | −5.56 | −0.18 | 5.39 | 4.02 | 0.96 | −3.06 | −1.54 | 0.78 | 2.32 |
| F456 | −0.91 | −0.06 | 0.84 | −1.96 | −0.47 | 1.49 | −2.87 | −0.53 | 2.34 |
| E484K | −0.57 | −2.10 | −1.53 | 2.35 | 0.56 | −1.79 | 1.78 | −1.54 | −3.32 |
| T500 | −0.75 | 0.35 | 1.10 | −1.66 | −0.40 | 1.27 | −2.42 | −0.05 | 2.37 |
| N501Y | 0.05 | −0.59 | −0.64 | −1.54 | −0.37 | 1.17 | −1.48 | −0.96 | 0.52 |
| Y505 | −0.33 | −0.15 | 0.17 | −3.63 | −0.87 | 2.76 | −3.96 | −1.02 | 2.94 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cavani, M.; Riofrío, W.A.; Arciniega, M. Molecular Dynamics and MM-PBSA Analysis of the SARS-CoV-2 Gamma Variant in Complex with the hACE-2 Receptor. Molecules 2022, 27, 2370. https://doi.org/10.3390/molecules27072370
Cavani M, Riofrío WA, Arciniega M. Molecular Dynamics and MM-PBSA Analysis of the SARS-CoV-2 Gamma Variant in Complex with the hACE-2 Receptor. Molecules. 2022; 27(7):2370. https://doi.org/10.3390/molecules27072370
Chicago/Turabian StyleCavani, Maurizio, Walter Arnaldo Riofrío, and Marcelino Arciniega. 2022. "Molecular Dynamics and MM-PBSA Analysis of the SARS-CoV-2 Gamma Variant in Complex with the hACE-2 Receptor" Molecules 27, no. 7: 2370. https://doi.org/10.3390/molecules27072370
APA StyleCavani, M., Riofrío, W. A., & Arciniega, M. (2022). Molecular Dynamics and MM-PBSA Analysis of the SARS-CoV-2 Gamma Variant in Complex with the hACE-2 Receptor. Molecules, 27(7), 2370. https://doi.org/10.3390/molecules27072370