
H-Bond Mediated Phase-Transfer Catalysis: Enantioselective Generating of Quaternary Stereogenic Centers in β-Keto Esters





Abstract
:1. Introduction
2. Results
3. Materials and Methods
3.1. Reagents and General Methods
3.2. Synthetic Procedures
3.2.1. Synthesis of (1S,2S,4S,5R)-5-Ethenyl-2-[(R)-hydroxy(7-methoxyquinolin-4-yl)methyl]-1-({[2-(naphthalen-2-yl)phenyl]carbamoyl}methyl)-1-azabicyclo [2.2.2]octan-1-ium bromide (M)
3.2.2. Synthesis of (1S,2S,4S,5R)-5-Ethenyl-2-[(R)-hydroxy(7-methoxyquinolin-4-yl)methyl]-1-({[2-(naphthalen-1-yl)phenyl]carbamoyl}methyl)-1-azabicyclo [2.2.2]octan-1-ium bromide (N)
3.2.3. (1S,2S,4S,5R)-5-Ethenyl-2-[(R)-hydroxy(7-methoxyquinolin-4-yl)methyl]-1-({[2-(quinolin-8-yl)phenyl]carbamoyl}methyl)-1-azabicyclo [2.2.2]octan-1-ium bromide (O)
3.2.4. Methyl 2-Benzyl-1-oxo-2,3-dihydro-1H-indene-2-carboxylate (2a)
3.2.5. Propan-2-yl 2-Benzyl-1-oxo-2,3-dihydro-1H-indene-2-carboxylate (2b)
3.2.6. Tert-Butyl 2-Benzyl-1-oxo-2,3-dihydro-1H-indene-2-carboxylate (2c)
3.2.7. Methyl 1-Benzyl-2-oxocyclopentane-1-carboxylate (2d)
3.2.8. Tert-Butyl 1-Benzyl-2-oxocyclopentane-1-carboxylate (2e)
3.2.9. Tert-butyl(2R)-2-[(2-methylphenyl)methyl]-1-oxo-2,3-dihydro-1H-indene-2-carboxylate (3)
3.2.10. Tert-Butyl 2-[(3-Methylphenyl)methyl]-1-oxo-2,3-dihydro-1H-indene-2-carboxylate (4)
3.2.11. Tert-Butyl 2-[(4-Methylphenyl)methyl]-1-oxo-2,3-dihydro-1H-indene-2-carboxylate (5)
3.2.12. Tert-Butyl 2-[(4-Chlorophenyl)methyl]-1-oxo-2,3-dihydro-1H-indene-2-carboxylate (6)
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Corey, E.J.; Guzman-Perez, A. The Catalytic Enantioselective Construction of Molecules with Quaternary Carbon Stereocenters. Angew. Chem. Int. Ed. 1998, 37, 388–401. [Google Scholar] [CrossRef]
- Christoffers, J.; Mann, A. Enantioselective Construction of Quaternary Stereocenters. Angew. Chem. Int. Ed. 2001, 40, 4591–4597. [Google Scholar] [CrossRef]
- Wu, G.; Xu, H.; Liu, Z.; Liu, Y.; Yang, X.; Zhang, X.; Huang, Y. Asymmetric Organocatalysis Combined with Palladium Catalysis: Synergistic Effect on Enantioselective Mannich/α-Allylation Sequential Reactions of Pyrazolones in Constructing Vicinal Quaternary Stereocenters. Org. Lett. 2019, 21, 7708–7712. [Google Scholar] [CrossRef] [PubMed]
- Sansinenea, E.; Martinez, E.F.; Ortiz, A. Organocatalytic Synthesis of Chiral Spirooxindoles with Quaternary Stereogenic Centers. Eur. J. Org. Chem. 2020, 2020, 5101–5118. [Google Scholar] [CrossRef]
- Wang, K.; Yu, J.; Shao, Y.; Tang, S.; Sun, J. Forming All-Carbon Quaternary Stereocenters by Organocatalytic Aminomethylation: Concise Access to β2,2-Amino Acids. Angew. Chem. Int. Ed. 2020, 59, 23516–23520. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.-J.; Zhu, Y.-N.; Ye, J.-L.; Huang, P.-Q. A versatile approach to functionalized cyclic ketones bearing quaternary carbon stereocenters via organocatalytic asymmetric conjugate addition of nitroalkanes to cyclic β-substituted α,β-Enones. Tetrahedron 2021, 84, 132005. [Google Scholar] [CrossRef]
- Vetica, F.; Chauhan, P.; Dochain, S.; Enders, D. Asymmetric organocatalytic methods for the synthesis of tetrahydropyrans and their application in total synthesis. Chem. Soc. Rev. 2017, 46, 1661–1674. [Google Scholar] [CrossRef]
- Xie, X.; Huang, W.; Peng, C.; Han, B. Organocatalytic Asymmetric Synthesis of Six-Membered Carbocycle-Based Spiro Compounds. Adv. Synth. Catal. 2018, 360, 194–228. [Google Scholar] [CrossRef]
- Lavios, A.; Sanz-Marco, A.; Vila, C.; Gonzalo, B.; Pedro, J.R. Asymmetric Organocatalytic Synthesis of aza-Spirocyclic Compounds from Isothiocyanates and Isocyanides. Eur. J. Org. Chem. 2021, 2021, 2268–2284. [Google Scholar] [CrossRef]
- Parella, R.; Jakkampudi, S.; Zhao, J.C.-G. Recent Applications of Asymmetrisc Organocatalytic Methods in Total Synthesis. ChemistrySelect 2021, 6, 2252–2280. [Google Scholar] [CrossRef]
- Tu, M.-S.; Chen, K.-W.; Wu, P.; Zhang, Y.-C.; Liu, X.-Q.; Shi, F. Advances in organocatalytic asymmetric reactions of vinylindoles: Powerful access to enantioenriched indole derivatives. Org. Chem. Front. 2021, 8, 2643–2672. [Google Scholar] [CrossRef]
- Xu, P.-W.; Yu, J.-S.; Chen, C.; Cao, Z.-Y.; Zhou, F.; Zhou, J. Catalytic Enantioselective Construction of Spiro Quaternary Carbon Stereocenters. ACS Catal. 2019, 9, 1820–1882. [Google Scholar] [CrossRef]
- Zhu, T.; Liu, Y.; Smetankova, M.; Zhuo, S.; Chi, Y.R.; Zhu, T.; Mou, C.; Chai, H.; Jin, Z. Carbene-Catalyzed Desymmetrization and Direct Construction of Arenes with All-Carbon Quaternary Chiral Center. Angew. Chem. Int. Ed. 2019, 58, 15778–15782. [Google Scholar] [CrossRef]
- Wang, Z.; Liu, J. All-carbon [3 + 2] cycloaddition in natural product synthesis. Beilstein J. Org. Chem. 2020, 16, 3015–3031. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z. Construction of all-carbon quaternary stereocenters by catalytic asymmetric conjugate addition to cyclic enones in natural product synthesis. Org. Chem. Front. 2020, 7, 3815–3841. [Google Scholar] [CrossRef]
- Wang, Z.; Yang, Z.-P.; Fu, G.C. Quaternary stereocentres via catalytic enantioconvergent nucleophilic substitution reactions of tertiary alkyl halides. Nat. Chem. 2021, 13, 236–242. [Google Scholar] [CrossRef]
- Xin, Z.; Wang, H.; He, H.; Gao, S. Recent advances in the total synthesis of natural products bearing the contiguous all-carbon quaternary stereocenters. Tetrahedron 2021, 71, 153029. [Google Scholar] [CrossRef]
- Benetti, S.; Romagnoli, R.; De Risi, C.; Spalluto, G.; Zanirato, V. Mastering. beta.-Keto Esters. Chem. Rev. 1995, 95, 1065–1114. [Google Scholar] [CrossRef]
- Kallepu, S.; Sanjeev, K.; Chegondi, R.; Mainkar, P.S.; Chandrasekhar, S. Benzyne Insertion onto β-Keto Esters of Polycyclic Natural Products: Synthesis of Benzo Octacyclo Scaffolds. Org. Lett. 2018, 20, 7121–7124. [Google Scholar] [CrossRef]
- Kazmierczak, J.C.; Cargnelutti, R.; Barcellos, T.; Silveira, C.C.; Schumacher, R.F. Selective synthesis of α-organylthio esters and α-organylthio ketones from β-keto esters and sodium S-organyl sulfurothioates under basic conditions. Beilstein J. Org. Chem. 2021, 17, 234–244. [Google Scholar] [CrossRef]
- Corral-Bautista, F.; Mayr, H. Quantification of the Nucleophilic Reactivities of Cyclic β-Keto Ester Anions. Eur. J. Org. Chem. 2015, 2015, 7594–7601. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, S.; Gao, Q.; Li, L.; Zhi, H.; Zhang, T.; Zhang, J. Transition-metal, organic solvent and base free α-hydroxylation of β-keto esters and β-keto amides with peroxides in water. Tetrahedron 2019, 75, 3856–3863. [Google Scholar] [CrossRef]
- Wang, Y.-F.; Jiang, Z.-H.; Chu, M.-M.; Qi, S.-S.; Yin, H.; Han, H.-T.; Xu, D.-Q. Asymmetric copper-catalyzed fluorination of cyclic β-keto esters in a continuous-flow microreactor. Org. Biomol. Chem. 2020, 18, 4927–4931. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Mo, M.; Zhu, K.; Zheng, C.; Zhang, H.; Wang, W.; Shao, Z. Asymmetric synthesis of syn-propargylamines and unsaturated β-amino acids under Bronsted base catalysis. Nat. Commun. 2015, 6, 8544. [Google Scholar] [CrossRef]
- Zhang, D.; Chen, X.; Zhang, R.; Yao, P.; Wu, Q.; Zhu, D. Development of β-amino acid dehydrogenase for the synthesis of β-amino acids via reductive amination of β-keto acids. ACS Catal. 2015, 5, 2220–2224. [Google Scholar] [CrossRef]
- Asano, T.; Moritani, M.; Nakajima, M.; Kotani, S. Chiral lithium binaphtholate for enantioselective amination of acyclic α-alkyl-β-keto esters: Application to the total synthesis of L-carbidopa. Tetrahedron 2017, 73, 5975–5982. [Google Scholar] [CrossRef]
- Bryliakov, K.P. Catalytic Asymmetric Oxygenations with the Environmentally Benign Oxidants H. Chem. Rev. 2017, 117, 11406–11459. [Google Scholar] [CrossRef] [Green Version]
- Tavakolian, M.; Vahdati-Khajeh, S.; Asgari, S. Recent Advances in Solvent-Free Asymmetric Catalysis. ChemCatChem 2019, 11, 2943–2977. [Google Scholar] [CrossRef]
- Trost, B.M.; Sacchi, K.L.; Schroeder, G.M.; Asakawa, N. Intramolecular Palladium-Catalyzed Allylic Alkylation: Enantio- and Diastereoselective Synthesis of [2.2.2] Bicycles. Org. Lett. 2002, 4, 3427–3430. [Google Scholar] [CrossRef]
- Nemoto, T.; Fukuda, T.; Matsumoto, T.; Hitomi, T.; Hamada, Y. Enantioselective Construction of All-Carbon Quaternary Stereocenters Using Palladium-Catalyzed Asymmetric Allylic Alkylation of γ-Acetoxy-α,β-unsaturated Carbonyl Compounds. Adv. Synth. Catal. 2005, 347, 1504–1506. [Google Scholar] [CrossRef]
- Zhou, H.; Wang, Y.; Zhang, L.; Cai, M.; Luo, S. Enantioselective Terminal Addition to Allenes by Dual Chiral Primary Amine/Palladium Catalysis. J. Am. Chem. Soc. 2017, 139, 3631–3634. [Google Scholar] [CrossRef] [PubMed]
- Verbicky, J.W., Jr.; O’Neil, E.A. Chiral phase-transfer catalysis. Enantioselective alkylation of racemic alcohols with a nonfunctionalized optically active phase-transfer catalyst. J. Org. Chem. 1985, 50, 1786–1787. [Google Scholar] [CrossRef]
- Ooi, T.; Takeuchi, M.; Kameda, M.; Maruoka, K. Practical catalytic enantioselective synthesis of α,α-dialkyl-α-amino acids by chiral phase-transfer catalysis. J. Am. Chem. Soc. 2000, 122, 5228–5229. [Google Scholar] [CrossRef]
- Arlt, A.; Toyama, H.; Takada, K.; Hashimoto, T.; Maruoka, K. Phase-transfer catalyzed asymmetric synthesis of α,β-unsaturated γ,γ-disubstituted γ-lactams. Chem. Commun. 2017, 53, 4779–4782. [Google Scholar] [CrossRef]
- Sicignano, M.; Rodriguez, R.I.; Capaccio, V.; Borello, F.; Cano, R.; De Riccardis, F.; Bernardi, L.; Diaz-Tendero, S.; Della Sala, G.; Aleman, J. Asymmetric trifluoromethylthiolation of azlactones under chiral phase transfer catalysis. Org. Biomol. Chem. 2020, 18, 2914–2920. [Google Scholar] [CrossRef]
- Woo, S.; Kim, Y.-G.; Lim, B.; Oh, J.; Lee, Y.; Gwon, H.; Nahm, K. Dimeric cinchona ammonium salts with benzophenone linkers: Enantioselective phase transfer catalysts for the synthesis of α-amino acids. RSC Adv. 2018, 8, 2157–2160. [Google Scholar] [CrossRef] [Green Version]
- Hu, B.; Deng, L. Direct Catalytic Asymmetric Synthesis of Trifluoromethylated γ-Amino Esters/Lactones via Umpolung Strategy. J. Org. Chem. 2019, 84, 994–1005. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Liu, Y.; Wei, Z.; Cao, J.; Liang, D.; Lin, Y.; Duan, H. Synthesis of 4-Azaindolines Using Phase-Transfer Catalysis via an Intramolecular Mannich Reaction. J. Org. Chem. 2020, 85, 4047–4057. [Google Scholar] [CrossRef] [PubMed]
- Majdecki, M.; Niedbała, P.; Tyszka-Gumkowska, A.; Jurczak, J. Assisted by Hydrogen-Bond Donors: Cinchona Quaternary Salts as Privileged Chiral Catalysts for Phase-Transfer Reactions. Synthesis 2021, 53, 2777–2786. [Google Scholar]
- Corey, E.J.; Xu, F.; Noe, M.C. A Rational Approach to Catalytic Enantioselective Enolate Alkylation Using a Structurally Rigidified and Defined Chiral Quaternary Ammonium Salt under Phase Transfer Conditions. J. Am. Chem. Soc. 1997, 119, 12414–12415. [Google Scholar] [CrossRef]
- Zhang, F.-Y.; Corey, E.J. Highly Enantioselective Michael Reactions Catalyzed by a Chiral Quaternary Ammonium Salt. Illustration by Asymmetric Syntheses of (S)-Ornithine and Chiral 2-Cyclohexenones. Org. Lett. 2000, 2, 1097–1100. [Google Scholar] [CrossRef] [PubMed]
- Manabe, K. Synthesis of novel chiral quaternary phosphonium salts with a multiple hydrogen-bonding site, and their application to asymmetric phase-transfer alkylation. Tetrahedron 1998, 54, 14465–14476. [Google Scholar] [CrossRef]
- Ooi, T.; Miki, T.; Taniguchi, M.; Shiraishi, M.; Takeuchi, M.; Maruoka, K. Highly Enantioselective Construction of Quaternary Stereocenters on β-Keto Esters by Phase-Transfer Catalytic Asymmetric Alkylation and Michael Reaction. Angew. Chem. Int. Ed. 2003, 42, 3796–3798. [Google Scholar] [CrossRef] [PubMed]
- Dehmlow, E.V.; Düttmann, S.; Neumann, B.; Stammler, H.-G. Monodeazacinchona Alkaloid Derivatives: Synthesis and Preliminary Applications as Phase-Transfer Catalysts. Eur. J. Org. Chem. 2002, 2002, 2087–2093. [Google Scholar] [CrossRef]
- Tarí, S.; Chinchilla, R.; Nájera, C.; Yus, M. Enantioselective alkylation of β-keto esters promoted by dimeric Cinchona-derived ammonium salts as recoverable organocatalysts. Arkivoc 2011, 7, 116–127. [Google Scholar] [CrossRef] [Green Version]
- Park, E.J.; Kim, M.H.; Kim, Y. Enantioselective Alkylation of β-Keto Esters by Phase-Transfer Catalysis Using Chiral Quaternary Ammonium Salts. J. Org. Chem. 2004, 69, 6897–6899. [Google Scholar] [CrossRef]
- Wang, Y.; Li, Y.; Lian, M.; Zhang, J.; Liu, Z.; Tang, X.; Yin, H.; Meng, Q. Asymmetric α-alkylation of cyclic β-keto esters and β-keto amides by phase-transfer catalysis. Org. Biomol. Chem. 2019, 17, 573–584. [Google Scholar] [CrossRef]
- Majdecki, M.; Niedbała, P.; Jurczak, J. Amide-Based Cinchona Alkaloids as Phase-Transfer Catalysts: Synthesis and Potential Application. Org. Lett. 2019, 21, 8085–8090. [Google Scholar] [CrossRef]
- Majdecki, M.; Niedbała, P.; Jurczak, J. Synthesis of C2 Hybrid Amide-Based PTC Catalysts and Their Comparison with Saturated Analogues. ChemistrySelect 2020, 5, 6424–6429. [Google Scholar] [CrossRef]
- Majdecki, M.; Tyszka-Gumkowska, A.; Jurczak, J. Highly Enantioselective Epoxidation of α,β-Unsaturated Ketones Using Amide-Based Cinchona Alkaloids as Hybrid Phase-Transfer Catalysts. Org. Lett. 2020, 22, 8687–8691. [Google Scholar] [CrossRef]
- Majdecki, M.; Grodek, P.; Jurczak, J. Stereoselective α-Chlorination of β-Keto Esters in the Presence of Hybrid Amide-Based Cinchona Alkaloids as Catalysts. J. Org. Chem. 2020, 86, 995–1001. [Google Scholar] [CrossRef] [PubMed]
- Wavefunction Inc. I.A. Spartan. 2018. Available online: www.wavefun.com (accessed on 6 March 2022).
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar]
- Kozuch, S.; Martin, J.M.L. Halogen Bonds: The Reliability of Pure and Hybrid DFT Methods Analysis. J. Chem. Theory Comput. 2013, 9, 1918–1931. [Google Scholar] [CrossRef]
- Bauzá, A.; Alkorta, I.; Frontera, A.; Elguero, J. On the Reliability of Pure and Hybrid DFT Methods for the Evaluation of Halogen, Chalcogen, and Pnicogen Bonds Involving Anionic and Neutral Electron Donors. J. Chem. Theory Comput. 2013, 9, 5201–5210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hohenstein, E.G.; Chill, S.T.; Sherrill, C.D. Assessment of the Performance of the M05−2X and M06−2X Exchange-Correlation Functionals for Noncovalent Interactions in Biomolecules. J. Chem. Theory Comput. 2008, 4, 1996–2000. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
 | |||||
|---|---|---|---|---|---|
| Entry | Solvent | Base | T (°C) | Yield b (%) | eec (%) |
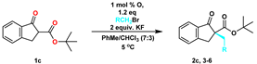
| 1 | PhMe/CHCl3 (7/3) | K2CO3 | 25 | 99 | 64 |
| 2 | PhMe/CHCl3 (7/3) | 50%aq K2CO3 | 25 | 99 | 62 |
| 3 | PhMe/CHCl3 (7/3) | KF | 25 | 99 | 67 |
| 4 | PhMe/CHCl3 (7/3) | 50%aq KF | 25 | 99 | 64 |
| 5 | PhMe/CHCl3 (7/3) | Na2CO3 | 25 | 95 | 65 |
| 6 | PhMe/CHCl3 (7/3) | 50%aq Na2CO3 | 25 | 94 | 61 |
| 7 | PhMe | KF | 25 | 99 | 67 |
| 8 | m-Xylene | KF | 25 | 99 | 66 |
| 9 | CH2Cl2 | KF | 25 | 99 | 66 |
| 10 | CHCl3 | KF | 25 | 99 | 65 |
| 11 | PhMe/CHCl3 (7/3) | KF | 10 | 99 | 71 |
| 12 | PhMe/CHCl3 (7/3) | KF | 5 | 99 | 72 |
| Entry | Catalyst | Time (h) | Yield b (%) | eec (%) | |||
|---|---|---|---|---|---|---|---|
| 1 | A | 5 | 99 | 60 | |||
| 2 | B | 6 | 98 | 55 | |||
| 3 | C | 5 | 99 | 52 | |||
| 4 | D | 5 | 99 | 60 | |||
| 5 | E | 5 | 99 | 61 | |||
| 6 | F | 4 | 99 | 64 | |||
| 7 | G | 5 | 98 | 31 | |||
| 8 | H | 6 | 96 | 49 | |||
| 9 | I | 7 | 98 | 42 | |||
| 10 | J | 7 | 97 | 56 | |||
| 11 | K | 6 | 98 | 57 | |||
| 12 | L | 4 | 99 | 68 | |||
| 13 | M | 4 | 99 | 70 | |||
| 14 | N | 4 | 99 | 73 | |||
| 15 | O | O’ | 3 | 99 | 99 | 80 | −84 |
 | |||
|---|---|---|---|
| Entry | Substrate | Yield b [%] | eec [%] |
| 1 |  | 99 | 80 |
| 2 |  | 98 | 85 |
| 3 | 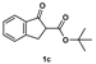 | 99 | 91 |
| 4 |  | 99 | 61 |
| 5 | 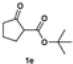 | 96 | 74 |
 | |||
|---|---|---|---|
| Entry | Substrate | Yield b (%) | ee c (%) |
| 1 |  | 99 | 91 |
| 2 | 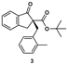 | 98 | 90 |
| 3 |  | 97 | 91 |
| 4 |  | 98 | 89 |
| 5 |  | 98 | 88 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Niedbała, P.; Majdecki, M.; Grodek, P.; Jurczak, J. H-Bond Mediated Phase-Transfer Catalysis: Enantioselective Generating of Quaternary Stereogenic Centers in β-Keto Esters. Molecules 2022, 27, 2508. https://doi.org/10.3390/molecules27082508
Niedbała P, Majdecki M, Grodek P, Jurczak J. H-Bond Mediated Phase-Transfer Catalysis: Enantioselective Generating of Quaternary Stereogenic Centers in β-Keto Esters. Molecules. 2022; 27(8):2508. https://doi.org/10.3390/molecules27082508
Chicago/Turabian StyleNiedbała, Patryk, Maciej Majdecki, Piotr Grodek, and Janusz Jurczak. 2022. "H-Bond Mediated Phase-Transfer Catalysis: Enantioselective Generating of Quaternary Stereogenic Centers in β-Keto Esters" Molecules 27, no. 8: 2508. https://doi.org/10.3390/molecules27082508
APA StyleNiedbała, P., Majdecki, M., Grodek, P., & Jurczak, J. (2022). H-Bond Mediated Phase-Transfer Catalysis: Enantioselective Generating of Quaternary Stereogenic Centers in β-Keto Esters. Molecules, 27(8), 2508. https://doi.org/10.3390/molecules27082508