
The Influence of Ionic Liquids Adsorption on the Electronic and Optical Properties of Phosphorene and Arsenene with Different Phases: A Computational Study


Abstract
:
1. Introduction
2. Results and Discussion
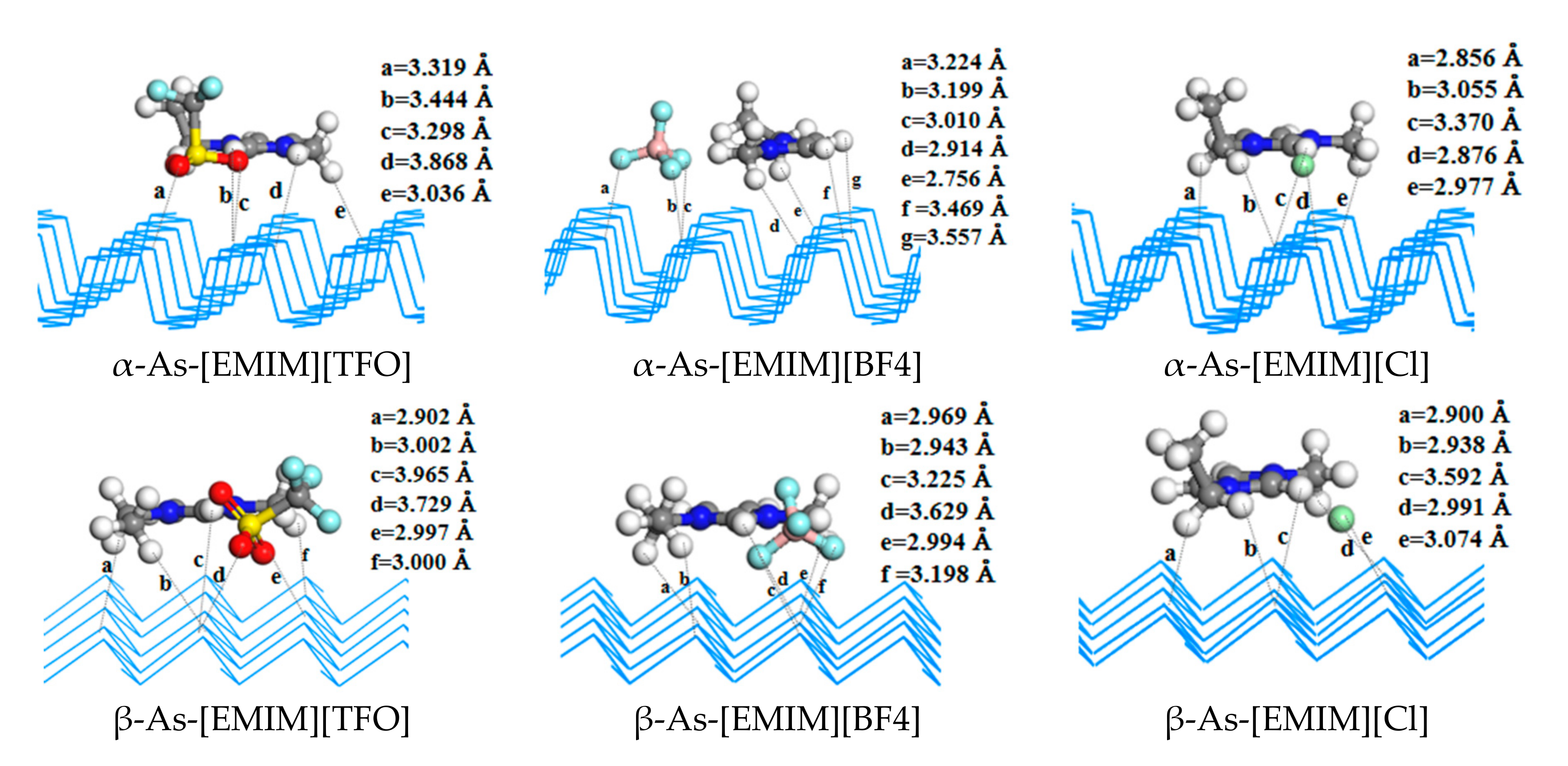
2.1. Structures and Energies of IL/2D Sheets
2.2. Valence Band Maximum, Conduction Band Minimum, and Density of States of IL/2D Sheets
2.3. Charge Transfer between Ionic Liquids and 2D Sheets
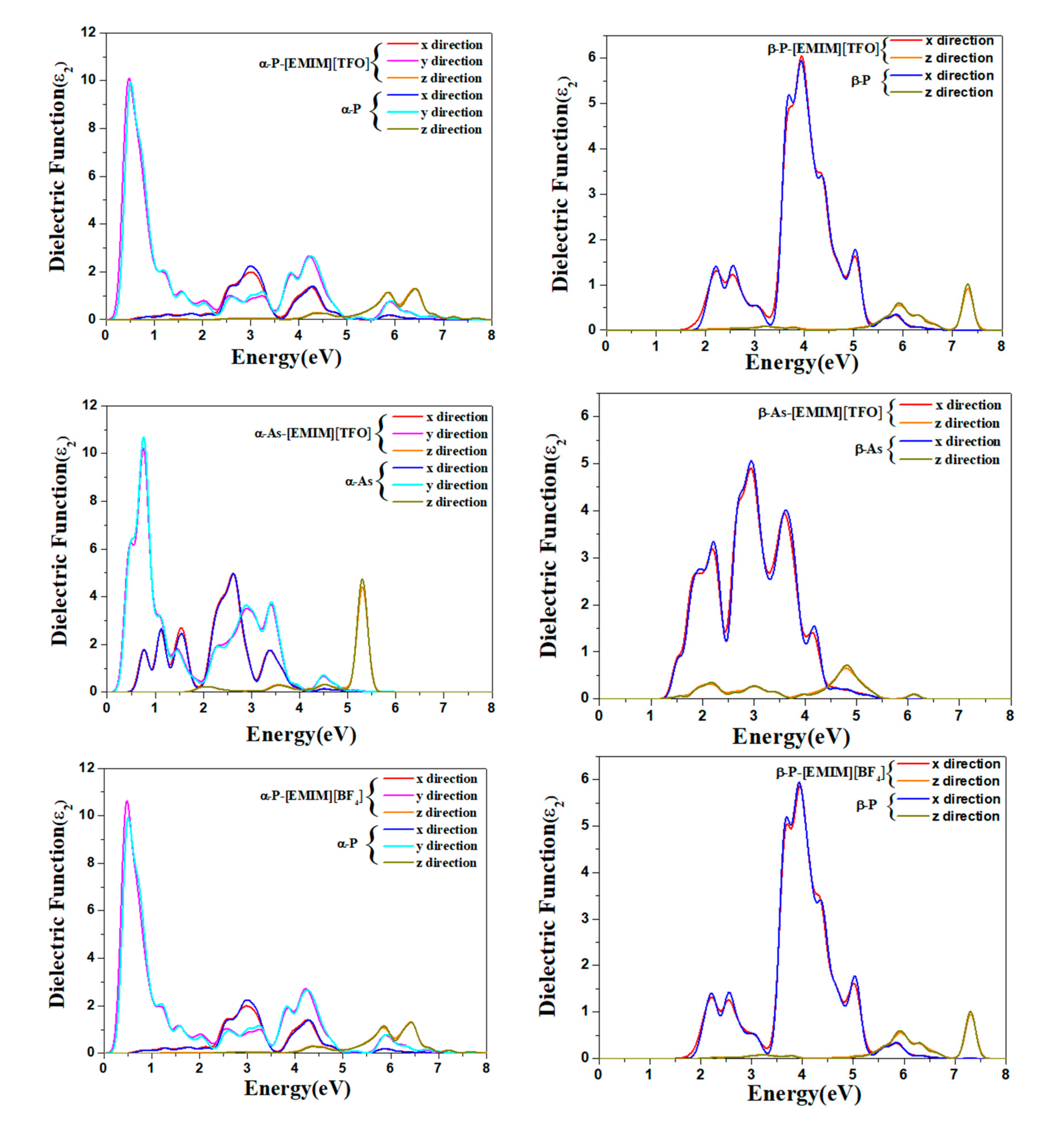
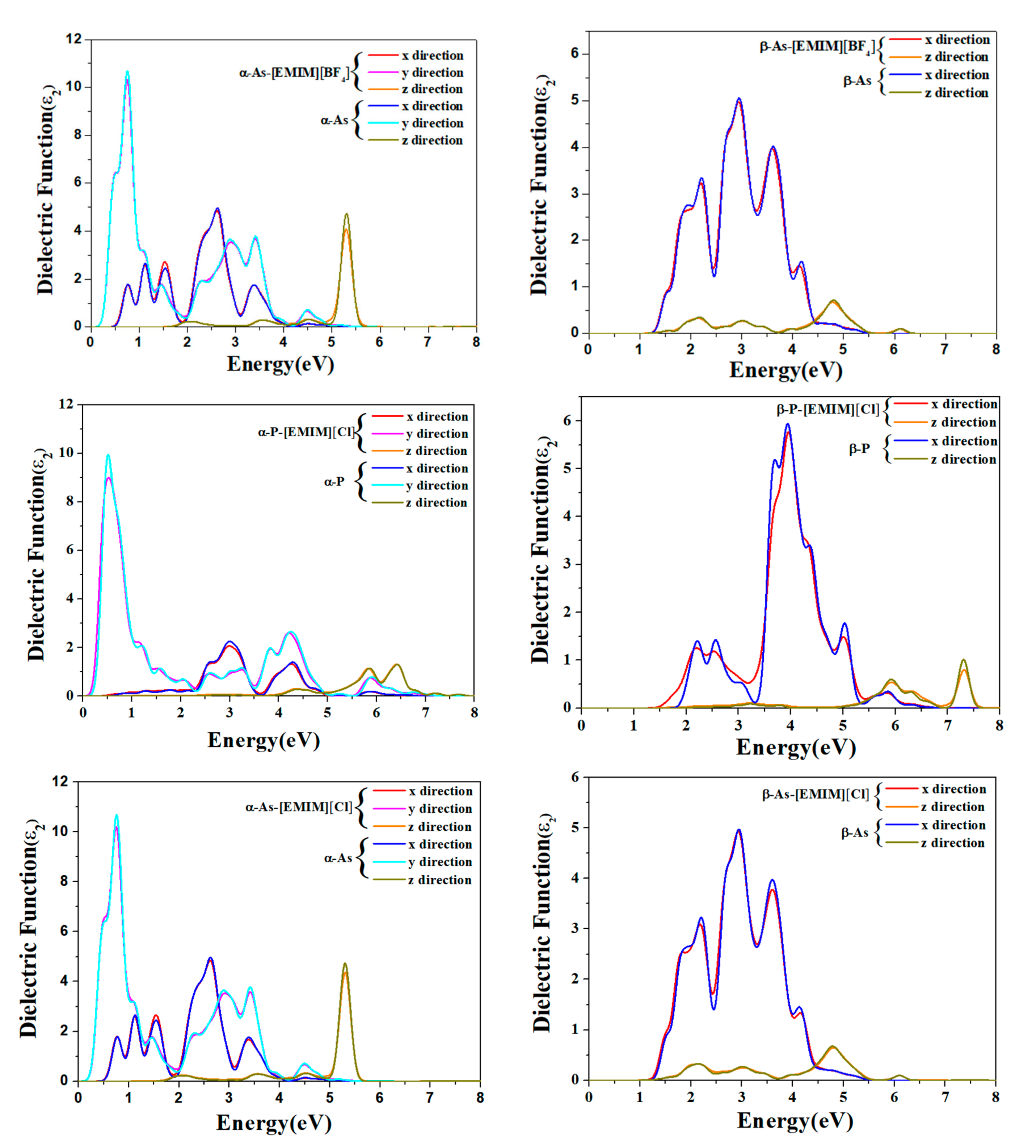
2.4. Optical Properties
3. Computational Details
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tan, C.; Cao, X.; Wu, X.J.; He, Q.; Yang, J.; Zhang, X.; Chen, J.; Zhao, W.; Han, S.; Nam, G.H.; et al. Recent Advances in Ultrathin Two-Dimensional Nanomaterials. Chem. Rev. 2017, 117, 6225–6331. [Google Scholar] [CrossRef]
- Zhang, S.; Xie, M.; Li, F.; Yan, Z.; Li, Y.; Kan, E.; Liu, W.; Chen, Z.; Zeng, H. Semiconducting Group 15 Monolayers: A Broad Range of Band Gaps and High Carrier Mobilities. Angew. Chem. Int. Ed. Engl. 2016, 55, 1666–1669. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Yan, Z.; Li, Y.; Chen, Z.; Zeng, H. Atomically Thin Arsenene and Antimonene: Semimetal-Semiconductor and Indirect-direct Band-gap Transitions. Angew. Chem. Int. Ed. Engl. 2015, 54, 3112–3115. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.Y.; Hu, X.M.; Zhou, W.H.; Liu, X.H.; Gao, Y.J.; Zhang, S.L.; Zhang, K.; Zhu, Z.; Zeng, H.B. Mechanistic Understanding of Two-Dimensional Phosphorus, Arsenic, and Antimony High-Capacity Anodes for Fast-Charging Lithium/Sodium Ion Batteries. J. Phys. Chem. C 2018, 122, 29559–29566. [Google Scholar] [CrossRef]
- Kong, X.; Liu, Q.; Zhang, C.; Peng, Z.; Chen, Q. Elemental Two-Dimensional nanosheets beyond graphene. Chem. Soc. Rev. 2017, 46, 2127–2157. [Google Scholar] [CrossRef]
- Ambrosi, A.; Chua, C.K.; Bonanni, A.; Pumera, M. Electrochemistry of Graphene and Related Materials. Chem. Rev. 2014, 114, 7150–7188. [Google Scholar] [CrossRef]
- Yu, X.; Liang, W.Y.; Xing, C.Y.; Chen, K.Q.; Chen, J.M.; Huang, W.C.; Xie, N.; Qiu, M.; Yan, X.B.; Xie, Z.J.; et al. Emerging 2D Pnictogens for catalytic applications: Status and Challenges. J. Mater. Chem. A 2020, 8, 12887–12927. [Google Scholar] [CrossRef]
- Huo, C.; Yan, Z.; Song, X.; Zeng, H. 2D Materials via Liquid Exfoliation: A Review on Fabrication and Applications. Sci. Bull. 2015, 60, 1994–2018. [Google Scholar] [CrossRef]
- Gui, R.J.; Jin, H.; Sun, Y.J.; Jiang, X.W.; Sun, Z.J. Two-Dimensional Group-VA Nanomaterials beyong Black Phosphorus: Synthetic Mathods, Properties, Functional Nanostructures and Applications. J. Mater. Chem. A 2019, 7, 25712–25771. [Google Scholar] [CrossRef]
- Wu, Z.; Qi, J.; Wang, W.; Zeng, Z.; He, Q. Emerging elemental two-dimensional materials for energy applications. J. Mater. Chem. A 2021, 9, 18793–18817. [Google Scholar] [CrossRef]
- Zhang, S.; Guo, S.; Chen, Z.; Wang, Y.; Gao, H.; Gomez-Herrero, J.; Ares, P.; Zamora, F.; Zhu, Z.; Zeng, H. Recent Progress in 2D Group-VA Semiconductors: From Theory to Experiment. Chem. Soc. Rev. 2018, 47, 982–1021. [Google Scholar] [CrossRef] [Green Version]
- Jellett, C.; Plutnar, J.; Pumera, M. Prospects for Functionalizing Elemental 2D Pnictogens: A Study of Molecular Models. ACS Nano 2020, 14, 7722–7733. [Google Scholar] [CrossRef]
- Esan, F.; Kecik, D.; Özçelik, V.O.; Kadioglu, Y.; Üzengi Aktürk, O.; Durgun, E.; Aktürk, E.; Ciraci, S. Two-dimensional pnictogens: A review of recent progresses and future research directions. Appl. Phys. Rev. 2019, 6, 021308. [Google Scholar] [CrossRef]
- Walia, S.; Balendhran, S.; Ahmed, T.; Singh, M.; El-Badawi, C.; Brennan, M.D.; Weerathunge, P. Ambient Protection of Few-Layer Black Phosphorus via Sequestration of Reactive Oxygen Species. Adv. Mater. 2017, 29, 1700152. [Google Scholar] [CrossRef]
- Sui, Y.; Zhou, J.; Wang, X.; Wu, L.; Zhong, S.; Li, Y. Recent Advances in Black-Phosphorus-Based Materials for electrochemical Energy Storage. Mater. Today 2021, 42, 117–136. [Google Scholar] [CrossRef]
- Zhu, Z.; Tomannek, D. Semiconducting Layered Blue Phosphorus: A Computational Study. Phys. Rev. Lett. 2014, 112, 176802. [Google Scholar] [CrossRef]
- Zhang, S.; Zhang, J.; Zhang, Y.; Deng, Y. Nanoconfined Ionic Liquids. Chem. Rev. 2017, 117, 6755–6833. [Google Scholar] [CrossRef]
- Fedorov, M.V.; Kornyshev, A.A. Ionic Liquids at Electrified Interfaces. Chem. Rev. 2014, 114, 2978–3036. [Google Scholar] [CrossRef] [Green Version]
- Valencia, H.; Kohyama, M.; Tanaka, S.; Matsumoto, H. Ab intio Study of EMIM-BF4 Crystal Interaction with a Li (100) surface as a Model for Ionic Liquid/Li Interfaces in Li-ion Batteries. J. Chem. Phys. 2009, 131, 244705. [Google Scholar] [CrossRef]
- Lahiri, A.; Borisenko, N.; Endres, F. Electrochemical Synthesis of Battery Electrode Materials from Ionic Liquids. Top. Curr. Chem. 2018, 376, 55–83. [Google Scholar]
- Atilhan, M.; Altamash, T.; Aparicio, S. Quantum Chemistry Insight into the Interactions Between Deep Eutectic Solvents and SO2. Molecules 2019, 24, 2963. [Google Scholar] [CrossRef] [Green Version]
- Hayes, R.; Warr, G.G.; Atkin, R. Structure and Nanostructure in Ionic Liquids. Chem. Rev. 2015, 115, 6357–6426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, M.; Li, S.; Feng, G. The Influence of Anion Shape on the Electrical Double Layer Microstructure and Capacitance of Ionic Liquids-Based Supercapacitors by Molecular Simulations. Molecules 2017, 22, 241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, K.; Liu, X.; Dong, H.; Zhang, X.; Zhang, S. Multiscale Studies on Ionic Liquids. Chem. Rev. 2017, 117, 6636–6695. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, M.; Thomas, M.L.; Zhang, S.; Ueno, K.; Yasuda, T.; Dokko, K. Application of Ionic Liquids to Energy Storage and Conversion Materials and Devices. Chem. Rev. 2021, 40, 402–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, L.; Geng, C.; Zhang, D.; Lan, Z.; Liu, C. Atomic Resolution Insights into the Structural Aggregations and Optical Properties of Neat Imidazolium-Based Ionic Liquids. J. Phys. Chem. B 2016, 120, 6721–6729. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Qin, L.; Mu, T.; Xue, Z.; Gao, G. Are Ionic Liquids Chemically Stable? Chem. Rev. 2017, 117, 7113–7131. [Google Scholar] [CrossRef] [PubMed]
- García, G.; Atilhan, M.; Aparicio, S. Theoretical Study on the Solvation of C60 Fullerene by Ionic Liquids II: DFT Analysis of the Interaction Mechanism. J. Phys. Chem. B 2014, 118, 11330–11340. [Google Scholar] [CrossRef]
- Garcia, G.; Atilhan, M.; Aparicio, S. Theoretical Study of Renewable Ionic Liquids in the Pure State and with Graphene and Carbon Nanotubes. J. Phys. Chem. B 2015, 119, 12224–12237. [Google Scholar] [CrossRef]
- Garcia, G.; Atilhan, M.; Aparicio, S. Adsorption of Choline Benzoate Ionic Liquid on Graphene, Silicene, Germanene and Boron-Nitride Nanosheets: A DFT Perspective. Phys. Chem. Chem. Phys. 2015, 17, 16315–16326. [Google Scholar] [CrossRef] [Green Version]
- Garcia, G.; Atilhan, M.; Aparicio, S. In Silico Rational Design of Ionic Liquids for the Exfoliation and Dispersion of Boron Nitride Nanosheets. Phys. Chem. Chem. Phys. 2016, 18, 1212–1224. [Google Scholar] [CrossRef]
- Bari, R.; Tamas, G.; Irin, F.; Aquino, A.J.A.; Green, M.J.; Quitevis, E.L. Direct Exfoliation of Graphene in Ionic Liquids with aromatic groups. Colloid Surf. A-Physicochem. Eng. Asp. 2014, 463, 63–69. [Google Scholar] [CrossRef]
- Shakourian-Fard, M.; Kamath, G.; Jamshidi, Z. Trends in Physisorption of Ionic Liquids on Boron-Nitride Sheets. J. Phys. Chem. C 2014, 118, 26003–26016. [Google Scholar] [CrossRef]
- Shakourian-Fard, M.; Jamshidi, Z.; Bayat, A.; Kamath, G. Meta-Hybrid Density Functional Theory Study of Adsorption of Imidazolium- and Ammonium-Based Ionic Liquids on Graphene Sheet. J. Phys. Chem. C 2015, 119, 7095–7108. [Google Scholar] [CrossRef]
- Shakourian-Fard, M.; Taimoory, S.M.; Semeniuchenko, V.; Kamath, G.; Trant, J.F. The effect of ionic liquid adsorption on the electronic and optical properties of flfluorographene nanosheets. J. Mol. Liq. 2018, 268, 206–214. [Google Scholar] [CrossRef]
- Shakourian-Fard, M.; Ghenaatian, H.R.; Kamath, G.; Taimoory, S.M. Unraveling the effect of nitrogen doping on graphene nanoflflakes and the adsorption properties of ionic liquids: A DFT study. J. Mol. Liq. 2020, 312, 113400. [Google Scholar] [CrossRef]
- Zhao, W.; Xue, Z.; Wang, J.; Jiang, J.; Zhao, X.; Mu, T. Large-Scale, Highly Efficient, and Green Liquid-Exfoliation of Black Phosphorus in Ionic Liquids. ACS Appl. Mater. Interfaces 2015, 7, 27608–27612. [Google Scholar] [CrossRef]
- Herrera, C.; Alcalde, R.; Atilhan, M.; Aparicio, S. Theoretical Study on Amino Acid-Based Ionic Pairs and Their Interaction with Carbon Nanostructures. J. Phys. Chem. C 2014, 118, 9741–9757. [Google Scholar] [CrossRef]
- Ruzanov, A.; Lembinen, M.; Ers, H.; García de la Vega, J.M.; Lage-Estebanez, I.; Lust, E.; Ivanistsev, V.B. Density Functional Theory Study of Ionic Liquid Adsorption on Circumcoronene Shaped Graphene. J. Phys. Chem. C 2018, 122, 2624–2631. [Google Scholar] [CrossRef] [Green Version]
- He, Y.; Guo, Y.; Yan, F.; Yu, T.; Liu, L.; Zhang, X.; Zheng, T. Density functional theory study of adsorption of ionic liquids on graphene oxide surface. Chem. Eng. Sci. 2021, 245, 116946. [Google Scholar] [CrossRef]
- Ghatee, M.H.; Moosavi, F. Physisorption of Hydrophobic and Hydrophilic 1-Alkyl-3-methylimidazolium Ionic Liquids on the Graphenes. J. Phys. Chem. C 2011, 115, 5626–5636. [Google Scholar] [CrossRef]
- Lalitha, M.; Lakshmipathi, S. Interface energetics of [Emim]+[X]− and [Bmim]+[X]− (X = BF4, Cl, PF6, TfO, Tf2N) based ionic liquids on graphene, defective graphene, and graphyne surfaces. J. Mol. Liq. 2017, 236, 124–134. [Google Scholar] [CrossRef]
- Talaei, R.; Khalili, B.; Mokhtary, M. Modulation of opto-electronic properties of the functionalized hexagonal boron nitride nanosheets with tunable aryl alkyl ionic liquids (TAAILs): Defect based analysis. J. Mol. Liq. 2020, 304, 112696. [Google Scholar] [CrossRef]
- Zhour, K.; Otero-Mato, J.M.; Hassan, F.E.H.; Fahs, H.; Vaezzadeh, M.; López-Lago, E.; Gallego, L.J.; Varela, L.M. Electronic and optical properties of borophene and graphene with an adsorbed ionic liquid: A density functional theory study. J. Mol. Liq. 2020, 316, 113803. [Google Scholar] [CrossRef]
- Lu, Y.; Xu, Y.; Lu, L.; Xu, Z.; Liu, H. Interfacial interactions and structures of protic ionic liquids on a graphite surface: A first-principles study and comparison with aprotic ionic liquids. Phys. Chem. Chem. Phys. 2021, 23, 18338–18348. [Google Scholar] [CrossRef]
- Anderson, E.; Grozovski, V.; Siinor, L.; Siimenson, C.; Ivaništšev, V.; Lust, K.; Kallip, S.; Lust, E. Influence of the electrode potential and in situ STM scanning conditionson the phase boundary structure of the single crystal Bi(111)|1-butyl-4-methylpyridinium tetrafluoroborate interface. J. Electroanal. Chem. 2013, 709, 46–56. [Google Scholar] [CrossRef]
- Siinor, L.; Siimenson, C.; Ivaništšev, V.; Lust, K.; Lust, E. Influence of cation chemical composition and structure on the double layer capacitance for Bi(111)|room temperature ionic liquid interface. J. Electroanal. Chem. 2012, 668, 30–36. [Google Scholar] [CrossRef]
- Lu, Y.; Hong, Y.; Xu, Z.; Liu, H. Interfacial interactions and structures of imidazolium-based ionic liquids on black phosphorus surface from first-principles. J. Mol. Liq. 2021, 335, 116562. [Google Scholar] [CrossRef]
- Tang, Y.; Huai, W.; Li, H.; Mao, X.; Xie, J.; Lee, J.Y.; Fu, A. Adsorption of [BF4]− Anion-Based Ionic Liquids on Phosphorene, Arsenene, and Antimonene: A Density Functional Theory Study. Int. J. Quantum Chem. 2021, 121, e26668. [Google Scholar] [CrossRef]
- Delley, B. From molecules to solids with the DMol3 approach. Chem. Phys. 2000, 113, 7756–7764. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef] [PubMed]
- López-Albarrán, P.; Navarro-Santos, P.; Garcia-Ramirez, M.A.; Ricardo-Chávez, J.L. Dibenzothiophene adsorption at boron doped carbon nanoribbons studied within density functional theory. J. Appl. Phys. 2015, 117, 234301. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Structures | VBM | CBM | Eg(△E) (eV) | △q(+)a | △q(−)b | △qsur | Ea (eV) | EaD2 (eV) |
|---|---|---|---|---|---|---|---|---|
| EMIMTFO | −0.1990 | −0.0799 | 3.24 | − | − | − | − | − |
| EMIMBF4 | −0.2497 | −0.0763 | 4.72 | − | − | − | − | − |
| EMIMCl | −0.1663 | −0.0395 | 3.45 | − | − | − | − | − |
| MPITFO | −0.1970 | −0.1499 | 1.28 | − | − | − | − | − |
| MPIBF4 | −0.2435 | −0.1372 | 2.89 | − | − | − | − | − |
| MPICl | −0.1868 | −0.0810 | 2.88 | − | − | − | − | − |
| TMATFO | −0.1980 | −0.0308 | 4.55 | − | − | − | − | − |
| TMABF4 | −0.2503 | −0.0295 | 6.01 | − | − | − | − | − |
| TMACl | −0.1521 | −0.0108 | 3.84 | − | − | − | − | − |
| α-P | −0.1819 | −0.1624 | 0.53 | − | − | − | − | − |
| β-P | −0.2241 | −0.1533 | 1.93 | − | − | − | − | − |
| α-As | −0.1552 | −0.1378 | 0.47 | − | − | − | − | − |
| β-As | −0.1936 | −0.1431 | 1.38 | − | − | − | − | − |
| α-P-EMIMTFO | −0.1796 | −0.1619 | 0.48 (−0.05) | 0.003 | 0.094 | −0.095 | 0.30 | 1.20 |
| β-P-EMIMTFO | −0.2132 | −0.1515 | 1.68 (−0.25) | −0.016 | 0.096 | −0.080 | 0.22 | 0.88 |
| α-As-EMIMTFO | −0.1549 | −0.1375 | 0.47 (0.00) | −0.040 | 0.045 | −0.007 | 0.24 | 1.31 |
| β-As-EMIMTFO | −0.1875 | −0.1428 | 1.22 (−0.16) | −0.084 | 0.060 | 0.018 | 0.31 | 1.12 |
| α-P-EMIMBF4 | −0.1798 | −0.1613 | 0.51 (−0.02) | 0.001 | 0.073 | −0.072 | 0.46 | 0.93 |
| β-P-EMIMBF4 | −0.2137 | −0.1498 | 1.74 (−0.19) | −0.018 | 0.085 | −0.067 | 0.37 | 0.83 |
| α-As-EMIMBF4 | −0.1549 | −0.1371 | 0.48 (0.01) | −0.057 | 0.067 | −0.012 | 0.31 | 1.18 |
| β-As-EMIMBF4 | −0.1892 | −0.1434 | 1.24 (−0.14) | −0.078 | 0.043 | 0.032 | 0.37 | 1.04 |
| α-P-EMIMCl | −0.1751 | −0.1577 | 0.47 (−0.06) | 0.252 | 0.108 | −0.359 | 0.86 | 1.54 |
| β-P-EMIMCl | −0.2008 | −0.1434 | 1.56 (−0.37) | 0.234 | 0.159 | −0.389 | 0.89 | 1.51 |
| α-As-EMIMCl | −0.1529 | −0.1354 | 0.48 (0.01) | 0.170 | 0.047 | −0.214 | 0.69 | 1.60 |
| β-As-EMIMCl | −0.1802 | −0.1376 | 1.16 (−0.22) | 0.160 | 0.090 | −0.250 | 0.88 | 1.52 |
| α-P-MPITFO | −0.1790 | −0.1617 | 0.47 (−0.06) | −0.016 | 0.072 | −0.058 | 0.23 | 1.12 |
| β-P-MPITFO | −0.2117 | −0.1516 | 1.64 (−0.29) | −0.014 | 0.110 | −0.096 | 0.25 | 0.87 |
| α-As-MPITFO | −0.1566 | −0.1412 | 0.42 (−0.05) | −0.168 | 0.031 | −0.139 | 0.14 | 1.12 |
| β-As-MPITFO | −0.1889 | −0.1465 | 1.15 (−0.23) | −0.103 | 0.037 | 0.065 | 0.25 | 1.04 |
| α-P-MPIBF4 | −0.1793 | −0.1627 | 0.45 (−0.08) | −0.042 | 0.082 | −0.041 | 0.29 | 1.10 |
| β-P-MPIBF4 | −0.2147 | −0.1528 | 1.69 (−0.24) | −0.021 | 0.077 | −0.057 | 0.17 | 0.72 |
| α-As-MPIBF4 | −0.1566 | −0.1407 | 0.43 (−0.04) | −0.183 | 0.043 | 0.141 | 0.27 | 1.18 |
| β-As-MPIBF4 | −0.1892 | −0.1471 | 1.15 (−0.23) | −0.134 | 0.061 | 0.074 | 0.32 | 0.99 |
| α-P-MPICl | −0.1748 | −0.1580 | 0.46 (−0.07) | 0.358 | −0.04 | −0.323 | 0.63 | 1.39 |
| β-P-MPICl | −0.2007 | −0.1487 | 1.41 (−0.52) | 0.392 | −0.012 | −0.384 | 0.59 | 1.17 |
| α-As-MPICl | −0.1539 | −0.1382 | 0.43 (−0.04) | 0.161 | −0.101 | −0.064 | 0.52 | 1.43 |
| β-As-MPICl | −0.1810 | −0.1450 | 0.98 (−0.40) | 0.270 | −0.055 | −0.214 | 0.54 | 1.22 |
| α-P-TMATFO | −0.1781 | −0.1603 | 0.49 (−0.04) | 0.027 | 0.061 | −0.083 | 0.35 | 0.98 |
| β-P-TMATFO | −0.2107 | −0.1483 | 1.70 (−0.23) | 0.004 | 0.080 | −0.083 | 0.24 | 0.74 |
| α-As-TMATFO | −0.1539 | −0.1364 | 0.48 (0.01) | −0.016 | 0.045 | −0.029 | 0.25 | 1.03 |
| β-As-TMATFO | −0.1850 | −0.1405 | 1.21 (−0.17) | −0.051 | 0.054 | −0.003 | 0.31 | 0.90 |
| α-P-TMABF4 | −0.1792 | −0.1614 | 0.49 (−0.04) | 0.006 | 0.065 | −0.072 | 0.14 | 0.75 |
| β-P-TMABF4 | −0.2124 | −0.1488 | 1.73 (−0.20) | −0.006 | 0.070 | −0.062 | 0.15 | 0.62 |
| α-As-TMABF4 | −0.1545 | −0.1369 | 0.48 (0.01) | −0.038 | 0.045 | −0.005 | 0.18 | 0.89 |
| β-As-TMABF4 | −0.1875 | −0.1416 | 1.25 (−0.13) | −0.053 | 0.038 | 0.015 | 0.24 | 0.79 |
| α-P-TMACl | −0.1729 | −0.1549 | 0.49 (−0.04) | 0.149 | 0.202 | −0.354 | 0.72 | 1.22 |
| β-P-TMACl | −0.1963 | −0.1398 | 1.54 (−0.39) | 0.148 | 0.231 | −0.380 | 0.70 | 1.13 |
| α-As-TMACl | −0.1507 | −0.1333 | 0.47 (0.00) | 0.098 | 0.129 | −0.227 | 0.48 | 1.10 |
| β-As-TMACl | −0.1770 | −0.1349 | 1.14 (−0.24) | 0.088 | 0.146 | −0.234 | 0.72 | 1.21 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, L.; Fu, A. The Influence of Ionic Liquids Adsorption on the Electronic and Optical Properties of Phosphorene and Arsenene with Different Phases: A Computational Study. Molecules 2022, 27, 2518. https://doi.org/10.3390/molecules27082518
Zhu L, Fu A. The Influence of Ionic Liquids Adsorption on the Electronic and Optical Properties of Phosphorene and Arsenene with Different Phases: A Computational Study. Molecules. 2022; 27(8):2518. https://doi.org/10.3390/molecules27082518
Chicago/Turabian StyleZhu, Lin, and Aiping Fu. 2022. "The Influence of Ionic Liquids Adsorption on the Electronic and Optical Properties of Phosphorene and Arsenene with Different Phases: A Computational Study" Molecules 27, no. 8: 2518. https://doi.org/10.3390/molecules27082518