
Identification of Novel Covalent XPO1 Inhibitors Based on a Hybrid Virtual Screening Strategy

Abstract
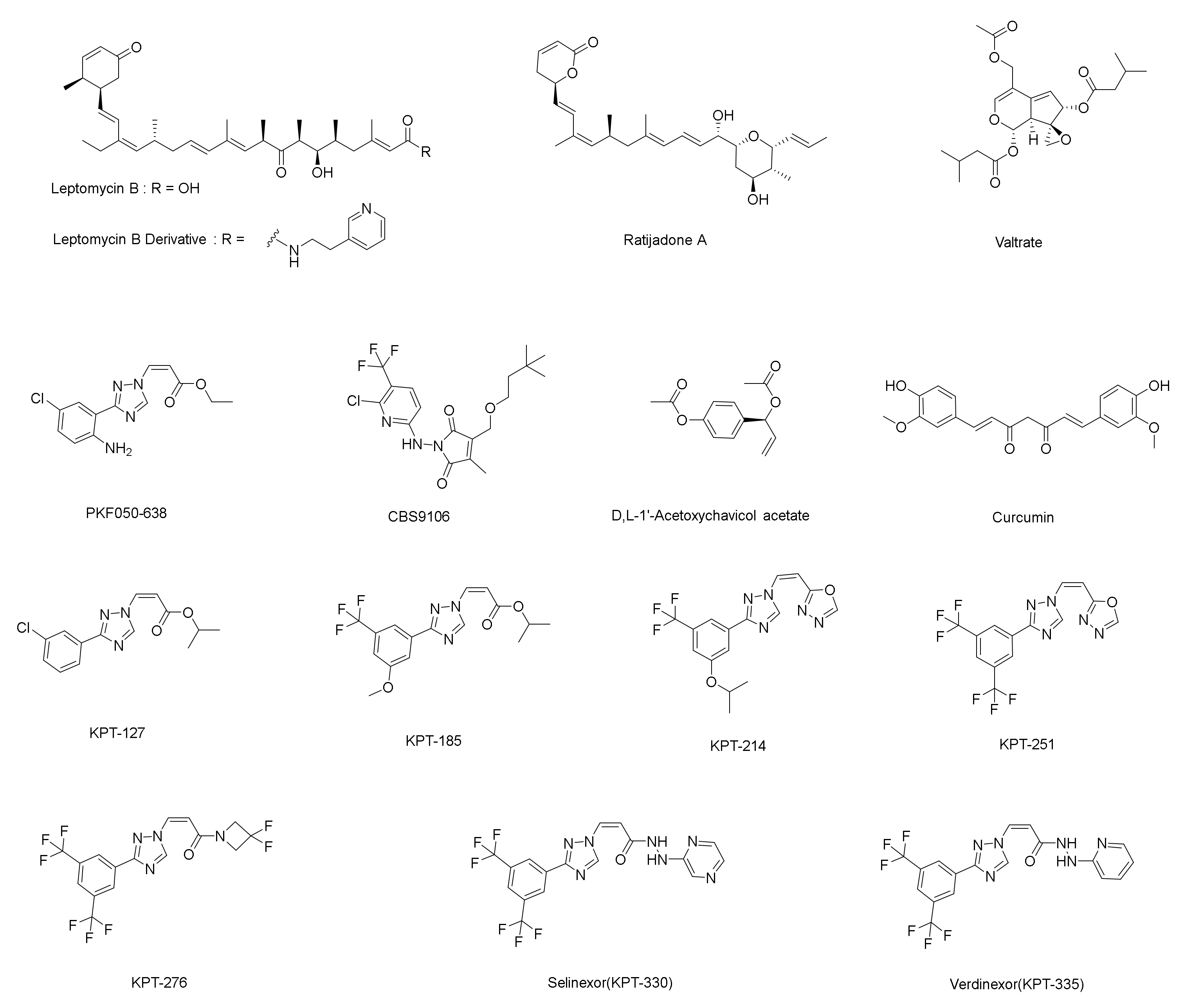
:1. Introduction
2. Results
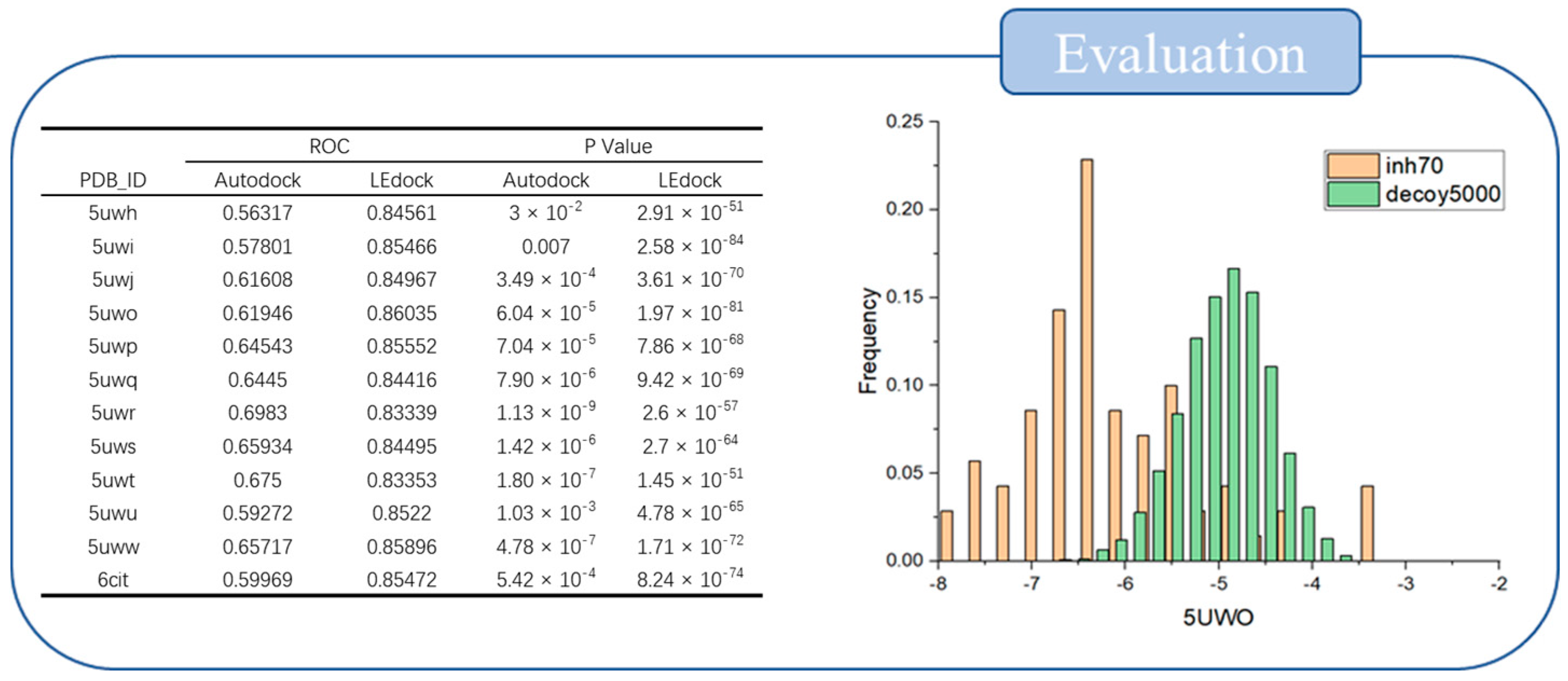
2.1. Evaluation of Screening Method
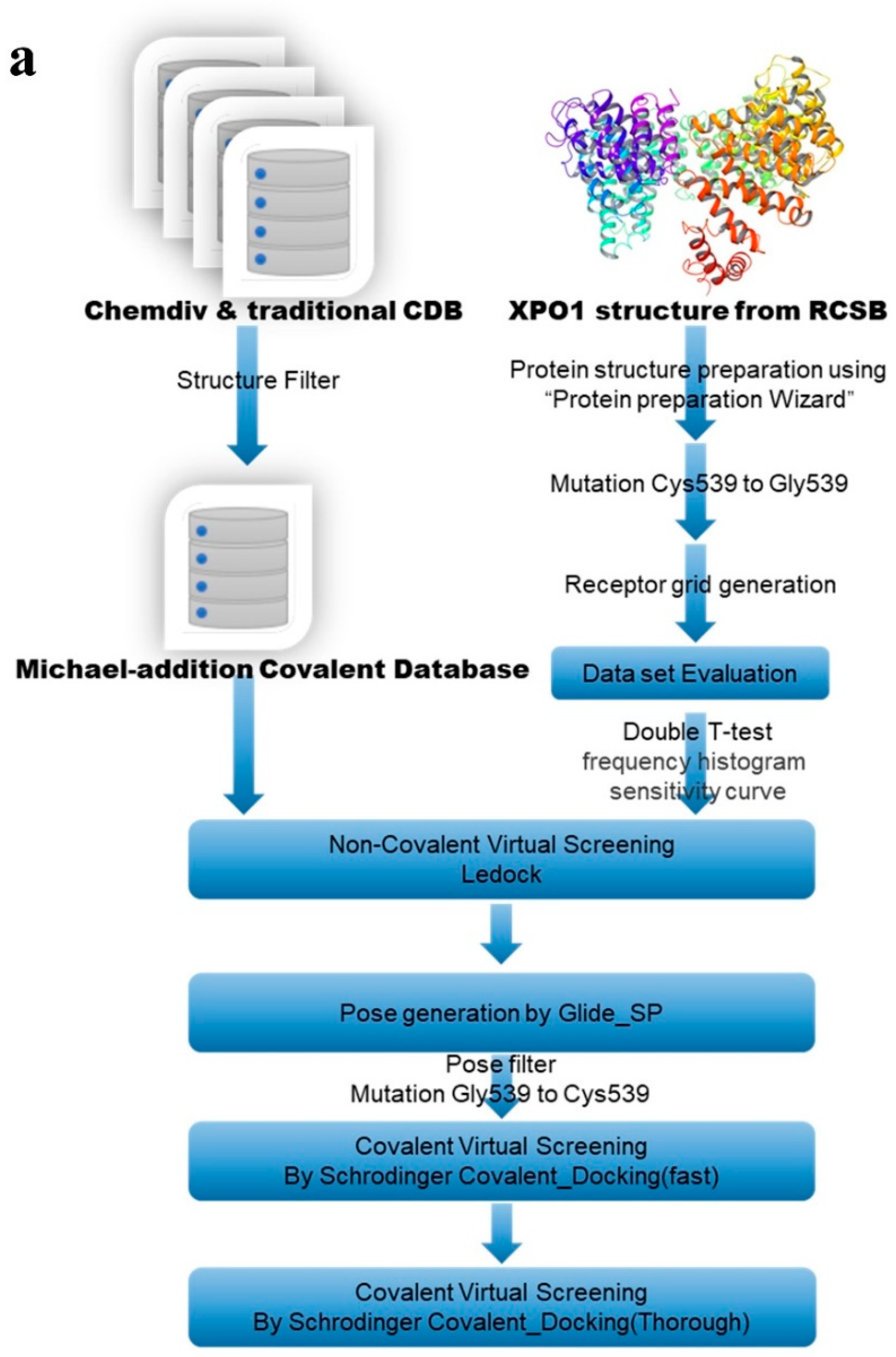
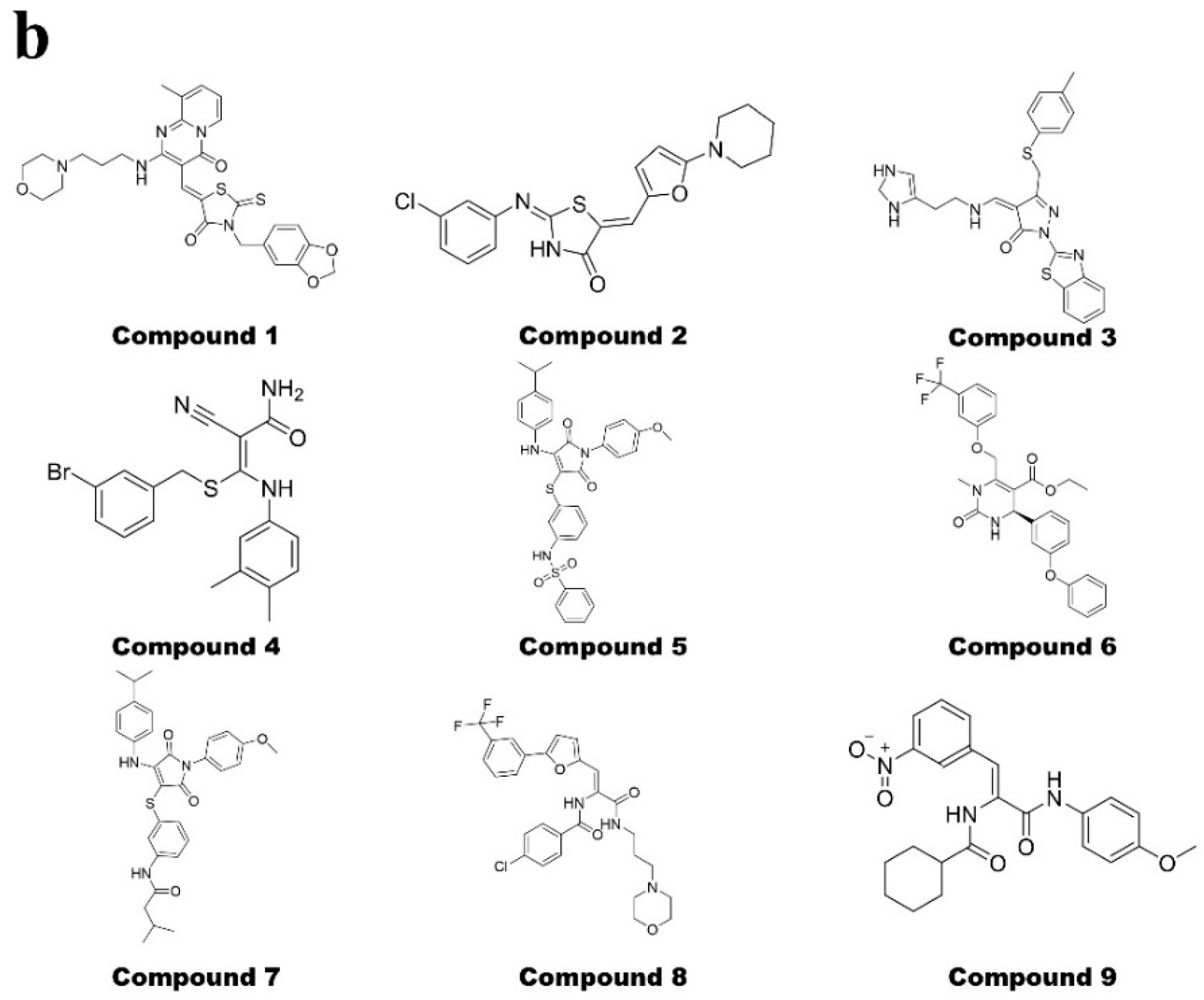
2.2. Hybrid Virtual Screening Process for XPO1 Covalent Inhibitor Discovery
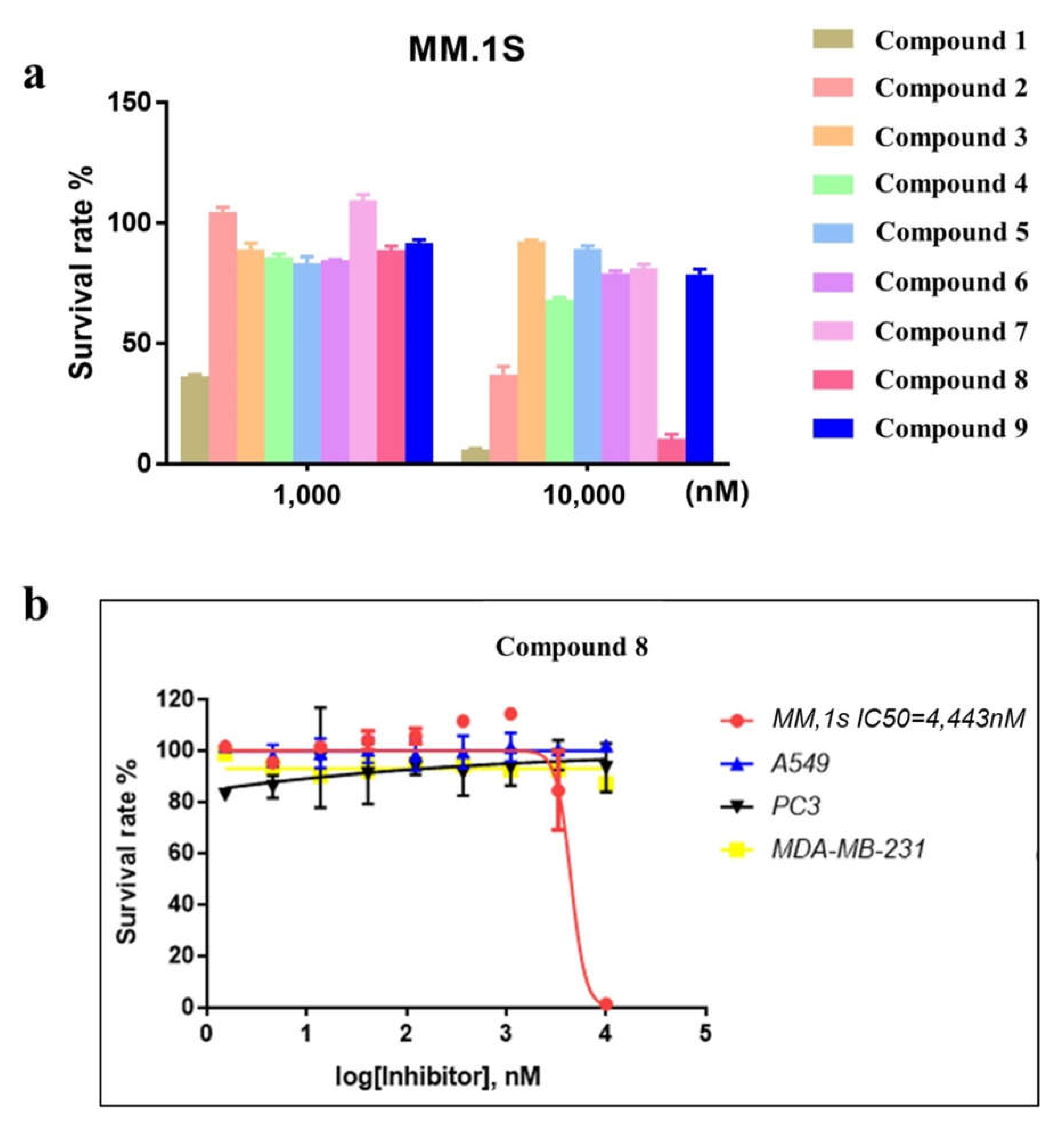
2.3. Compound 8 Shows Promising Anti-Proliferation Activity
2.4. Compound 8 Shows a Moderate Inhibitory Effect on Nuclear Export
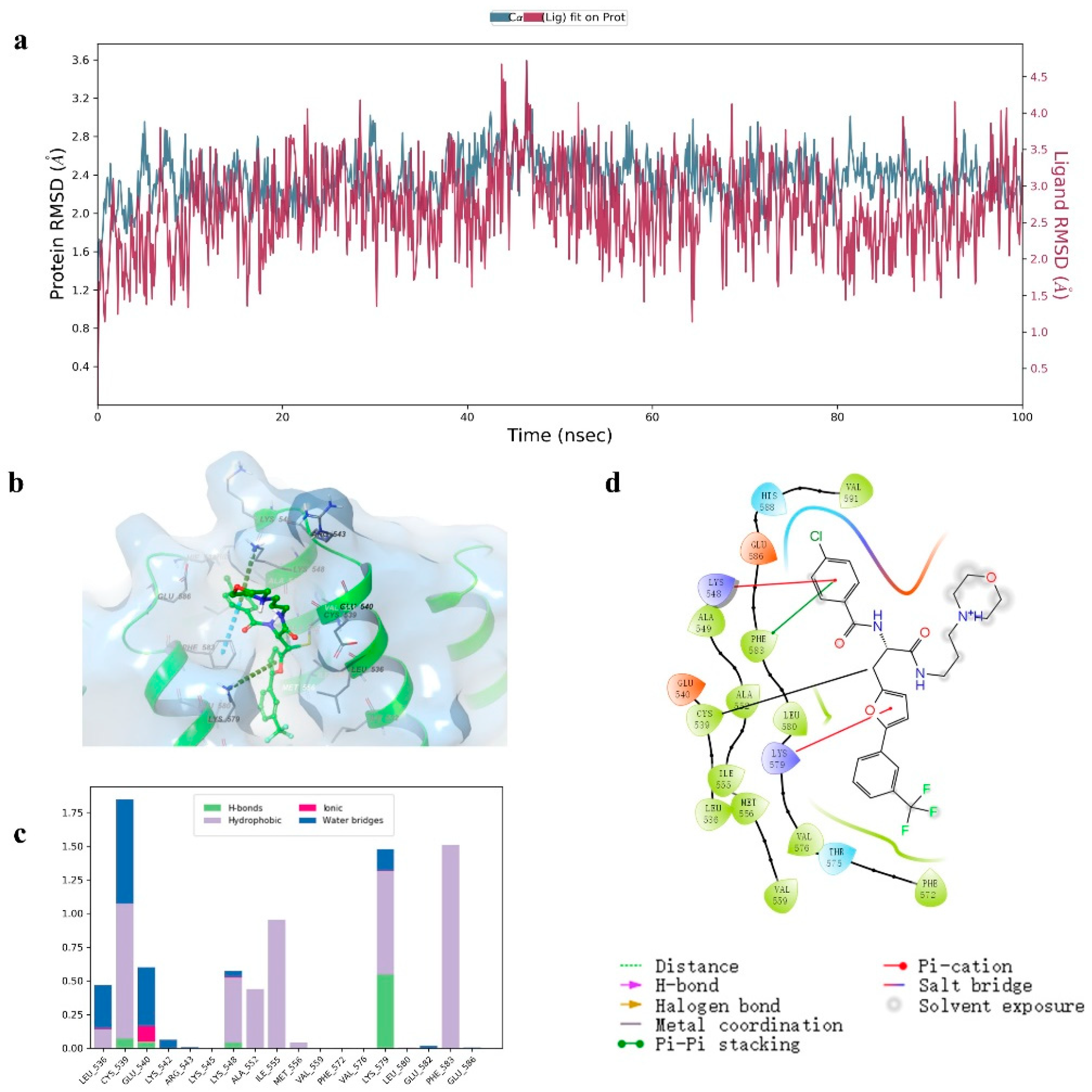
2.5. Molecular Dynamics Investigation
3. Discussion
4. Materials and Methods
4.1. Covalent Compound Library Production
4.2. Protein and Software Evaluation
4.3. Non-Covalent and Covalent-Coupling Workflow
4.4. Molecular Dynamics
4.5. Cell Proliferation Assays
4.6. Immunofluorescence Staining of RanBP1
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Wang, A.Y.; Liu, H. The past, present, and future of CRM1/XPO1 inhibitors. Stem Cell Investig. 2019, 6, 6. [Google Scholar] [CrossRef] [PubMed]
- Azizian, N.G.; Li, Y. XPO1-dependent nuclear export as a target for cancer therapy. J. Hematol. Oncol. 2020, 13, 61. [Google Scholar] [CrossRef] [PubMed]
- Hutten, S.; Kehlenbach, R.H. CRM1-mediated nuclear export: To the pore and beyond. Trends Cell Biol. 2007, 17, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Parikh, K.; Cang, S.; Sekhri, A.; Liu, D. Selective inhibitors of nuclear export (SINE)—A novel class of anti-cancer agents. J. Hematol. Oncol. 2014, 7, 78. [Google Scholar] [CrossRef] [Green Version]
- Chanukuppa, V.; Paul, D.; Taunk, K. XPO1 is a critical player for bortezomib resistance in multiple myeloma: A quantitative proteomic approach. J. Proteom. 2019, 209, 103504. [Google Scholar] [CrossRef]
- Uddin, M.d.H.; Zonder, J.A.; Azmi, A.S. Exportin 1 inhibition as antiviral therapy. Drug Discov. Today 2020, 25, 1775–1781. [Google Scholar] [CrossRef]
- Taylor, J.; Sendino, M.; Gorelick, A.N. Altered Nuclear Export Signal Recognition as a Driver of Oncogenesis. Cancer Discov. 2019, 9, 1452–1467. [Google Scholar] [CrossRef] [Green Version]
- Muqbil, I.; Azmi, A.S.; Mohammad, R.M. Nuclear Export Inhibition for Pancreatic Cancer Therapy. Cancers 2018, 10, 138. [Google Scholar] [CrossRef] [Green Version]
- Lei, Y.; An, Q.; Shen, X.F. Structure-Guided Design of the First Noncovalent Small-Molecule Inhibitor of CRM1. J. Med. Chem. 2021, 64, 6596–6607. [Google Scholar] [CrossRef]
- Ferreira, B.I.; Cautain, B.; Grenho, I.; Link, W. Small Molecule Inhibitors of CRM1. Front. Pharmacol. 2020, 11, 625. [Google Scholar] [CrossRef]
- Conforti, F.; Wang, Y.; Rodriguez, J.A.; Alberobello, A.T.; Zhang, Y.W.; Giaccone, G. Molecular Pathways: Anticancer Activity by Inhibition of Nucleocytoplasmic Shuttling. Clin. Cancer Res. 2015, 21, 4508–4513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hing, Z.A.; Fung, H.Y.J.; Ranganathan, P. Next-generation XPO1 inhibitor shows improved efficacy and in vivo tolerability in hematological malignancies. Leukemia 2016, 30, 2364–2372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azmi, A.S.; Mohammad, R.M. Targeting Cancer at the Nuclear Pore. J. Clin. Oncol. 2016, 34, 4180–4182. [Google Scholar] [CrossRef] [Green Version]
- Chari, A.; Vogl, D.T.; Gavriatopoulou, M. Oral Selinexor–Dexamethasone for Triple-Class Refractory Multiple Myeloma. N. Engl. J. Med. 2019, 381, 727–738. [Google Scholar] [CrossRef]
- Gravina, G.L.; Tortoreto, M.; Mancini, A. XPO1/CRM1-Selective Inhibitors of Nuclear Export (SINE) reduce tumor spreading and improve overall survival in preclinical models of prostate cancer (PCa). J. Hematol. Oncol. 2014, 7, 46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, Y.; Ke, X.; Deng, T.; Qin, Q. The Second-Generation XPO1 Inhibitor Eltanexor Inhibits Human Cytomegalovirus (HCMV) Replication and Promotes Type I Interferon Response. Front. Microbiol. 2021, 12, 675112. [Google Scholar] [CrossRef] [PubMed]
- Hays, J.; Zhang, J.; Berlin, J.D. Eltanexor (KPT-8602), a second-generation selective inhibitor of nuclear export (SINE) compound, in patients with metastatic colorectal cancer (mCRC). Ann. Oncol. 2018, 29, viii716. [Google Scholar] [CrossRef]
- Verbeke, D.; Demeyer, S.; Prieto, C. The XPO1 Inhibitor KPT-8602 Synergizes with Dexamethasone in Acute Lymphoblastic Leukemia. Clin. Cancer Res. 2020, 26, 5747–5758. [Google Scholar] [CrossRef]
- Azmi, A.S.; Uddin, M.H.; Mohammad, R.M. The nuclear export protein XPO1—From biology to targeted therapy. Nat. Rev. Clin. Oncol. 2021, 18, 152–169. [Google Scholar] [CrossRef]
- Gravina, G.L.; Mancini, A.; Sanita, P. KPT-330, a potent and selective exportin-1 (XPO-1) inhibitor, shows antitumor effects modulating the expression of cyclin D1 and survivin in prostate cancer models. BMC Cancer 2015, 15, 941. [Google Scholar] [CrossRef] [Green Version]
- Soulère, L.; Barbier, T.; Queneau, Y. Docking-based virtual screening studies aiming at the covalent inhibition of SARS-CoV-2 MPro by targeting the cysteine 145. Comput. Biol. Chem. 2021, 92, 107463. [Google Scholar] [CrossRef]
- London, N.; Miller, R.M.; Krishnan, S. Covalent Docking of Large Libraries for the Discovery of Chemical Probes. Nat. Chem. Biol. 2014, 10, 1066–1072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scarpino, A.; Bajusz, D.; Proj, M. Discovery of Immunoproteasome Inhibitors Using Large-Scale Covalent Virtual Screening. Molecules 2019, 24, 2590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Resnick, E.; Bradley, A.; Gan, J. Rapid Covalent-Probe Discovery by Electrophile-Fragment Screening. J. Am. Chem. Soc. 2019, 141, 8951–8968. [Google Scholar] [CrossRef] [Green Version]
- Nnadi, C.I.; Jenkins, M.L.; Gentile, D.R. Novel K-Ras G12C Switch-II Covalent Binders Destabilize Ras and Accelerate Nucleotide Exchange. J. Chem. Inf. Model. 2018, 58, 464–471. [Google Scholar] [CrossRef]
- Toledo Warshaviak, D.; Golan, G.; Borrelli, K.W.; Zhu, K.; Kalid, O. Structure-Based Virtual Screening Approach for Discovery of Covalently Bound Ligands. J. Chem. Inf. Model. 2014, 54, 1941–1950. [Google Scholar] [CrossRef]
- Ouyang, X.; Zhou, S.; Su, C.T.T.; Ge, Z.; Li, R.; Kwoh, C.K. CovalentDock: Automated covalent docking with parameterized covalent linkage energy estimation and molecular geometry constraints. J. Comput. Chem. 2013, 34, 326–336. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Wu, X.; Gao, J. Identification of new IDH2R140Q inhibitors by discriminatory analysis-based molecular docking and biological evaluation. Arch Pharm. 2021, 354, 2000063. [Google Scholar] [CrossRef] [PubMed]
- Cheng, G.; Mei, X.B.; Yan, Y.Y. Identification of new NIK inhibitors by discriminatory analysis-based molecular docking and biological evaluation. Arch Pharm. 2019, 352, 1800374. [Google Scholar] [CrossRef]
- Tong, L.; Song, P.; Jiang, K. Discovery of (R)-5-((5-(1-methyl-1H-pyrazol-4-yl)-4-(methylamino)pyrimidin-2-yl)amino)-3-(piperidin-3-yloxy)picolinonitrile, a novel CHK1 inhibitor for hematologic malignancies. Eur. J. Med. Chem. 2019, 173, 44–62. [Google Scholar] [CrossRef]
- Dong, X.W.; Zhang, J.K.; Xu, L. Covalent docking modelling-based discovery of tripeptidyl epoxyketone proteasome inhibitors composed of aliphatic-heterocycles. Eur. J. Med. Chem. 2019, 164, 602–614. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.Q.; Li, K.; Yao, T.T.; Hu, Y.Z.; Ying, H.Z.; Dong, X.W. Integrating docking scores and key interaction profiles to improve the accuracy of molecular docking: Towards novel B-RafV600E inhibitors. MedChemComm 2017, 8, 1835–1844. [Google Scholar] [CrossRef] [PubMed]
- Yao, T.T.; Xie, J.F.; Liu, X.G. Integration of pharmacophore mapping and molecular docking in sequential virtual screening: Towards the discovery of novel JAK2 inhibitors. RSC Adv. 2017, 7, 10353–10360. [Google Scholar] [CrossRef] [Green Version]
- Zhan, W.; Li, D.; Che, J. Integrating docking scores, interaction profiles and molecular descriptors to improve the accuracy of molecular docking: Toward the discovery of novel Akt1 inhibitors. Eur. J. Med. Chem. 2014, 75, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Yan, J.; Du, L. Pharmacophore identification, docking and “in silico” screening for novel CDK1 inhibitors. J. Mol. Graph Model. 2012, 37, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.; Liu, J.; Chen, S. Discover Novel Covalent Inhibitors Targeting FLT3 through Hybrid Virtual Screening Strategy. Biol. Pharm. Bull. 2021, 44, 1872–1877. [Google Scholar] [CrossRef] [PubMed]
- Fung, H.Y.J.; Fu, S.C.; Chook, Y.M. Nuclear export receptor CRM1 recognizes diverse conformations in nuclear export signals. ELife 2017, 6, e23961. [Google Scholar] [CrossRef]
- Fu, S.C.; Fung, H.Y.J.; Cağatay, T.; Baumhardt, J.; Chook, Y.M. Correlation of CRM1-NES affinity with nuclear export activity. Mol. Biol. Cell. 2018, 29, 2037–2044. [Google Scholar] [CrossRef]
- Gargantilla, M.; López-Fernández, J.; Camarasa, M.-J.; Persoons, L.; Daelemans, D.; Priego, E.-M.; Pérez-Pérez, M.-J. Inhibition of XPO-1 Mediated Nuclear Export through the Michael-Acceptor Character of Chalcones. Pharmaceuticals 2021, 14, 1131. [Google Scholar] [CrossRef]
- Schmidt, J.; Braggio, E.; Kortuem, K. Genome-wide studies in multiple myeloma identify XPO1/CRM1 as a critical target validated using the selective nuclear export inhibitor KPT-276. Leukemia 2013, 27, 2357–2365. [Google Scholar] [CrossRef]
- Plafker, K.; Macara, I.G. Facilitated nucleocytoplasmic shuttling of the Ran binding protein RanBP1. Mol. Cell. Biol. 2000, 20, 3510–3521. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Title | Force Field | Prime Energy Solvent Model | RXN | Docking Score | MM-GBSA dG Bind |
|---|---|---|---|---|---|---|
| Compound 1 | 4578–1143 | OPLS3e | VSGB2.1 | Michael Addition | −7.333 | −59.68 |
| Compound 2 | 5090–1053 | OPLS3e | VSGB2.1 | Michael Addition | −7.314 | −58.60 |
| Compound 3 | Y600–2961 | OPLS3e | VSGB2.1 | Michael Addition | −7.493 | −59.50 |
| Compound 4 | K291–0098 | OPLS3e | VSGB2.1 | Michael Addition | −7.116 | −51.65 |
| Compound 5 | V029–9645 | OPLS3e | VSGB2.1 | Michael Addition | −6.902 | −69.82 |
| Compound 6 | V003–8338 | OPLS3e | VSGB2.1 | Michael Addition | −7.451 | −59.33 |
| Compound 7 | V025–8829 | OPLS3e | VSGB2.1 | Michael Addition | −8.072 | −60.33 |
| compound 8 | 4476–4961 | OPLS3e | VSGB2.1 | Michael Addition | −7.164 | −61.77 |
| Compound 9 | 2733–3746 | OPLS3e | VSGB2.1 | Michael Addition | −7.135 | −63.64 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shen, Z.; Zhuang, W.; Li, K.; Guo, Y.; Qu, B.; Chen, S.; Gao, J.; Liu, J.; Xu, L.; Dong, X.; et al. Identification of Novel Covalent XPO1 Inhibitors Based on a Hybrid Virtual Screening Strategy. Molecules 2022, 27, 2543. https://doi.org/10.3390/molecules27082543
Shen Z, Zhuang W, Li K, Guo Y, Qu B, Chen S, Gao J, Liu J, Xu L, Dong X, et al. Identification of Novel Covalent XPO1 Inhibitors Based on a Hybrid Virtual Screening Strategy. Molecules. 2022; 27(8):2543. https://doi.org/10.3390/molecules27082543
Chicago/Turabian StyleShen, Zheyuan, Weihao Zhuang, Kang Li, Yu Guo, Bingxue Qu, Sikang Chen, Jian Gao, Jing Liu, Lei Xu, Xiaowu Dong, and et al. 2022. "Identification of Novel Covalent XPO1 Inhibitors Based on a Hybrid Virtual Screening Strategy" Molecules 27, no. 8: 2543. https://doi.org/10.3390/molecules27082543
APA StyleShen, Z., Zhuang, W., Li, K., Guo, Y., Qu, B., Chen, S., Gao, J., Liu, J., Xu, L., Dong, X., Che, J., & Li, Q. (2022). Identification of Novel Covalent XPO1 Inhibitors Based on a Hybrid Virtual Screening Strategy. Molecules, 27(8), 2543. https://doi.org/10.3390/molecules27082543