
Green Strategies for the Preparation of Enantiomeric 5–8-Membered Carbocyclic β-Amino Acid Derivatives through CALB-Catalyzed Hydrolysis




Abstract
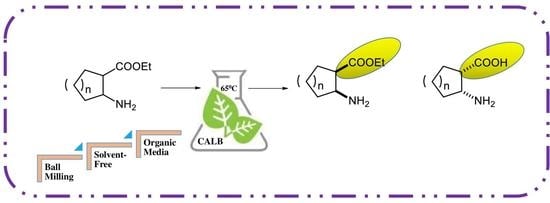
:
1. Introduction
2. Results and Discussion
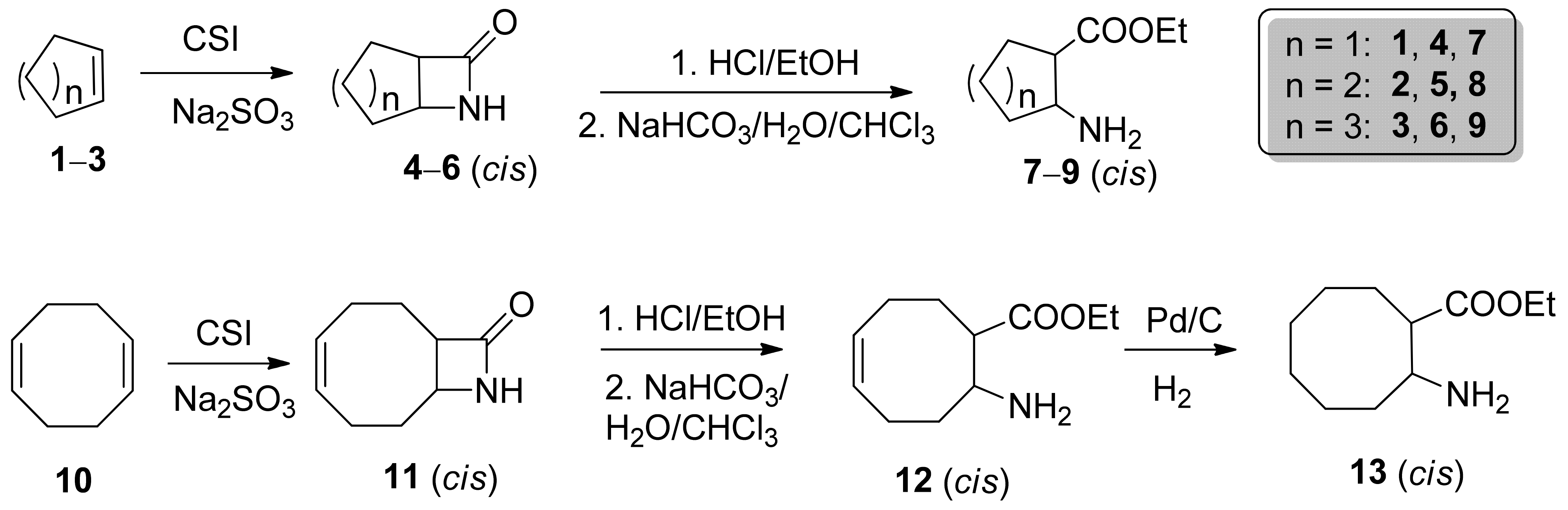
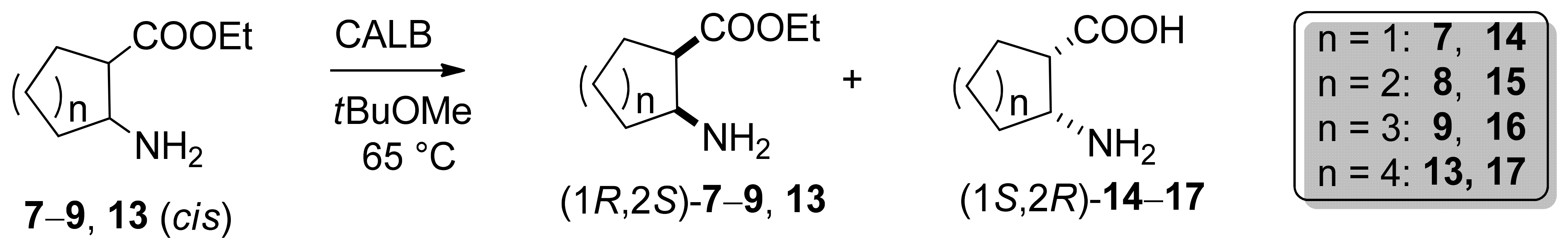
2.1. Synthesis of cis-Amino Esters 7–9 and 13
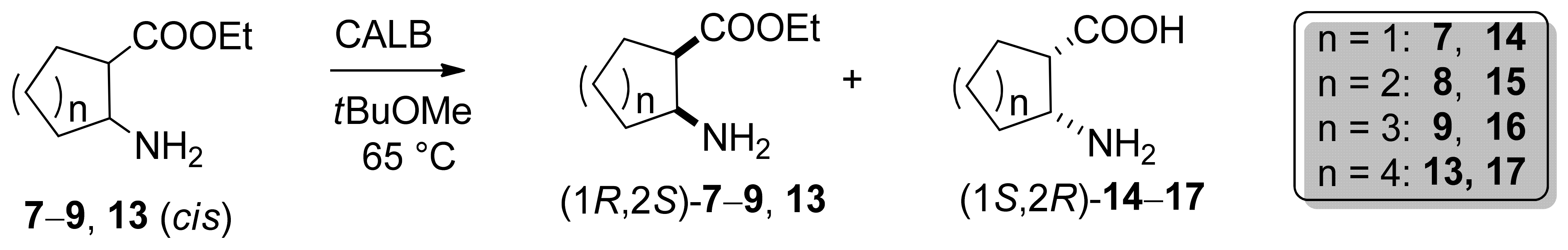
2.2. Enzyme-Catalyzed Hydrolysis of Carbocyclic cis β-Amino Esters 7–9 and 13
2.2.1. Preliminary Experiments
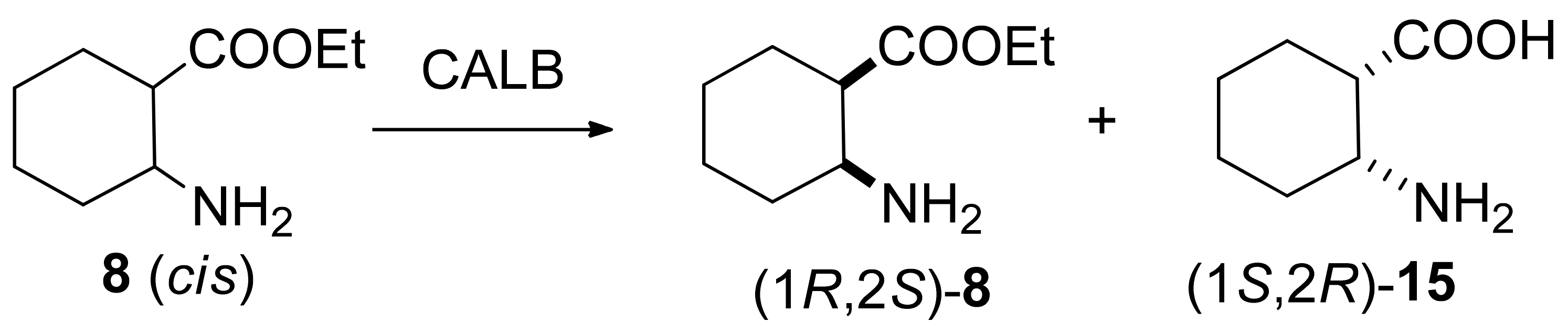
2.2.2. Preparative-Scale Resolutions of cis 7–9, and 13
2.2.3. Determination of Absolute Configurations
3. Materials and Methods
3.1. Procedure for the Synthesis of 7–9 and 13
3.2. Derivatization Process
3.3. Procedure for the Preparative-Scale Hydrolysis of (±) cis-5–8-Membered Amino Esters
3.3.1. (1R,2S)-Ethyl 2-Aminocyclopentanecarboxylate (7)
3.3.2. (1R,2S)-Ethyl 2-Aminocyclohexanecarboxylate (8)
3.3.3. (1R,2S)-Ethyl 2-Aminocycloheptanecarboxylate (9)
3.3.4. (1R,2S)-Ethyl 2-Aminocyclooctanecarboxylate (13)
3.3.5. (1R,2S)-2-Aminocyclopentanecarboxylic Acid (14)
3.3.6. (1R,2S)-2-Aminocyclohexanecarboxylic Acid (15)
3.3.7. (1R,2S)-2-Aminocycloheptanecarboxylic Acid (16)
3.3.8. (1R,2S)-2-Aminocyclooctanecarboxylic Acid (17)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Gardiner, J.; Anderson, K.H.; Downard, A.; Abell, A.D. Synthesis of cyclic β-amino acid esters from methionine, allylglycine, and serine. J. Org. Chem. 2004, 69, 3375–3382. [Google Scholar] [CrossRef]
- Bell, A.D.; Gardiner, J. Synthesis of substituted cyclohexenyl-based β-amino acids by ring-closing metathesis. Org. Lett. 2002, 4, 3663–3666. [Google Scholar]
- Fülöp, F. The chemistry of 2-aminocycloalkanecarboxylic acids. Chem. Rev. 2001, 101, 2181–2204. [Google Scholar] [CrossRef]
- Fülöp, F. The chemistry of 2-aminocyclopentanecarboxylic acid. In Studies in Natural Product Chemistry; Atta-ur, R., Ed.; Elsevier Science Publishers: New York, NY, USA, 2000; Volume 22, pp. 273–306. [Google Scholar]
- Park, K.; Kurth, M.J. Cyclic amino acid derivatives. Tetrahedron 2002, 58, 8629–8659. [Google Scholar] [CrossRef]
- Mittendorf, J.; Benet-Buch-holz, J.; Fey, P.; Mohrs, K.H. Efficient asymmetric synthesis of β-amino acid BAY 10-8888/PLD-118, a novel antifungal for the treatment of yeast infections. Synthesis 2003, 1, 136–140. [Google Scholar] [CrossRef]
- Kuhl, A.; Hahn, M.G.; Dumic, M.; Mittendorf, J. Alicyclic beta-amino acids in medicinal chemistry. Amino Acids 2005, 29, 89–100. [Google Scholar] [CrossRef]
- Fülöp, F.; Martinek, T.A.; Tóth, G.K. Application of alicyclic β-amino acids in peptide chemistry. Chem. Soc. Rev. 2006, 35, 323–334. [Google Scholar] [CrossRef] [PubMed]
- Petraitiene, R.; Petraitis, V.; Kelaher, A.M.; Sarafandi, A.A.; Mickiene, D.; Groll, A.H.; Sein, T.; Bacher, J.; Walsh, T.J. Efficacy, plasma pharmacokinetics, and safety of icofungipen, an inhibitor of candida isoleucyl-tRNA synthetase, in treatment of experimental disseminated candidiasis in persistently neutropenic rabbits. Antimicrob. Agents. Chemother. 2005, 49, 2084–2092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steer, D.L.; Lew, R.A.; Perlmutter, P.; Smith, A.I.; Aguilar, M.I. β-amino acids: Versatile peptidomimetics. Curr. Med. Chem. 2002, 9, 811–822. [Google Scholar] [CrossRef] [PubMed]
- Forró, E.; Fülöp, F. Cispentacin-enzymatic highlights of its 25-year history. Mini. Rev. Org. Chem. 2016, 13, 219–226. [Google Scholar] [CrossRef]
- Forró, E.; Fülöp, F. The first direct enzymatic hydrolysis of alicyclic β-amino esters: A route to enantiopure cis and trans β-amino acids. Chem. Eur. J. 2007, 13, 6397–6401. [Google Scholar] [CrossRef]
- Forró, E.; Fülöp, F. Vapour-assisted enzymatic hydrolysis of β-lactams in a solvent-free system. Tetrahedron Asymmetry 2008, 19, 1005–1009. [Google Scholar] [CrossRef]
- Hernández, J.G.; Bolm, C. Altering product selectivity by mechanochemistry. J. Org. Chem. 2017, 82, 4007–4019. [Google Scholar] [CrossRef]
- Rodríguez, B.; Rantanen, T.; Bolm, C. Solvent-free asymmetric organocatalysis in a ball mill. Angew. Chem. Int. Ed. 2006, 45, 6924–6926. [Google Scholar] [CrossRef]
- Declerck, V.; Nun, P.; Martinez, J.; Lamaty, F. Solvent-free synthesis of peptides. Angew. Chem. Int. Ed. 2009, 48, 9318–9321. [Google Scholar] [CrossRef]
- Bonnamour, J.; Métro, T.-X.; Martinez, J.; Lamaty, F. Environmentally benign peptide synthesis using liquid-assisted ball-milling: Application to the synthesis of Leuenkephalin. Green Chem. 2013, 15, 1116–1120. [Google Scholar] [CrossRef]
- Baig, R.B.N.; Varma, R.S. Alternative energy input: Mechanochemical, microwave and ultrasound-assisted organic synthesis. Chem. Soc. Rev. 2012, 41, 1559–1584. [Google Scholar] [CrossRef]
- Jones, W.; Eddleston, M.D. Introductory lecture: Mechanochemistry, a versatile synthesis strategy for new materials. Faraday Discuss. 2014, 170, 9–34. [Google Scholar] [CrossRef] [Green Version]
- Hernández, J.G.; Friščić, T. Metal-catalyzed organic reactions using mechanochemistry. Tetrahedron Lett. 2015, 56, 4253–4265. [Google Scholar] [CrossRef]
- Lawrenson, S.B.; Arav, R.; North, M. The greening of peptide synthesis. Green Chem. 2017, 19, 1685–1691. [Google Scholar] [CrossRef] [Green Version]
- Hernández, J.G.; Juaristi, E. Recent efforts directed to the development of more sustainable asymmetric organocatalysis. Chem. Commun. 2012, 48, 5396–5409. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, R.; Stolle, A.; Ondruschka, B. Aromatic substitution in ball mills: Formation of aryl chlorides and bromides using potassium peroxomonosulfate and NaX. Green Chem. 2012, 14, 1673–1679. [Google Scholar] [CrossRef]
- Hernández, J.G.; Macdonald, N.A.J.; Mottillo, C.; Butler, I.S.; Friščić, T. A mechanochemical strategy for oxidative addition: Remarkable yields and stereoselectivity in the halogenation of organometallic Re(I) complexes. Green Chem. 2014, 16, 1087–1092. [Google Scholar] [CrossRef]
- Machuca, E.; Juaristi, E. Ball Milling Towards Green Synthesis: Applications, Projects, Challenges; Ranu, B., Stolle, A., Eds.; Royal Society of Chemistry: Cambridge, UK, 2015; pp. 81–95. [Google Scholar]
- McKissic, K.S.; Caruso, J.T.; Blair, R.G.; Mack, J. Comparison of shaking versus baking: Further understanding the energetics of a mechanochemical reaction. Green Chem. 2014, 16, 1628–1632. [Google Scholar] [CrossRef]
- Schmidt, R.; Burmeister, C.F.; Baláž, M.; Kwade, A.; Stolle, A. Effect of reaction parameters on the synthesis of 5-arylidene barbituric acid derivatives in ball mills. Org. Process Res. Dev. 2015, 19, 427–436. [Google Scholar] [CrossRef]
- Venegas, M.P.; Juaristi, E. Mechanoenzymology: State of the art and challenges towards highly sustainable biocatalysis. ChemSusChem 2021, 14, 2682–2688. [Google Scholar] [CrossRef]
- Bolm, C.; Hernandez, J.G. From synthesis of amino acids and peptides to enzymatic catalysis: A bottom-up approachin mechanochemistry. ChemSusChem 2018, 11, 1410–1420. [Google Scholar] [CrossRef]
- Ortiz, C.G.A.; Venegas, M.P.; Caporali, J.V.; Juaristi, E. Recent applications of mechanochemistry in enantioselective synthesis. Tetrahedron Lett. 2019, 60, 1749–1757. [Google Scholar] [CrossRef]
- Venegas, M.P.; Juaristi, E. Mechanoenzymatic resolution of racemic chiral amines, a green technique for the synthesis of pharmaceutical building blocks. Tetrahedron 2018, 74, 6453–6458. [Google Scholar] [CrossRef]
- Venegas, M.P.; Treviño, A.M.R.; Juaristi, E. Dual mechanoenzymatic kinetic resolution of (±)-ketorolac. ChemCatChem 2020, 12, 1782–1788. [Google Scholar] [CrossRef]
- Venegas, M.P.; Cruz, M.M.T.; Feria, O.S.; Munguia, A.L.; Castillo, E.; Juaristi, E. Thermal and mechanical stability of immobilized Candida antarctica lipase B: An approximation to mechanochemical energetics in enzyme catalysis. ChemCatChem 2020, 12, 803–811. [Google Scholar] [CrossRef]
- Venegas, M.P.; Rangel, G.R.; Neri, A.; Escalante, J.; Juaristi, E. Mechanochemical enzymatic resolution of N-benzylated-β3-amino esters. Beilstein J. Org. Chem. 2017, 13, 1728–1734. [Google Scholar] [CrossRef] [Green Version]
- Moriconi, E.J.; Meyer, W.C. The reaction of dienes with chlorosulfonyl isocyanate. J. Org. Chem. 1971, 36, 2841–2849. [Google Scholar] [CrossRef]
- Dragovich, P.S.; Murphy, D.E.; Dao, K.; Kim, S.H.; Li, L.S.; Ruebsam, F.; Chinh, Z.S.; Tran, V.; Xiang, A.X.; Zhou, Y. Efficient synthesis of (1R,2S) and (1S,2R)-2-aminocyclopentanecarboxylic acid ethyl ester derivatives in enantiomerically pure form. Tetrahedron Asymmetry 2008, 19, 2796–2803. [Google Scholar] [CrossRef]
- Viña, D.; Santana, L.; Uriarte, E.; Quezada, E.; Valencia, L. Synthesis of 1-[2-(hydroxymethyl) cyclohexyl] pyrimidine analogues of nucleosides: A comparative study. Synthesis 2004, 15, 2517–2522. [Google Scholar] [CrossRef]
- Palkó, M.; Benedek, G.; Forró, E.; Wéber, E.; Hänninen, M.; Sillanpää, R.; Fülöp, F. Synthesis of mono- and dihydroxy-substituted 2-aminocyclooctanecarboxylic acid enantiomers. Tetrahedron Asymmetry 2010, 21, 957–961. [Google Scholar] [CrossRef]
- Forró, E.; Árva, J.; Fülöp, F. Preparation of (1R,8S)-and (1S,8R)-9-azabicyclo[6.2.0]dec-4-en-10-one: Potential starting compounds for the synthesis of anatoxin-α. Tetrahedron Asymmetry 2001, 12, 643–649. [Google Scholar] [CrossRef]
- Forró, E. New gas chromatographic method for the enantioseparation of β-amino acids by a rapid double derivatization technique. J. Chromatogr. A 2009, 1216, 1025–1029. [Google Scholar] [CrossRef]
- Forró, E.; Fülöp, F. New enzymatic two-step cascade reaction for the preparation of a key intermediate for the Taxol side-chain. Eur. J. Org. Chem. 2010, 2010, 3074–3079. [Google Scholar] [CrossRef]
- Straathof, A.J.J.; Rekels, J.L.L.; Heijnen, J.J. Mass balancing in kinetic resolution: Calculating yield and enantiomeric excess using chiral balance. Biotechnol. Bioeng. 1995, 45, 536–538. [Google Scholar] [CrossRef]
- Chen, C.S.; Fujimoto, Y.; Girdaukas, G.; Sih, C.J. Quantitative analyses of biochemical kinetic resolutions of enantiomers. J. Am. Chem. Soc. 1982, 104, 7294–7299. [Google Scholar] [CrossRef]
- Friščić, T.; Childs, S.L.; Rizvi, S.A.A.; Jones, W. The role of solvent in mechanochemical and sonochemical cocrystal formation: A solubility-based approach for predicting cocrystallisation outcome. CrystEngComm 2009, 11, 418–426. [Google Scholar] [CrossRef]
- Forró, E.; Kiss, L.; Árva, J.; Fülöp, F. Efficient enzymatic routes for the synthesis of new eight-membered cyclic β-amino acid and β-lactam. Molecules 2017, 22, 2211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanerva, L.T.; Csomós, P.; Sundholm, O.; Bernáth, G.; Fülöp, F. Approach to highly enantiopure β-amino acid esters by using lipase catalysis in organic media. Tetrahedron Asymmetry 1996, 7, 1705–1716. [Google Scholar] [CrossRef]
- Forró, E.; Fülöp, F. Lipase-catalyzed enantioselective ring opening of unactivated alicyclic-fused β-lactams in an organic solvent. Org. Lett. 2003, 5, 1209–1212. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Solvent (mL) | ees (%) b | eep (%) c | Conv. (%) d | E e |
|---|---|---|---|---|---|
| 1 | iPr2O | 60 | 95 | 39 | 66 |
| 2 | tBuOMe | 63 | >99 | 39 | 133 |
| 3 | EtOAc | - | - | - | - |
| 4 | Propylene carbonate | 30 | >99 | 23 | 73 |
| 5 | 2-Me-THF | 14 | >99 | 12 | 74 |
| 6 | 2M-2B | 6 | >99 | 6 | 65 |
| CALB (mg mL−1) | ees (%) b | eep (%) c | Conv. (%) d | E e |
|---|---|---|---|---|
| Once used | 78 | 97 | 45 | 180 |
| Twice used | 58 | 96 | 37 | 82 |
| 3 times used | 43 | 96 | 31 | 71 |
| Entry | Temp (°C) | ees (%) b | eep (%) c | Conv. (%) d | E e |
|---|---|---|---|---|---|
| 1 | 23 | 11 | 95 | 11 | 45 |
| 2 | 65 | 66 | 94 | 41 | 70 |
| 3 | 70 | 72 | 72 | 50 | 13 |
| 4 | 80 | 91 | 57 | 62 | 11 |
| Entry | Frequency (Hz) | ees (%) c | eep (%) d | Conv. (%) e | E f |
|---|---|---|---|---|---|
| 1 | 25 | 2 | 69 | 3 | 6 |
| 2 | 15 | 3 | 90 | 3 | 19 |
| 3 | 10 | 5 | 87 | 5 | 16 |
| 4 | 8 | 5 | 91 | 5 | 21 |
| 5 | 3 | 15 | 98 | 14 | 147 |
| 6 b | 3 | 16 | 97 | 14 | 89 |
| Entry | CALB (mg) | ees (%) b | eep (%) c | Conv. (%) d | E e |
|---|---|---|---|---|---|
| 1 | 30 | 24 | >99 | 20 | >200 |
| 2 | 10 | 13 | 81 | 14 | 11 |
| Entry | Rt (hours) | ees (%) d | eep (%) e | Conv. (%) f | E g |
|---|---|---|---|---|---|
| 1 a | 23 | 96 | >99 | 50 | >200 |
| 2 b | 2 (22) | 35 (>99) | 96 (69) | 27 (59) | 58 (27) |
| 3 c | 8 (67) | 20 (98) | >99 (48) | 14 (67) | 163 (11) |
| β-Amino Esters: (1R,2S)-7–9, 13 | β-Amino Acids: (1S,2R)-14–17 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| (±) | Time (hours) | Conv. (%) | Yield (%) | Isomer | eese (%) | (EtOH) | Yield (%) | Isomer | eepf (%) | (H2O) |
| 7a | 4 (24) | 36 (75) | 31 | (1R,2S)-7 | 98 | −6.94 g | 25 | (1S,2R)-14 | 96 | +9.41 g |
| 8b | 23 | 50 | 27 | (1R,2S)-8 | 96 | −11.13 g | 33 | (1S,2R)-15 | 98 | +19.84 h |
| 9c | 23 (3d) | 20 (69) | 30 | (1R,2S)-9 | 91 | −4.09 i | 32 | (1S,2R)-16 | 98 | +6.54 h |
| 13d | 23 (20d) | 20 (62) | 27 | (1R,2S)-13 | 62 | +20.92 j | 28 | (1S,2R)-17 | >99 | −19.15 k |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shahmohammadi, S.; Faragó, T.; Palkó, M.; Forró, E. Green Strategies for the Preparation of Enantiomeric 5–8-Membered Carbocyclic β-Amino Acid Derivatives through CALB-Catalyzed Hydrolysis. Molecules 2022, 27, 2600. https://doi.org/10.3390/molecules27082600
Shahmohammadi S, Faragó T, Palkó M, Forró E. Green Strategies for the Preparation of Enantiomeric 5–8-Membered Carbocyclic β-Amino Acid Derivatives through CALB-Catalyzed Hydrolysis. Molecules. 2022; 27(8):2600. https://doi.org/10.3390/molecules27082600
Chicago/Turabian StyleShahmohammadi, Sayeh, Tünde Faragó, Márta Palkó, and Enikő Forró. 2022. "Green Strategies for the Preparation of Enantiomeric 5–8-Membered Carbocyclic β-Amino Acid Derivatives through CALB-Catalyzed Hydrolysis" Molecules 27, no. 8: 2600. https://doi.org/10.3390/molecules27082600
APA StyleShahmohammadi, S., Faragó, T., Palkó, M., & Forró, E. (2022). Green Strategies for the Preparation of Enantiomeric 5–8-Membered Carbocyclic β-Amino Acid Derivatives through CALB-Catalyzed Hydrolysis. Molecules, 27(8), 2600. https://doi.org/10.3390/molecules27082600