
Total Synthesis of Eliglustat via Diastereoselective Amination of Chiral para-Methoxycinnamyl Benzyl Ether

 , , and
, , and
Abstract
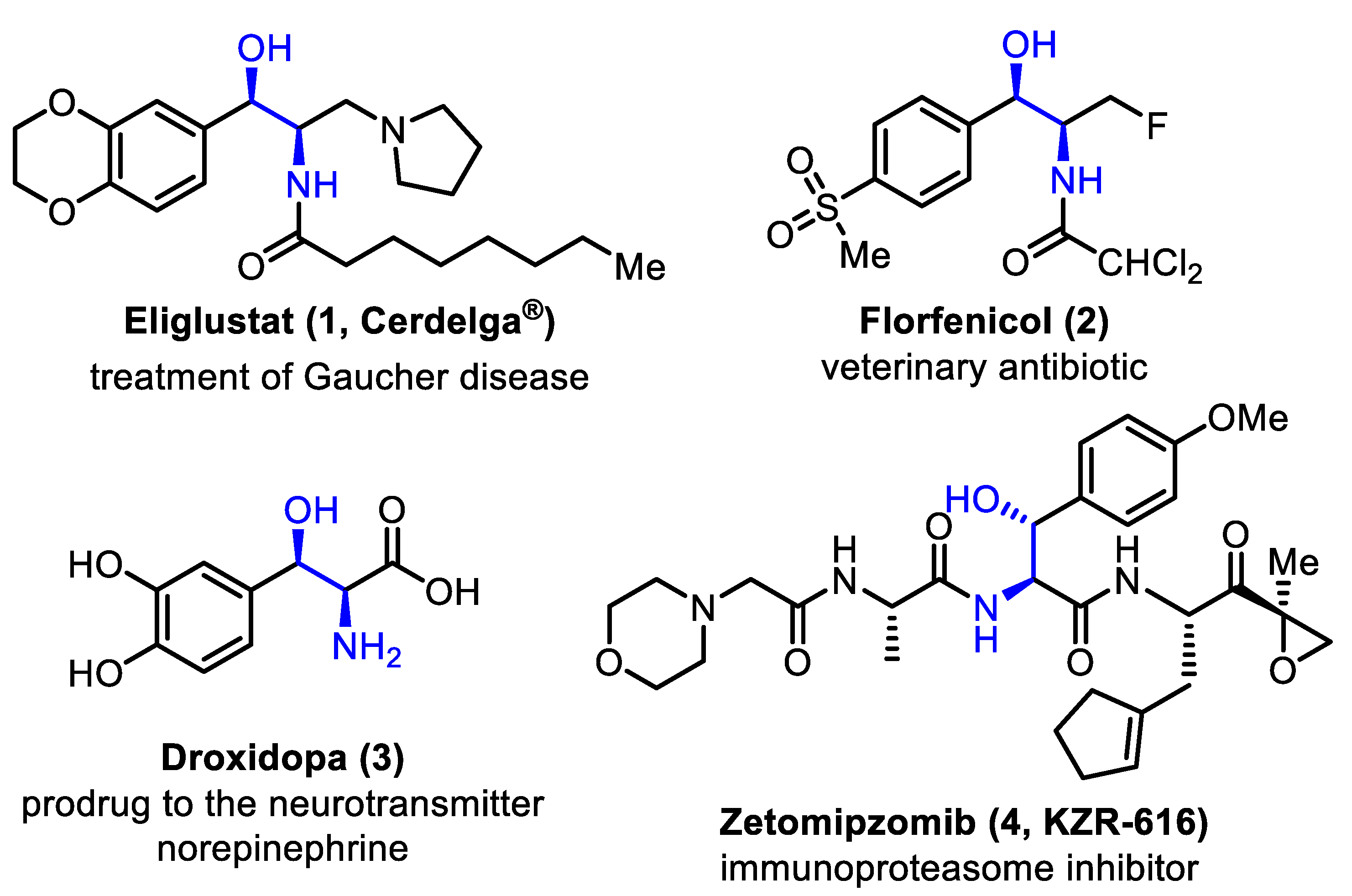
:1. Introduction
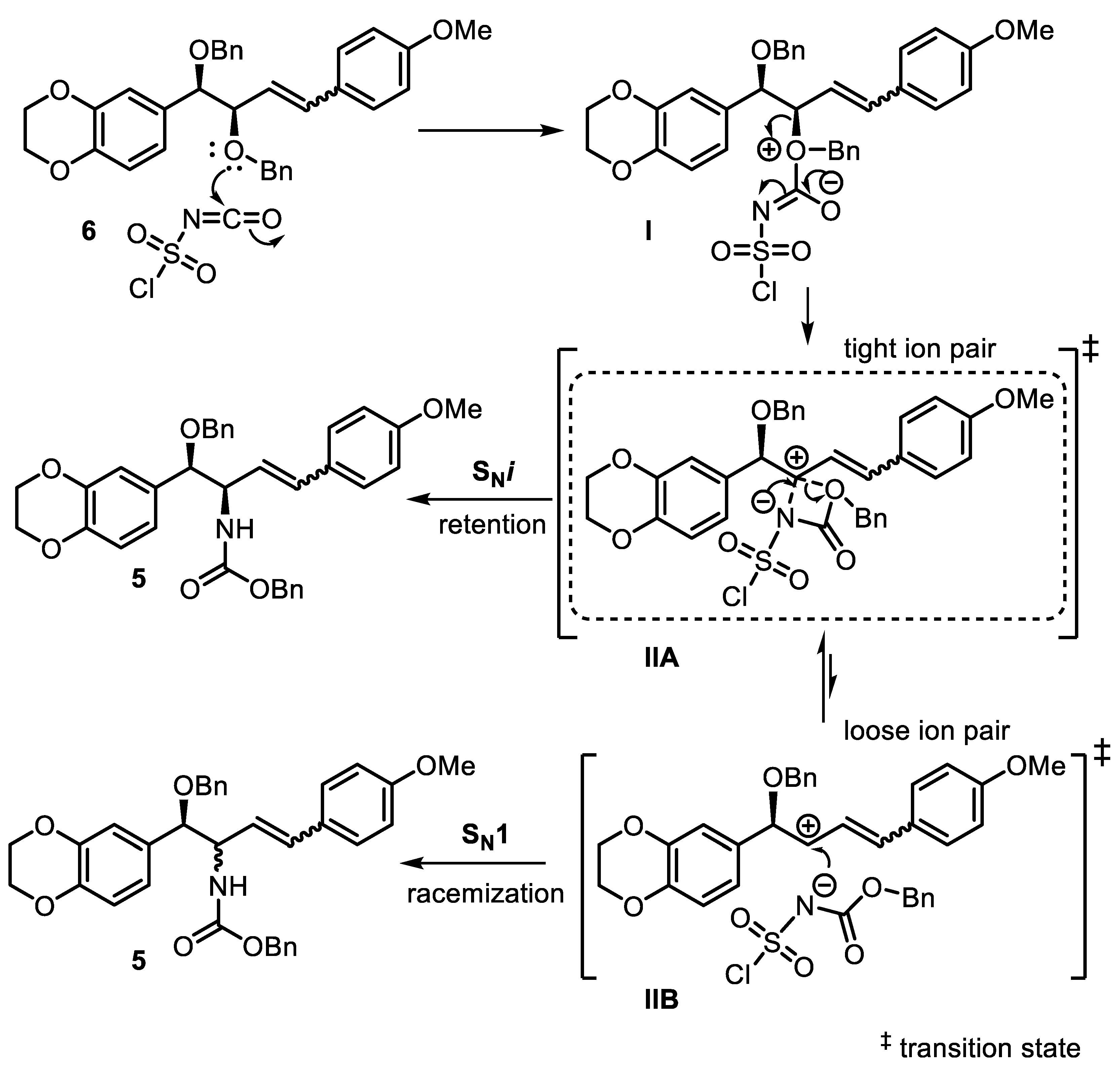
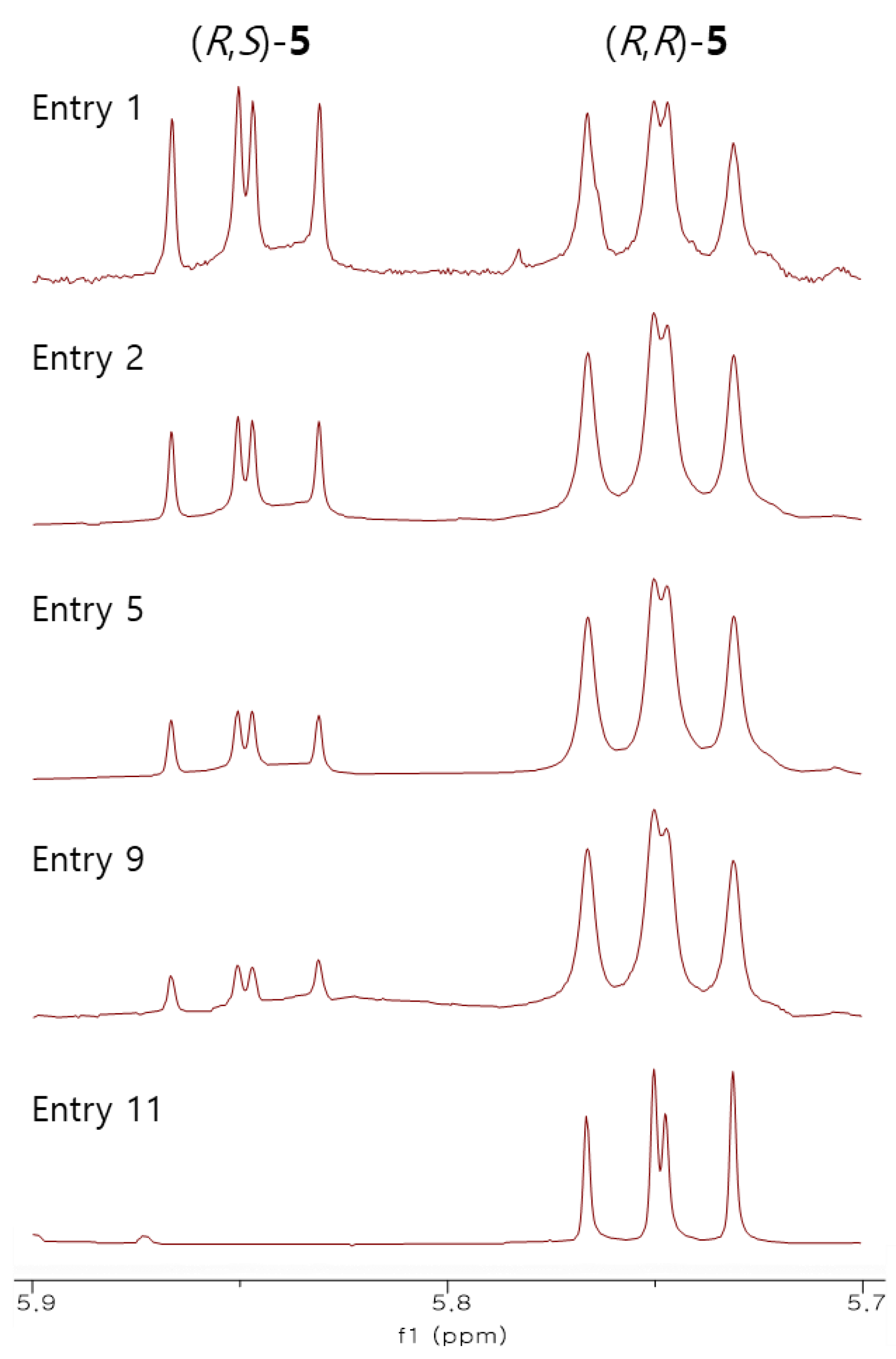
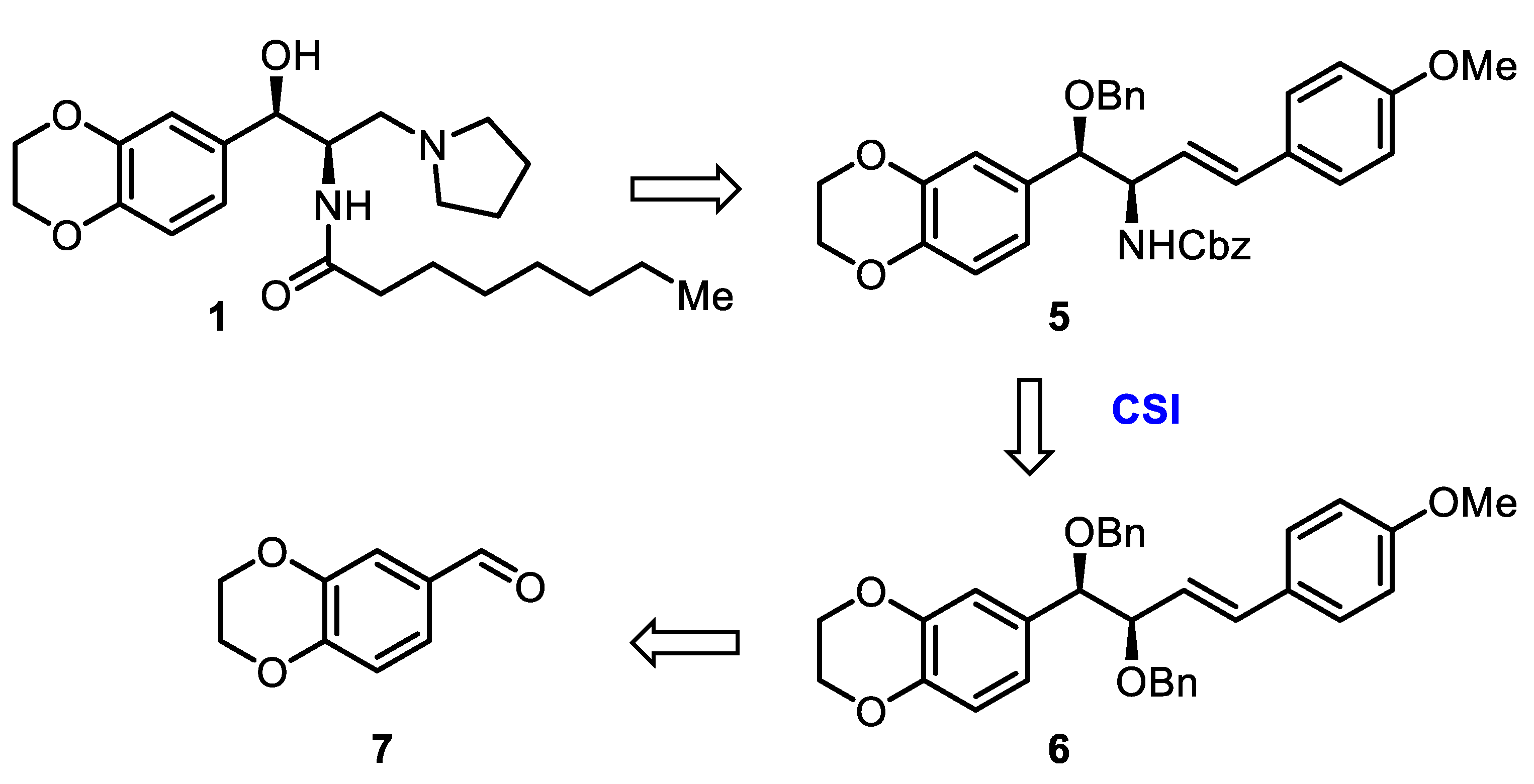
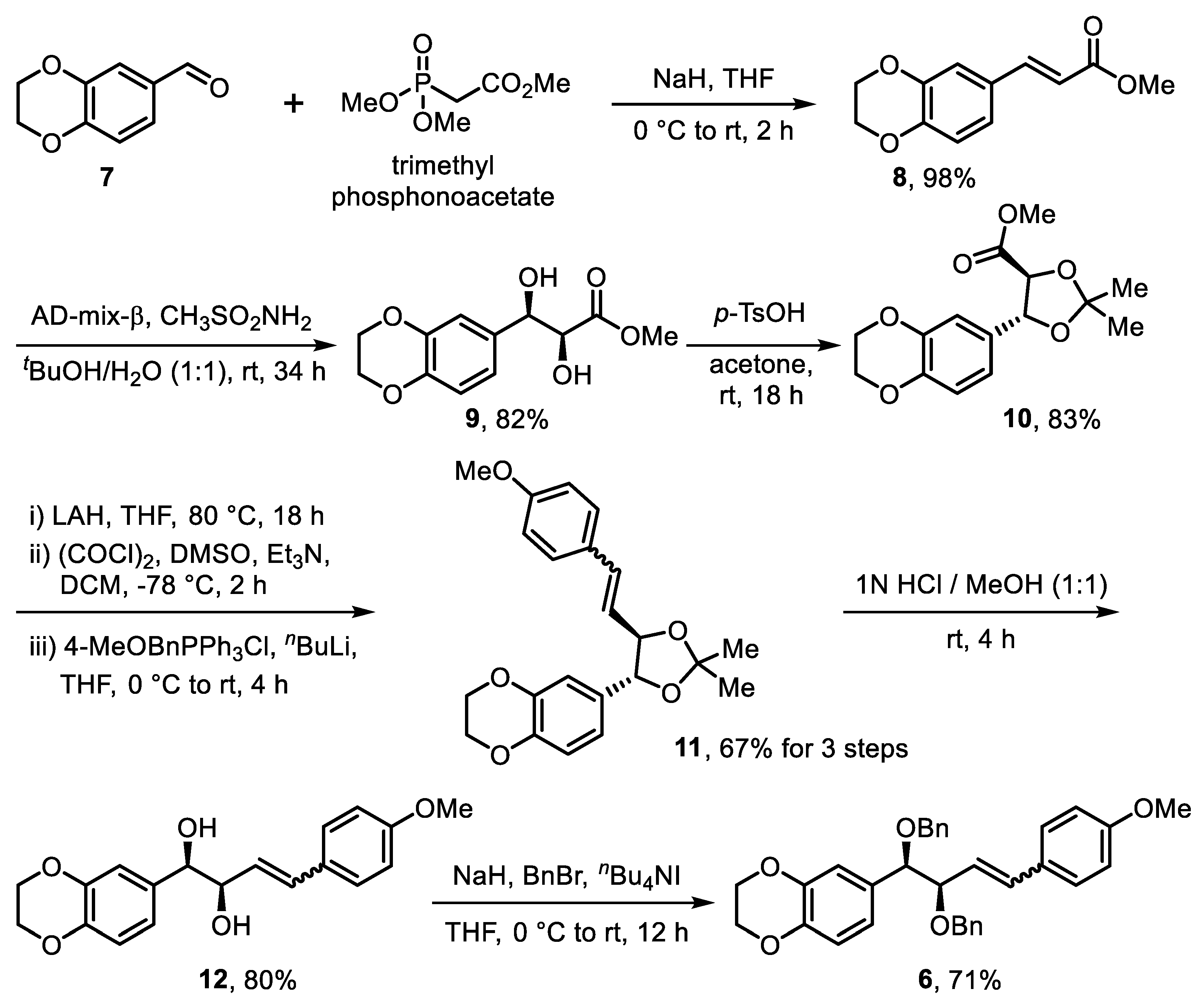
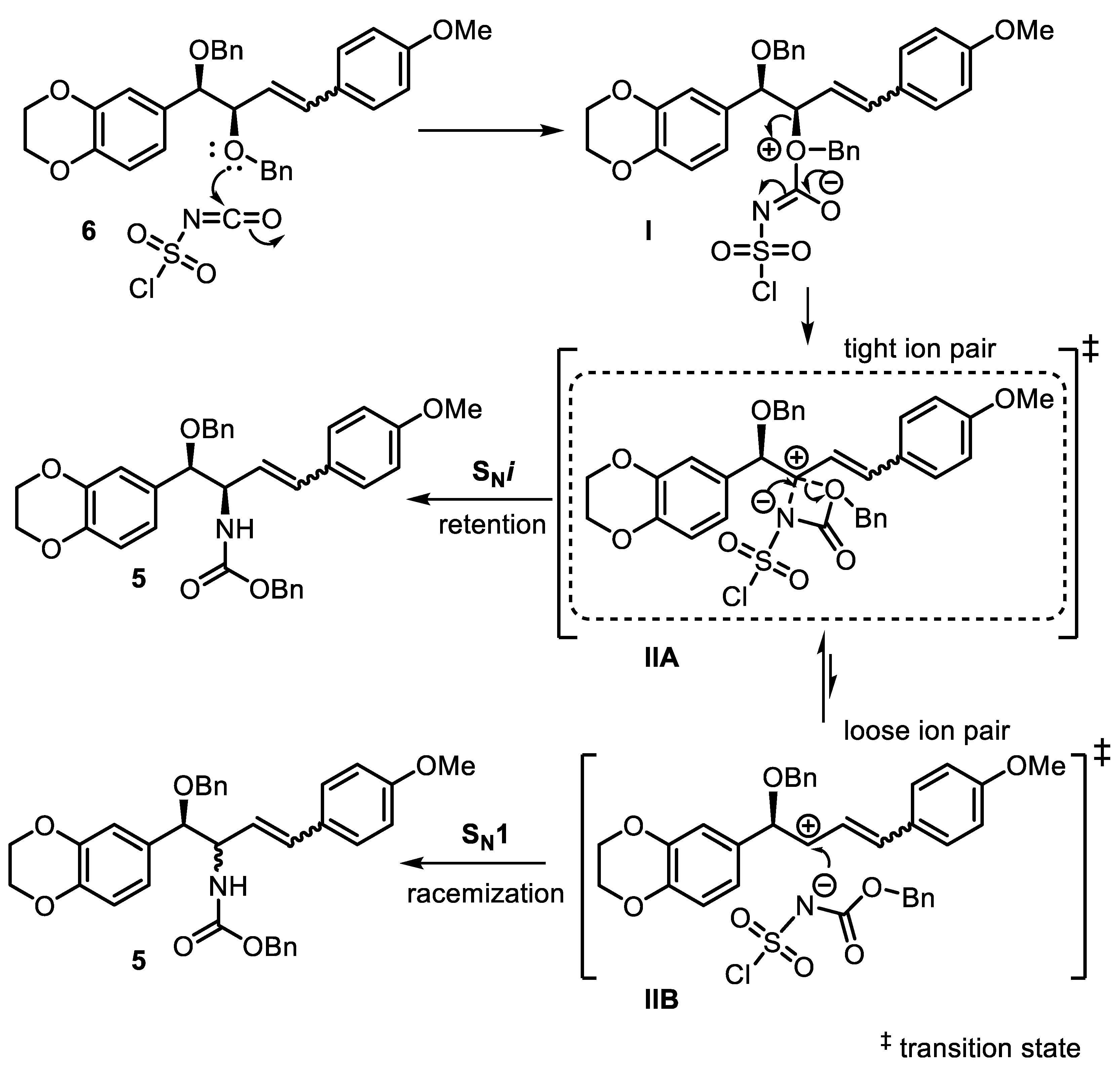
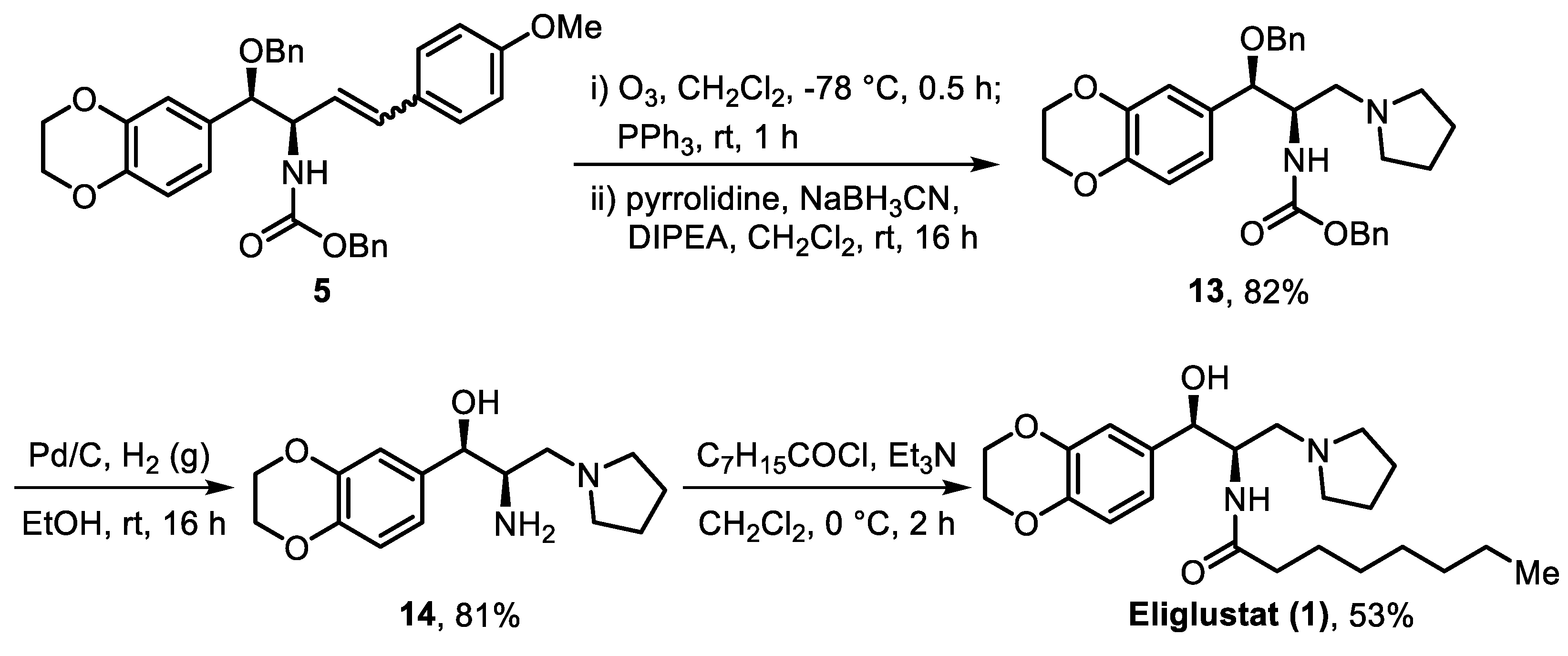
2. Results and Discussion
3. Materials and Methods
3.1. General Information
3.2. Experimental Procedure
3.2.1. Synthesis of (E)-Methyl 3-(2,3-Dihydrobenzo[b][1,4]dioxin-6-yl)acrylate (8)
3.2.2. Synthesis of Methyl (2S,3R)-3-(2,3-Dihydrobenzo[b][1,4]dioxin-6-yl)-2,3-Dihydroxypropanoate (9)
3.2.3. Synthesis of Methyl (4S,5R)-5-(2,3-Dihydrobenzo[b][1,4]dioxin-6-yl)-2,2-Dimethyl-1,3-Dioxolane-4-Carboxylate (10)
3.2.4. Synthesis of 6-((4R,5R)-5-(4-Methoxystyryl)-2,2-Dimethyl-1,3-Dioxolan-4-yl)-2,3-Dihydrobenzo[b][1,4]di-Oxine (11)
3.2.5. Synthesis of (1R,2R)-1-(2,3-Dihydrobenzo[b][1,4]dioxin-6-yl)-4-(4-Methoxyphenyl)but-3-ene-1,2-diol (12)
3.2.6. Synthesis of 6-((1R,2R)-1,2-Bis(benzyloxy)-4-(4-methoxyphenyl)but-3-en-1-yl)-2,3-dihydrobenzo[b][1,4]di-oxine (6)
3.2.7. Synthesis of Benzyl ((1R,2R)-1-(Benzyloxy)-1-(2,3-dihydrobenzo[b][1,4]dioxin-6-yl)-4-(4-Methoxyphenyl)-but-3-en-2-yl)carbamate (5)
3.2.8. Synthesis of Benzyl ((1R,2R)-1-(Benzyloxy)-1-(2,3-Dihydrobenzo[b][1,4]dioxin-6-yl)-3-(Pyrrolidin-1-yl)-Propan-2-yl)carbamate (13)
3.2.9. Synthesis of (1R,2R)-2-Amino-1-(2,3-Dihydrobenzo[b][1,4]dioxin-6-yl)-3-(Pyrrolidin-1-yl)propan-1-ol (14)
3.2.10. Synthesis of N-((1R,2R)-1-(2,3-Dihydrobenzo[b][1,4]dioxin-6-yl)-1-Hydroxy-3-(Pyrrolidin-1-yl)propan-2-yl)octanamide, Eliglustat (1)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bergmeier, S.C. The Synthesis of Vicinal Amino Alcohols. Tetrahedron 2000, 56, 2561–2576. [Google Scholar] [CrossRef]
- Ager, D.J.; Prakash, I.; Schaad, D.R. 1,2-Amino Alcohols and Their Heterocyclic Derivatives as Chiral Auxiliaries in Asymmetric Synthesis. Chem. Rev. 1996, 96, 835–876. [Google Scholar] [CrossRef]
- Paleo, M.R.; Cabeza, I.; Sardina, F.J. Enantioselective addition of doethylzinc to aldehydes catalyzed by N-(9-phenylfluoren-9-yl) beta-amino alcohols. J. Org. Chem. 2000, 65, 2108–2113. [Google Scholar] [CrossRef] [PubMed]
- Shayman, J.A. Eliglustat tartrate. Drugs Future 2010, 35, 613–620. [Google Scholar] [CrossRef]
- Bennett, L.L.; Turcotte, K. Eliglustat tartrate for the treatment of adults with type 1 Gaucher disease. Drug Des. Dev. Ther. 2015, 9, 4639–4647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poole, R.M. Eliglustat: First global approval. Drugs 2014, 74, 1829–1836. [Google Scholar] [CrossRef]
- Sehl, T.; Maugeri, Z.; Rother, D. Multi-step synthesis strategies towards 1,2-amino alcohols with special emphasis on phenylpropanolamines. J. Mol. Catal. B Enzym. 2015, 114, 65–71. [Google Scholar] [CrossRef]
- Syriopoulou, V.P.; Harding, A.L.; Goldmann, D.A.; Smith, A.L. In vitro antibacterial activity of fluorinated analogs of chloramphenicol and thiamphenicol. Antimicrob Agents Chemother. 1981, 19, 294–297. [Google Scholar] [CrossRef] [Green Version]
- Shen, J.; Wu, X.; Hu, D.; Jiang, H. Pharmacokinetics of florfenicol in healthy and Escherichia coil-infected broiler chickens. Res Vet. Sci. 2002, 73, 137–140. [Google Scholar] [CrossRef]
- Gaunt, P.S.; Langston, C.; Wrzesinski, C.; Gao, D.; Adams, P.; Crouch, L.; Sweeney, D.; Endris, R. Multidose pharmacokinetics of orally administered florfenicol in the channel catfish (Ictalurus punctatus). J. Vet. Pharmacol. Ther. 2013, 36, 502–506. [Google Scholar] [CrossRef]
- Goldstein, D.S. L-Dihydroxyphenylserine (L-DOPS): A norepinephrine prodrug. Cardiovasc. Drug Rev. 2006, 24, 189–203. [Google Scholar] [CrossRef] [PubMed]
- Mathias, C.J. L-dihydroxyphenylserine (Doxidopa) in the treatment of orthostatic hypotension. Clin. Auton. Res. 2008, 18, 25–29. [Google Scholar] [CrossRef]
- Kaufmann, H.; Freeman, R.; Biaggioni, I.; Low, P.; Pedder, S.; Hewitt, L.A.; Mauney, J.; Feirtag, M.; Mathias, C.J. Droxidopa for neurogenic orthostatic hypotension; a randomized, placebo-controlled, phase 3 trial. Neurology 2014, 83, 328–335. [Google Scholar] [CrossRef] [PubMed]
- Johnson, H.W.B.; Lowe, E.; Anderl, J.L.; Fan, A.; Muchamuel, T.; Bowers, S.; Moebius, D.C.; Kirk, C.; McMinn, D.L. Required Immunoproteasome Subunit Inhibition Profile for AntiInflammatory Efficacy and Clinical Candidate KZR-616 ((2S,3R)-N-((S)-3-(Cyclopent-1-en-1-yl)-1-((R)-2-methyloxiran-2-yl)-1-oxopropan-2-yl)-3-hydroxy-3-(4-methoxyphenyl)-2-((S)-2-(2- morpholinoacetamido)propanamido)propenamide). J. Med. Chem. 2018, 61, 11127–11143. [Google Scholar] [PubMed]
- Fang, Y.; Johnson, H.; Anderl, J.L.; Muchamuel, T.; McMinn, D.; Morisseau, C.; Hammock, B.D.; Kirk, C.; Wang, J. Role of Epoxide Hydrolases and Cytochrome P450s on Metabolism of KZR-616, a First-in-Class Selective Inhibitor of the Immunoproteasome. Drug Metab. Dispos. 2021, 49, 810–821. [Google Scholar] [CrossRef] [PubMed]
- Wang, B. N-(1-Hydroxy-3-(pyrrolidinyl)propan-2-yl)pyrrolidine-3-carboxamide Derivatives as Glucosylceramide Synthase Inhibitors. WO Patent WO2015065937 A1, 7 May 2015. [Google Scholar]
- Inokuchi, J.I.; Yoshimura, Y.; Go, S. Glucosylceramide synthase inhibitor. WO Patent WO2017204319 A1, 30 November 2017. [Google Scholar]
- Van den Berg, R.J.B.H.N.; Van Rijssel, E.R.; Ferraz, M.J.; Houben, J.; Strijland, A.; Donker-Koopman, W.E.; Wennekes, T.; Bonger, K.M.; Ghisaidoobe, A.B.T.; Hoogendoorn, S.; et al. Synthesis and Evaluation of Hybrid Structures Composed of Two Glucosylceramide Synthase Inhibitors. ChemMedChem. 2015, 10, 2042–2062. [Google Scholar] [CrossRef]
- Hirth, B.H.; Siegel, C. Synthesis of Udp-Glucose: N-Acylsphingosine Glucosyltransferase Inhibitors. WO Patent WO2003008399 A1, 30 January 2003. [Google Scholar]
- Liu, X.; Li, X.; Yang, H.; Shi, X.; Yang, F.; Jiao, X.; Xie, P. Concise and efficient synthesis of eliglustat. Synth. Commun. 2018, 48, 594–599. [Google Scholar] [CrossRef]
- Xu, X.N. Preparation Method of Eliglustat. CN Patent CN104557851 A, 29 April 2015. [Google Scholar]
- Kim, I.S.; Jung, Y.H. Recent advances in the total synthesis of indolizidine iminosugars. Heterocycles 2011, 83, 2489–2507. [Google Scholar] [CrossRef] [Green Version]
- Dong, G.R.; Hong, S.M.; Kim, S.I.; Kim, I.S.; Jung, Y.H. Stereoselective Synthesis of (–)-α-Conhydrine and Its Pyrrolidine Analogue. Eur. J. Org. Chem. 2012, 2012, 4200–4205. [Google Scholar] [CrossRef]
- Kim, S.J.; Jeon, T.H.; Min, I.S.; Kim, I.S.; Jung, Y.H. Asymmetric total synthesis of (S)-dapoxetine. Tetrahedron Lett. 2012, 53, 3680–3682. [Google Scholar] [CrossRef]
- Lee, S.H.; Park, S.J.; Kim, I.S.; Jung, Y.H. Asymmetric total synthesis of (þ)-indatraline via diastereoselective amination of chiral ethers using chlorosulfonyl isocyanate. Tetrahedron 2013, 69, 1877–1880. [Google Scholar] [CrossRef]
- Ma, S.-H.; Kim, Y.S.; Jung, J.M.; Boggu, P.R.; Kim, I.S.; Jung, Y.H. Total synthesis of chromanol 293B and cromakalim via stereoselective amination of chiral benzylic ethers. Tetrahedron Lett. 2020, 61, 151431. [Google Scholar] [CrossRef]
- Junttila, M.H.; Hormi, O.O.E. Methanesulfonamide: A Cosolvent and a General Acid Catalyst in Sharpless Asymmetric Dihydroxylations. J. Org. Chem. 2009, 74, 3038–3047. [Google Scholar] [CrossRef]
- Lee, S.H.; Kim, I.S.; Li, Q.R.; Dong, G.R.; Jeong, L.S.; Jung, Y.H. Stereoselective Amination of Chiral Benzylic Ethers Using Chlorosulfonyl Isocyanate: Total Synthesis of (+)-Sertraline. J. Org. Chem. 2011, 76, 10011–10019. [Google Scholar] [CrossRef] [PubMed]
- Weaver, D.F.; Campbell, A.J. Heterocyclic Anti-Epileptogenic Agents and Methods of Use Thereof. U.S. Patent US20030114441 A1, 19 June 2003. [Google Scholar]
- Yang, R.; Lv, M.; Xu, H. Synthesis of Piperine Analogs Containing Isoxazoline/Pyrazoline Scaffold and Their Pesticidal Bioactivities. J. Agric. Food Chem. 2018, 66, 11254–11264. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||
| Entry | Solvent | T (°C) | Yield b (%) | dr c |
| 1 | CH2Cl2 | 0 | 29 | 1.2:1 |
| 2 | CH2Cl2 | −40 | 27 | 2.3:1 |
| 3 | CH2Cl2 | −78 | 34 | 5.4:1 |
| 4 | n-Hexane | 0 | 28 | 1.3:1 |
| 5 | n-Hexane | −40 | 56 | 3.7:1 |
| 6 | n-Hexane | −78 | 59 | 5:1 |
| 7 | Toluene | 0 | 24 | 2.1:1 |
| 8 | Toluene | −40 | 51 | 5.1:1 |
| 9 | Toluene | −78 | 54 | 5.6:1 |
| 10 | Toluene/n-Hexane | −40 | 58 | 12:1 |
| 11 | Toluene/n-Hexane | −78 | 62 | >20:1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kong, Y.; Boggu, P.R.; Park, G.M.; Kim, Y.S.; An, S.H.; Kim, I.S.; Jung, Y.H. Total Synthesis of Eliglustat via Diastereoselective Amination of Chiral para-Methoxycinnamyl Benzyl Ether. Molecules 2022, 27, 2603. https://doi.org/10.3390/molecules27082603
Kong Y, Boggu PR, Park GM, Kim YS, An SH, Kim IS, Jung YH. Total Synthesis of Eliglustat via Diastereoselective Amination of Chiral para-Methoxycinnamyl Benzyl Ether. Molecules. 2022; 27(8):2603. https://doi.org/10.3390/molecules27082603
Chicago/Turabian StyleKong, Younggyu, Pulla Reddy Boggu, Gi Min Park, Yeon Su Kim, Seong Hwan An, In Su Kim, and Young Hoon Jung. 2022. "Total Synthesis of Eliglustat via Diastereoselective Amination of Chiral para-Methoxycinnamyl Benzyl Ether" Molecules 27, no. 8: 2603. https://doi.org/10.3390/molecules27082603
APA StyleKong, Y., Boggu, P. R., Park, G. M., Kim, Y. S., An, S. H., Kim, I. S., & Jung, Y. H. (2022). Total Synthesis of Eliglustat via Diastereoselective Amination of Chiral para-Methoxycinnamyl Benzyl Ether. Molecules, 27(8), 2603. https://doi.org/10.3390/molecules27082603