Abstract
Ternary potassium-iron sulfide, KFeS2, belongs to the family of highly anisotropic quasi-one-dimensional antiferromagnets with unusual “anti-Curie–Weiss” susceptibility, quasi-linearly growing with a rising temperature up to 700 K, an almost vanishing magnetic contribution to the specific heat, drastically reduced magnetic moment, etc. While some of the measurements can be satisfactorily described, the deficiency of the entropy changes upon the magnetic transition and the spin state of the iron ion remains a challenge for the further understanding of magnetism. In this work, high-quality single-crystalline samples of KFeS2 were grown by the Bridgman method, and their stoichiometry, crystal structure, and absence of alien magnetic phases were checked, utilizing wave-length dispersive X-ray electron-probe microanalysis, powder X-ray diffraction, and 57Fe Mössbauer spectroscopy, respectively. An ab initio approach was developed to calculate the thermodynamic properties of KFeS2. The element-specific phonon modes and their density of states (PDOS) were calculated applying the density functional theory in the DFT + U version, which explicitly takes into account the on-site Coulomb repulsion U of electrons and their exchange interaction J. The necessary calibration of the frequency scale was carried out by comparison with the experimental iron PDOS derived from the inelastic nuclear scattering experiment. The infrared absorption measurements confirmed the presence of two high-frequency peaks consistent with the calculated PDOS. The calibrated PDOS allowed the calculation of the lattice contribution to the specific heat of KFeS2 by direct summation over the phonon modes without approximations and adjustable parameters. The temperature-dependent magnetic specific heat evaluated by subtraction of the calculated phonon contribution from the experimental specific heat provides a lower boundary for estimating the reduced spin state of the iron ion.
1. Introduction
Ternary potassium-iron sulfide, KFeS2, belongs to the family of quasi-one-dimensional compounds with the general chemical composition AFeCh2 (where A–K or Rb; Ch–S or Se) [1,2]. These compounds share a common structural motif, i.e., a chain of edge-sharing chalcogen tetrahedra with iron ions inside (see Figure 1) [1,2,3]. Because of the strong intra-chain antiferromagnetic exchange interaction between the iron ions, these compounds are considered as model ones to study the magnetic properties and thermal behavior of a one-dimensional antiferromagnetic chain. KFeS2 is the most extensively investigated compound of this family. Nevertheless, so far, no consensus has been reached on the magnetic ground state of this compound, and the magnetic moment values presented in the literature are different from each other [4,5,6,7]. Several investigations argue that the spin state of the iron ions in this compound is low, S = 1/2 [4,5]. Therefore, in [6], it was noticed that the spin-1/2 one-dimensional Heisenberg model provides a good fit for the temperature dependence of the susceptibility of KFeS2. In contrast, based on the X-ray absorption spectroscopy study, the spin state was estimated as S = 5/2 [7]. Moreover, the magnetic moment of the iron ions in KFeS2 of about 1.9 μB obtained from neutron diffraction studies in [1] suggests that the intermediate spin state S = 3/2 is quite probable.
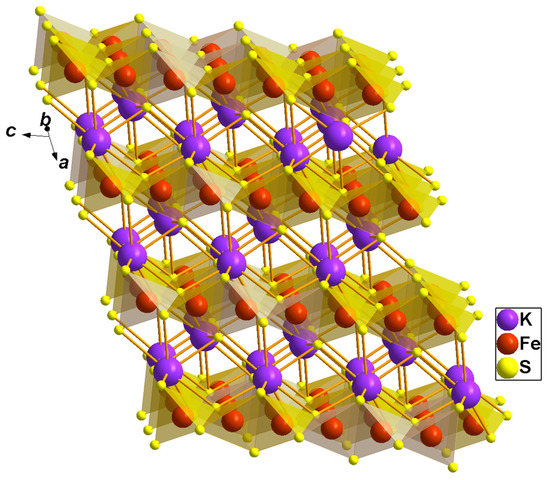
Figure 1.
Crystal structure of KFeS2. The FeS4 tetrahedra, with Fe drawn as a red sphere in the center and S as a yellow sphere at the corners and highlighted in a transparent orange color. Violet large spheres denote K.
Neutron diffraction and heat capacity studies [1,8,9] find the compound as an antiferromagnet with a Néel temperature of 250 K. The spins of the iron ions are antiferromagnetically coupled along the chain, thereby creating alternating ferromagnetic sheets [1]. However, the magnetic order of the compound is not fully established. Another study [10], based on magnetic susceptibility measurements, a molecular-field approximation, and the temperature-dependent Green’s function method, suggests that in KFeS2 the three-dimensional antiferromagnetic state sets in at a Néel temperature of 12.5 K rather than at 250 K, as is usually assumed. Hence, it was proposed to perform low-temperature total and magnetic specific heat measurements to further elucidate the nature of the magnetic ordering and the values of the exchange constants in this interesting quasi-one-dimensional system [10].
The temperature dependence of the heat capacity of the magnetic subsystem of KFeS2 could also shed light on the problem of estimating the Néel temperature and spin state of the iron ions in this compound. If the magnetic contribution to the heat capacity is determined over a wide temperature range, the change in the entropy of the magnetic subsystem contained in the transition from complete magnetic disorder in the paramagnetic state at high temperatures to an ordered state at low temperatures can be estimated and then compared with the values that are predicted for specific spin states.
The standard approach associates the magnetic contribution to the total heat capacity with the lambda anomaly at the temperature of the onset of magnetic order. The temperature dependence of the heat capacity beyond the lambda anomaly is usually treated as the lattice contribution, which can be fitted by a combination of Debye and Einstein contributions. The resulting difference between the total measured specific heat and the modeled lattice specific heat is ascribed to the magnetic contribution. However, sometimes in the case of a weak lambda anomaly, the accumulated entropy change appears to be an order of magnitude smaller than the lowest one, Rln2 = 5.76 J mol−1 K−1, expected when assuming an S = 1/2 state of Fe3+ [9,11,12,13,14,15,16,17].
In [17], doubts were expressed about the applicability of the standard description of phonons in compounds with a complex unit cell and significant anisotropy in the framework of the Debye and Einstein models. Indeed, ab initio calculations for phonon spectra and their density of states (PDOS) in the quasi-one-dimensional chain compound RbFeSe2 have shown a complicated structure of PDOS [16]. After summation over all phonon modes in the entire frequency range, the calculated lattice heat capacity appeared to be significantly different from the phenomenological one. It was shown that the magnetic contribution to the heat capacity, taken as a difference between the measured and calculated ones, extends from zero temperature to temperatures far above the Néel temperature of 250 K. The associated change in the magnetic entropy required a reasonable low-boundary value ΔSM = 6.03 J mol−1 K−1, which is in between the 5.76 J mol−1 K−1 and 11.52 J mol−1 K−1 expected for the S = 1/2 and S = 3/2 spin states, respectively. However, the temperature limit of 290 K of our equipment did not allow us to give an unambiguous answer about the spin state of the Fe3+ ions in RbFeSe2.
Applying the above-mentioned approach to KFeS2, it can be assumed that an accurate analysis of the temperature dependence of the heat capacity can make progress in resolving the existing contradictions in studies regarding the magnetic properties of this compound. To start with, we notice that KFeS2 is a poor conductor with resistivity above 103 Ω∙cm at temperatures below 250 K [18] and a semiconductor-type temperature dependence towards low temperatures. This allows the contribution of the charge carriers to the specific heat to be neglected.
Ab initio density functional theory calculations allow calculation of the phonon frequencies and their density of states [19,20]. Nevertheless, due to systematic over- or underestimation of inter-ion forces, it is known that the frequency scale of the calculated phonon density of states (PDOS) needs to be corrected [20,21].
Some experimental methods, sensitive and susceptible to the vibrational properties of solids, such as infrared (IR) absorption, Raman spectroscopy, Mössbauer spectroscopy, nuclear inelastic scattering, neutron inelastic scattering, etc., can be used for calibration of the calculated PDOS. Our calibration for RbFeSe2 was realized using IR absorption and Mössbauer spectroscopy techniques [18]. It was an implicit procedure from a qualitative comparison with the frequency dependence of the IR absorption intensity and the best fit of the temperature dependence of the Lamb–Mössbauer factor. In the present work, the nuclear inelastic scattering (NIS) method yields the partial phonon density of states in an output that can be directly compared with the calculated one. This allows the necessary frequency correction factor to be found [22,23,24]. The only limitation of this method is that the compound must contain Mössbauer nuclei, which is the case in our study (Fe in KFeS2).
The goals of the present study were: (i) to synthesize high-quality single-crystalline samples of KFeS2, (ii) to perform 57Fe nuclear inelastic scattering measurements to obtain the partial PDOS of the iron ions in this compound, (iii) to perform IR-absorption measurements, (iv) to measure the total specific heat of the single-crystalline samples of KFeS2, (v) to calculate the total and partial phonon densities of states within the DFT + U approach and check their agreement with NIS and IR-absorption experimental data, (vi), to calculate from total PDOS the lattice contribution to the specific heat of KFeS2, and (vii), eventually, to obtain the temperature dependence of the magnetic specific heat of KFeS2 as the difference between the total heat capacity and the lattice specific heat.
2. Synthesis, Structural Characterization, and Experimental Methods
KFeS2 single crystals were grown using the Bridgman method. They had a needle-like shape as was observed for the related compound RbFeSe2 [18]. Such a shape indicates the quasi-one-dimensional structure of these compounds. The elemental composition was determined using wave-length dispersive X-ray electron-probe microanalysis (WDX EPMA, Cameca SX50, Industriële Veiling Eindhoven, Eindhoven, the Netherlands), which was 24.72 at% for K, 24.72 at% for Fe, and 50.56 at% for S, respectively, resulting in the composition of K0.99Fe0.99S2.02, indicating insignificant deviation from the exact stoichiometry of KFeS2. The structural parameters of the crystals were investigated employing conventional X-ray diffraction (XRD) on powdered single crystals at room temperature using a STOE STADI P diffractometer (Darmstadt, Germany) with Cu-Kα radiation. A typical XRD pattern of our KFeS2 sample is displayed in Figure 2. The pattern reveals a narrow width of peaks that indicates a good quality of the crystals. The data were analyzed by a standard Rietveld refinement using the FullProf software [25]. The XRD pattern was described within a quasi-one-dimensional monoclinic structure with C2/c space symmetry group and lattice parameters a = 7.092(2) Å, b = 11.348(2) Å, c = 5.398(2) Å, and β = 113.2(1)°. This description correlates well with the crystallographic data reported for KFeS2 using neutron diffraction at room temperature [1]. Since all diffraction peaks were well described within the known KFeS2 structure model, it can be argued that the samples contain no impurity phases. The refinement does not fully describe the observed peak intensities. It can result from a combined effect of strain of the sample when crushed into powder and texture. Thus, from the microprobe and XRD studies, we conclude that the composition of our KFeS2 samples exhibits only small deviations from the stoichiometry and no foreign phases.
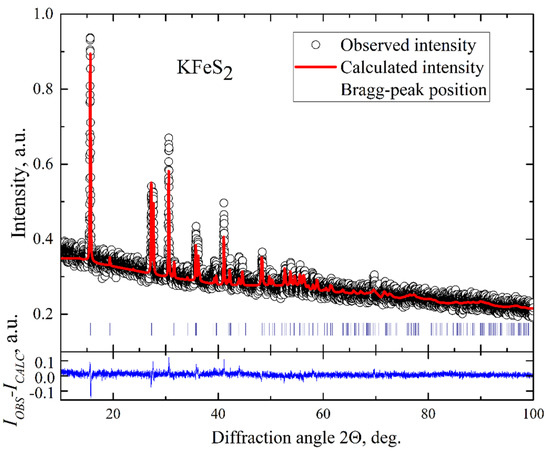
Figure 2.
Powder X-ray diffraction pattern of KFeS2. The empty circles represent the measured intensities. The solid red line is the refined pattern. The Bragg peak positions are indicated by vertical blue bars. The blue line represents the difference between the measured spectrum and the fit.
The data analysis reveals two short (2.097 Å) and two long (2.299 Å) Fe-S distances, as well as four different S-Fe-S angles, which indicate a strong distortion of the FeS4 tetrahedra. The intra-chain Fe-Fe distance of 2.699 Å exceeds the Fe-Fe distance in metallic iron by about 9%.
To exclude the possibility of sample contamination caused by iron-containing impurities with an amount below what can be detected by microprobe and XRD, we used conventional Mössbauer spectroscopy to characterize the samples. Because of strong magnetism of iron, these contaminants can complicate and compromise an accurate analysis of the magnetic properties of theKFeS2samples. The Mössbauer spectra were recorded at room temperature and a temperature of 4.2 K on a conventional constant acceleration spectrometer (Wissel, Germany) equipped with a room-temperature rhodium-matrix cobalt-57 gamma-radiation source. The observed spectra were least-square fitted with the assumption that the line shapes are Lorentzian. Beforehand, the spectrometer was calibrated at room temperature with an α-iron foil.
The room temperature spectrum is presented in Figure 3. It is an asymmetric doublet with the QS and IS values of 0.50 mm/s and 0.17 mm/s, respectively, which are in good agreement with the previously reported Mössbauer data for KFeS2 [26]. The asymmetry of the spectrum is a consequence of the quasi-one-dimensional structure of KFeS2 and the needle-like shape of the samples, which complicates the preparation of a fully isotropic powder sample. We did not detect any additional peaks above the background.
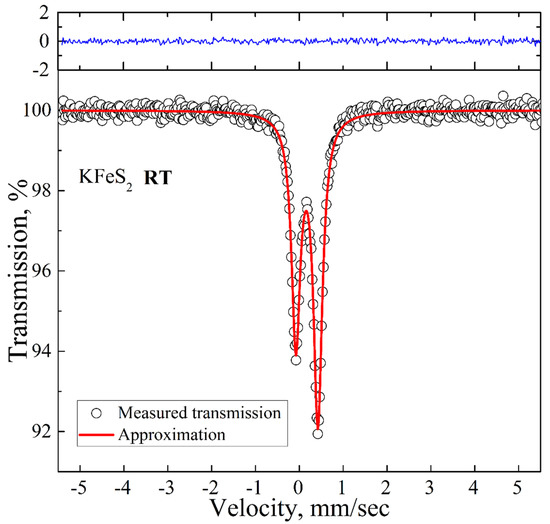
Figure 3.
Room temperature 57Fe Mössbauer spectrum of KFeS2 (empty circles). Solid red lines represent the best fitting of the spectrum obtained by the least-squares fit (red line) under the assumption that the line shapes are Lorentzian. The blue line represents the difference between the measured spectrum and the fit.
The low-temperature spectrum is presented in Figure 4. It is a magnetic sextet with the QS, IS, and HF values of 0.57 mm/s, 0.09 mm/s, and 225 kOe, respectively. The experimental spectrum is well described within a model with a single contribution of a magnetic sextet due to the long-range magnetic order already established in the compound at this temperature. The hyperfine field value of 225 kOe is in good agreement with the reported previous results on KFeS2 [27] (i.e., 237 kOe extrapolated to T = 0 K).
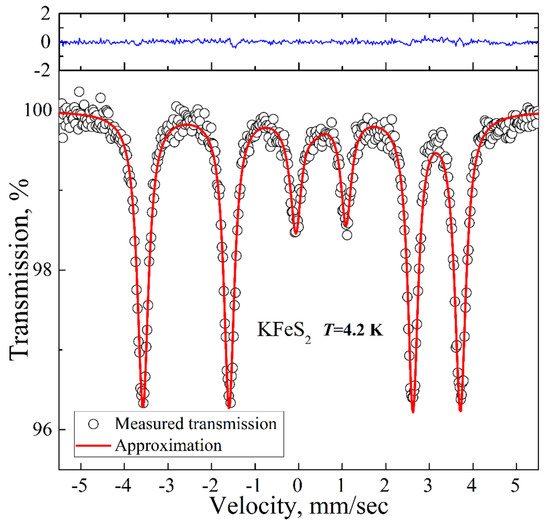
Figure 4.
Low-temperature 57Fe Mössbauer spectrum of KFeS2 (empty circles). Solid red lines represent the best fitting of the spectrum obtained by the least-squares fit (red line) under the assumption that the line shapes are Lorentzian. The blue line represents the difference between the measured spectrum and the fit.
The hyperfine parameters of KFeS2 correlate well with the Mössbauer results for the related iron chalcogenide RbFeSe2 [17]. The value of the hyperfine field is significantly smaller than the values for the high and intermediate spin states of Fe3+ in ionic oxide compounds (about 500 kOe and 300 kOe, respectively [28]). Such a low value of the hyperfine field indicates a strong reduction in the local spin moment of the iron ions. We should note that both room temperature and low-temperature spectra are well described by models with a single contribution of an asymmetric doublet and a magnetic sextet, respectively. Both model spectra are in good quantitative agreement with the previously reported results on KFeS2. Summarizing the XRD and Mössbauer spectroscopy results, we conclude that our sample is a high-quality KFeS2 crystal without iron-containing impurity phases.
The NIS [22,23] experiment was carried out by the Dynamics Beamline P01 of PETRA III synchrotron (DESY, Hamburg, Germany) [29]. The measurements utilizing the nuclear gamma-resonance of 57Fe at 14.413 keV were performed with an inline high-resolution monochromator providing an energy bandwidth of 0.9 meV full width at half maximum (FWHM). The sample with a natural abundance of 57Fe was measured at 295 K.
The IR absorption was obtained in the frequency range 4–14 THz at a Bruker IFS 113v spectrometer (Bruker Analytik GmbH, Karlsruhe, Germany). KFeS2 was ground and dispersed in CsI powder in the ratio of 1.35 mg:100 mg. This mixture was cold-pressed under vacuum to obtain a disk-like pellet with a diameter of 13 mm.
The specific heat of single-crystalline samples of KFeS2 was measured by a relaxation method using a PPMS (Quantum Design) in the temperature range 1.8 ≤ T ≤ 300 K.
3. Experimental Results
The Fe NIS spectrum of KFeS2 is shown in inset A of Figure 5. The partial Fe PDOS was evaluated from the NIS spectrum using the procedure described in [23] and is presented in the mainframe of Figure 5. The experimental infrared absorption spectrum is depicted in inset B of Figure 5.
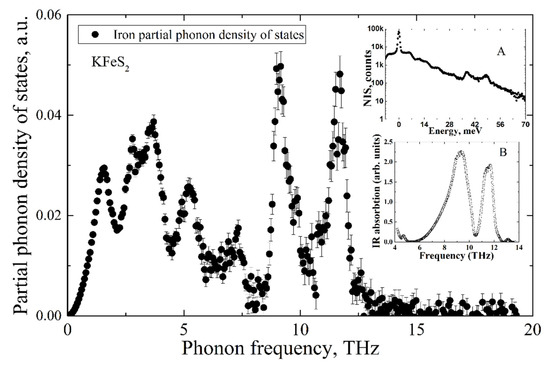
Figure 5.
Partial iron PDOS of KFeS2 evaluated from the nuclear inelastic scattering spectrum of KFeS2 (presented in the inset A) and IR-absorption spectrum (presented in inset B).
While the IR-absorption spectrum allows estimation of the non-zero PDOS only for optical phonon modes, the NIA technique measures the density of states of all phonon modes but only for iron ions. As is predicted by the classical Debye model, in the low-frequency range, the partial PDOS of iron ions depends on vibrational frequency by cubic law.
Notably, both methods of NIS and IR-absorption show two peaks of high intensity in the density of states of optical phonons in the frequency ranges of 8–10 THz and 10–12 THz. Such a good agreement of two independent methods allows for arguing for the high accuracy of the results.
4. Density Functional Theory Calculations for the Phonon Density of States of KFeS2
Density functional theory (DFT) ab initio calculations were performed by means of the Vienna Ab initio Simulation Package (VASP 5.4) [30,31,32,33]. The electron–ion interactions were taken into account using the projector-augmented wave (PAW) method, i.e., a frozen-core method including the exact shape of the wave functions of the valence electrons instead of pseudo-wave functions [34]. The Perdew–Burke–Ernzerhof (PBE) generalized gradient approximation (GGA) was applied for the exchange and correlation corrections [35]. The K (3p6 4s1), Fe (3d6 4s2), and S (3s2 3p4) electrons of the valence shell were treated explicitly, whereas the remaining electrons of the cores were taken into account by using pseudopotentials. The maximum energy for the plane-wave basis was selected to be equal to 500 eV. The k-point mesh was a 5 × 5 × 6 Monkhorst–Pack grid, which corresponds to the actual spacing of 0.227 × 0.228 × 0.221 per Å [36]. Equilibrium geometry was obtained after several stages of full structural relaxation including atomic positions, cell shape, and cell volume. The force tolerance was 0.2 eV/nm and the energy tolerance for the self-consistency loop was 10−5 eV. The phonon dispersion and density of states were obtained utilizing a direct approach of harmonic approximation making use of the MedeA Phonon commercial software [37,38]. The so-called direct approach to the lattice dynamics is based on the ab initio evaluation of forces on all atoms by a set of finite displacements of a few atoms within an otherwise perfect crystal. The phonon frequencies are calculated from the eigenvalues of the resulting dynamical matrix. The totally optimized equilibrium crystal structure was used for the calculation of the phonon dispersion. The lattice parameters obtained after the lattice relaxation are given by a = 6.89 Å, b = 11.02 Å, c = 5.10 Å, and the angle β = 111.56°. A slight deviation in the calculated lattice parameters from the experimental ones [1], of about 1–5 percent, is typical for DFT calculations [39].
All calculations accounted for the spin polarization due to the antiferromagnetic state of KFeS2. The antiferromagnetic spin pattern was set in accordance with the magnetic structure obtained previously by neutron diffraction data [13].
We published the calculated PDOS of KFeS2 within the DFT approach in [40]. The partial PDOS of the iron ions is not in good agreement with the NIS experimental data presented in the current study (Figure 5). The PDOS published in [40] was obtained with preliminary calculations within a simple DFT approach, which ignores any Hubbard-like U parameter [41]. It was mentioned that the magnetic moment of iron ions obtained in [40] is not in agreement with the neutron diffraction experimental results [1]. This may point to the strong delocalization of Fe d-electrons, and the DFT + U approach is necessary for a proper description of the system [40,42].
A simplified DFT approach corrects some of the inadequacies connected to the DFT treatment of localized states but suffers from the dependence of the results on the value of Ueff = U − J (the difference between intra-atomic Coulomb and exchange energies [43]), which de facto is an empirical parameter [44].
In [42], it was shown that the set of parameters U = 1.5 eV and J = 2 eV (Ueff = −0.5 eV) provides a correct description of the magnetic properties (magnetic moment) and electronic properties (bandgap value) of KFeS2 simultaneously. Thus, the calculations presented in this paper were carried out within the DFT + U scheme with the above-mentioned set of parameters.
Figure 6 illustrates the calculated total and element-specific PDOS as a function of frequency for KFeS2. It exhibits a complex structure distributed over two distinct frequency ranges. The vibrational modes of potassium atoms have relatively low frequencies, thus dominating the frequency range of 1–5 THz (see Figure 6, the top bar chart in blue color). Iron atoms possess the highest vibrational frequencies and constitute a substantial part of the high-frequency range (9–13 THz, see Figure 6, the second bar chart from the top in red color). Sulfur atoms show vibrational modes in the whole frequency range under consideration (see the third bar chart from the top of Figure 6). The Debye-type PDOS of the K atoms manifests vibrations almost independently of the iron atoms in the chain, as expected for weakly bound K atoms.
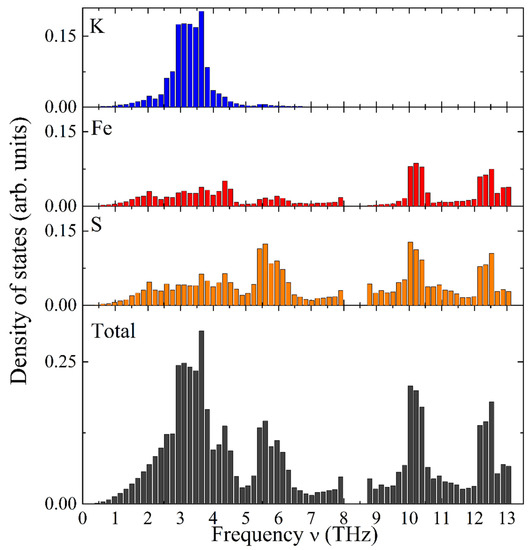
Figure 6.
Calculated PDOS as a function of frequency in KFeS2: element-specific (K, Fe, and S atoms from top towards bottom) and the total PDOS (bottom).
One more feature of the calculation method needs to be clarified before we pass to the temperature dependence of the specific heat. As we mentioned and have shown in [18], the DFT approach usually under- or overestimates vibrational frequencies of the phonon modes. A frequency correction factor is usually established by comparison of the calculated and experimental phonon spectra [18]. In the present study, the factor is obtained from a comparison of the calculated results with the experimental data on NIS and IR absorption.
The calculated PDOS is a discrete set of contributions from phonon modes, while the experimental data is a quasi-continuous sequence determined by the settings of the beamline setup, i.e., mainly by the monochromator resolution of 0.9 meV. To compare the experiment with the ab initio calculated iron partial PDOS, the latter was convoluted with a Gaussian profile with an FWHM of 0.9 meV, matching the resolution window of the monochromator. The comparison of the convolution with the experimental data is presented in Figure 7 and shows adequate agreement between the PDOS evaluated from our NIS data and the ab initio calculated PDOS with the frequency scale corrected by a factor of 0.92. The calculated pattern quantitatively describes all features of the iron PDOS within the entire frequency range of vibrations in the KFeS2 lattice.
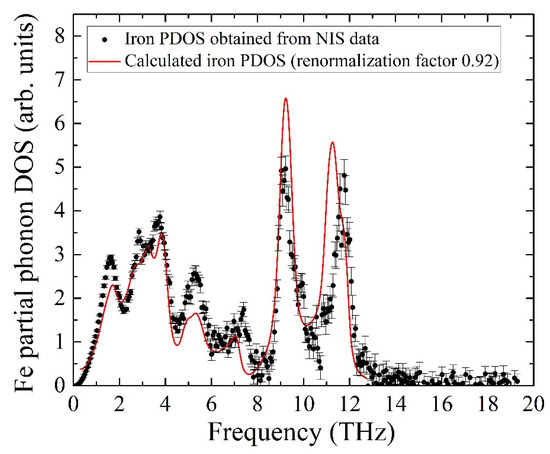
Figure 7.
Partial PDOS for iron atoms of KFeS2 obtained from the NIS spectrum (black dots) and from ab initio calculations (red line, see description in the body text).
The insignificant disagreement in amplitudes should be the result of a fast oxidation tendency of KFeS2 in the air atmosphere, which must lead to crystal structure defects. The considered sample was exposed to air for about 10 min on the probe preparation, and during the measurements, the sample was protected by Kapton scotch tape, which does not provide the best protection. Moreover, the theoretical PDOS is calculated for the case of a fully isotropic sample, while KFeS2 consists of small needle-like crystallites in the powdered sample that prevents the preparation of an isotropic sample. The last fact is important because the PDOS of KFeS2 is anisotropic.
The experimental infrared absorption spectrum shows two broad lines, which are compared with the total PDOS in Figure 8. The best agreement for the resonant frequencies of the two experimental absorption peaks with the calculated ones was obtained for the frequency correction factor of 0.92. Notably, this is the same value that we obtained from the comparison of the iron PDOS with the NIS data.
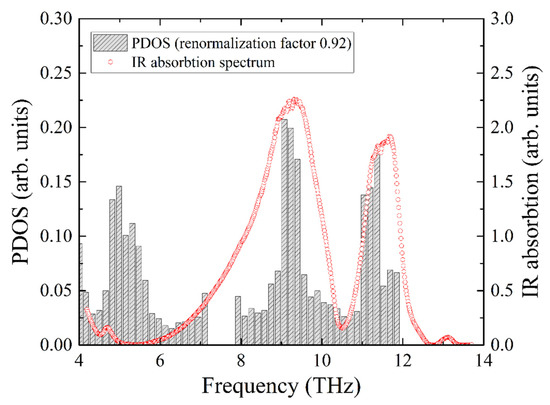
Figure 8.
Infrared absorption spectrum of KFeS2 compared with the calculated PDOS of KFeS2. The frequency scale of the PDOS is corrected to obtain the best coincidence of the peaks of PDOS maxima with the IR absorption maxima (shaded bar charts).
5. Specific Heat Analysis
The experimentally measured total specific heat C(T) of the KFeS2 single crystal is presented in Figure 9 (see the legend). An anomaly is observed in the C(T) data at TN = 247 K. The phonon contribution to the heat capacity reaches a value of 95 J mol−1 K−1 at room temperature. This value does not exceed Dulong–Petit law’s limit (about 99 J mol−1 K−1).
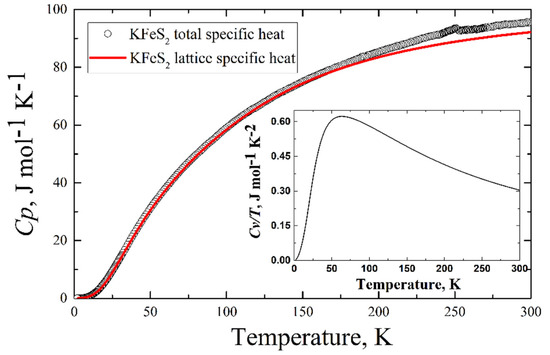
Figure 9.
Temperature dependence of the specific heat CP of KFeS2 (black circles)—experimental data, red line—calculated lattice contribution to the specific heat at constant pressure CP = CV + 0.003 [J/mol K2]T. Inset—the calculated specific heat at constant volume drawn as CV(T)/T.
The total specific heat is assumed to originate from two contributions: a lattice contribution due to acoustic and optical phonons, and a magnetic contribution determined by the thermal population of the excited magnetic states. We can estimate the magnetic specific heat as the difference between the total experimental heat capacity and the calculated vibrational part. Knowledge of the PDOS enables direct calculation of the lattice contribution to the specific heat by using the harmonic approximation [45]. In the harmonic approximation the lattice heat capacity at constant volume CV can be determined as follows [46]
where D is the number of degrees of freedom in the unit cell (three in our case), and gT(ω) is the total PDOS. Actually, direct experimental methods typically measure the specific heat at constant ambient pressure Cp. Because the specific heat is measured in a wide temperature range, the thermal expansion cannot be neglected. As was discussed in [18], from the general thermodynamic approach, Cp(T) is always larger than CV(T), and for solids, the difference can be expressed as
where α denotes the thermal expansion coefficient, V0 the molar volume, and B is the bulk modulus. The bulk modulus of KFeS2 is estimated from the second derivative of the total energy as a function of the unit cell volume by ab initio calculations: B = 10.1 GPa. The total energy per unit cell volume itself was obtained from a polynomial fit, which was truncated after the fourth-order term. The thermal expansion coefficient α is estimated from the crystal structure data c for room temperature and 14 K in reference [1]. Hence, CP ≈ CV + 0.003 (J/mol K2)T was used to calculate the specific heat shown in Figure 9 (see the legend).
The comparison of the calculated lattice contribution to the specific heat and experimentally obtained total specific heat is in Figure 9. Below 25 K, the calculated lattice contribution demonstrates a cubic dependence on temperature (the inset in Figure 9), which is predicted by the Debye model of the specific heat of solids.
The magnetic specific heat of KFeS2 is shown in Figure 10. The corresponding magnetic entropy change can be calculated from the experimentally measured magnetic specific heat as . Integrating from zero temperature to 300 K, we obtain ΔSM = 2.98 J mol−1 K−1 (see Figure 10), which is an estimation of the lower boundary, because of the temperature limit (T ≤ 300 K) of our equipment. For quasi-one-dimensional systems, one expects magnetic fluctuations to persist even significantly above the Néel temperature. This must be compared to the theoretical expectation for the possible cases of low-spin 1/2, intermediate spin 3/2, and high-spin 5/2 states for Fe3+ (3d5) atoms in KFeS2, where the corresponding entropy change ΔSM at the antiferromagnet-paramagnet order-disorder transition is given by Rln2 = 5.76 J mol−1 K−1, Rln4 = 11.52 J mol−1 K−1, and Rln6 = 14.89 J mol−1 K−1, respectively. Considering the temperature limitation of 300 K of our PPMS-9 setup, compared with the thermal stability temperature ~900–1000 K of the crystal [4], the experimentally obtained value of the magnetic entropy change ΔSM should be considered as the lower boundary, suggesting as most probable the low-spin state S = 1/2, but not excluding S = 3/2.
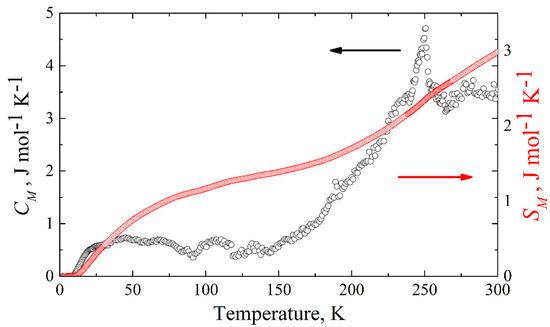
Figure 10.
Temperature dependence of the magnetic heat capacity (black empty circles) and change of the magnetic entropy (red empty squares) of KFeS2 obtained as the difference between the experimentally measured specific heat and the calculated lattice contribution.
6. Conclusions
We have performed element-specific ab initio density functional theory calculations of the vibrational properties of the covalent antiferromagnetic chain compound KFeS2. The calculations are based on the DFT + U approach to the KFeS2 band structure and magnetic moment per iron ion. Experimentally, the high-quality single-crystalline KFeS2 samples were grown utilizing the Bridgman method. The crystal structure and absence of the alien crystallographic and magnetic phases were confirmed with the powder XRD and 57Fe Mössbauer spectroscopy measurements. The temperature dependence of the specific heat of KFeS2 was measured. Then, nuclear inelastic scattering measurements on KFeS2 samples were performed and partial PDOS were evaluated from the NIS spectrum. The PDOS obtained from NIS experiments was used to calibrate the frequency scale of the DFT + U calculations by comparison of the measured and calculated iron PDOS. The IR absorption measurements showed the presence of two high-frequency peaks consistent with the calculated PDOS. The ab initio results for the total PDOS with the corrected frequency scale were used to estimate the temperature dependence of the lattice specific heat of KFeS2. The magnetic specific heat was obtained as a difference between the experimentally measured heat capacity and the calculated lattice one in the temperature range of 2–300 K. The resulting magnetic contribution to the entropy change, ΔSM(300 K) ≈ 2.98 J mol−1 K−1, obtained by integrating the magnetic specific heat, is not far below the 5.76 J mol−1 K−1 expected for the pure low-spin state of the iron ion at the disorder-order transition. It is clear that there is enough room up to the crystal stability temperature above 900 K to collect further entropy change; however, at high temperatures, the approximations made upon the lattice specific heat calculations (harmonic vibrations, independence of the phonon eigenfrequencies on temperature, thermodynamic accounting for thermal expansion, etc.) break down. The estimation of the magnetic entropy change provides, therefore, a lower boundary for the statements about the reduced value of the iron spin state. Nevertheless, such verification of the degree of correctness of the calculations on one hand, and on the other hand, obtaining information about the possible parent spin state of the iron ion, leading to the observed magnetic moment, provides motivation for the development of advanced theoretical models of spin chains, generalizing the known Bonner-Fischer model of S = 1/2 linear magnetic chains with anisotropic coupling [47].
Another important result of this study is the building of a consistent picture of the physical properties of the KFeS2 quasi-one-dimensional compound within the DFT + U approximation. Indeed, it is well known that GGA + U functionals may underestimate bandgaps, therefore, hybrid functionals [48,49,50,51] or self-interaction-corrected DFT [52,53] techniques have been used to obtain more accurate results. We used a strategy of finding U and J (U = 1.5 eV and J = 2 eV [42]) within the DFT + U route, which provides the experimentally measured magnetic moment per iron ion (1.9 μB) and band gap (0.35 eV). Then, the phonon frequencies were calculated from the eigenvalues of the dynamical matrix. The latter involves the evaluation of interatomic forces in all directions acting on atoms of supercells in the amount of 18 for the particular KFeS2 lattice. Thus, in our case for each supercell containing 96 atoms the dynamic matrix accumulates 96 × 18 × 3 = 5184 force constants. The resulting PDOS for iron ions is compared with the experimentally measured PDOS obtained from nuclear inelastic scattering and shows quite good agreement, thus, opening prospects for the analysis of thermal properties. The overall consistency of the calculated magnetic moment, bandgap, and iron PDOS with the experimental results make the DFT + U scheme promising for studying the physical properties of the KFeS2-family compound by the low-cost ab initio calculation methods in the light of recent attempts to search for high-temperature superconductivity in such compounds [54].
Author Contributions
Conceptualization, A.K., Z.S. and H.-A.K.v.N.; data curation, A.K.; formal analysis, A.K., M.K. and H.-A.K.v.N.; investigation, A.K., D.C., V.T., F.M., S.W., F.V. and L.T.; software, A.K.; validation, A.K. and D.T.; visualization, A.K., M.K., I.F. and L.T.; writing—original draft, A.K.; writing—review and editing, A.K., H.-A.K.v.N., Z.S., D.T. and L.T.; funding acquisition, D.T.; project administration, D.T. All authors have read and agreed to the published version of the manuscript.
Funding
The reported study was funded by RFBR, projects №19-52-12068, and DFG according to project #KR2254/3-1. V.T., I.F. and D.C. acknowledge the support from the project ANCD 20.80009.5007.19 (Moldova).
Data Availability Statement
Not applicable.
Acknowledgments
NIS measurements were performed at the beamlines P01 (PETRA-III, DESY), and we thank PETRA-III, DESY, for the provision of beamtime; we are especially grateful for the scientific support from Hans-Christian Wille, Ilya Sergueev, and Olaf Leupold.
Conflicts of Interest
The authors declare no conflict of interest.
Sample Availability
Not applicable.
References
- Bronger, W.; Kyas, A.; Müller, P. The antiferromagnetic structures of KFeS2, RbFeS2, KFeSe2, and RbFeSe2 and the correlation between magnetic moments and crystal field calculations. J. Solid State Chem. 1987, 70, 262–270. [Google Scholar] [CrossRef]
- Harrison, M.R.; Francesconi, M.G. Mixed-metal one-dimensional sulfides—A class of materials with differences and similarities to oxides. Coord. Chem. Rev. 2011, 255, 451–458. [Google Scholar] [CrossRef]
- Liang, D.; Chen, Y.; Gao, Y.; Sun, T.; Ri, K.; Xie, X. Low-temperature conversion of Fe-rich sludge to KFeS2 whisker: A new flocculant synthesis from laboratory scale to pilot scale. Sustain. Environ. Res. 2021, 31, 25. [Google Scholar] [CrossRef]
- Tiwary, S.K.; Vasudevan, S. Regular versus alternating [FeS4]n chains: Magnetism in KFeS2 and CsFeS2. Phys. Rev. B 1997, 56, 7812. [Google Scholar] [CrossRef]
- Tiwary, S.K.; Vasudevan, S. Spin state and exchange in the quasi-one-dimensional antiferromagnet KFeS2. Pramana J. Phys. 1997, 48, 1123–1124. [Google Scholar] [CrossRef]
- Souza, A.M.C.; Oliveira Neto, S.R.; Macedo, C.A. Magnetic behavior of the KFeS2. J. Magn. Magn. Mater. 2004, 272–276, 521–522. [Google Scholar] [CrossRef]
- Atanasov, M.; Potze, R.H.; Sawatzky, G.A. Electronic structure of tetrahedral iron(III)—Sulfur clusters in alkaline thioferrates: An X-ray absorption study. J. Solid State Chem. 1995, 119, 380–393. [Google Scholar] [CrossRef]
- Nissen, H.P.; Nagorny, K. Mossbauer spectroscopic and magnetic studies of alkaline thioferrates (iii). Z. Phys. Chem. 1975, 95, 301–304. [Google Scholar] [CrossRef]
- Johnston, D.C.; Mraw, S.C.; Jacobson, A.J. Observation of the antiferromagnetic transition in the linear chain compound KFeS2 by magnetic susceptibility and heat capacity measurements. Solid State Commun. 1982, 44, 255–258. [Google Scholar] [CrossRef]
- Mauger, A.; Escorne, M.; Taft, C.A.; Furtado, N.C.; Arguello, Z.P.; Arsenio, T.P. Exchange interactions in the quasi-linear-chain antiferromagnet KFeS2. Phys. Rev. B 1984, 30, 5300. [Google Scholar] [CrossRef]
- Aldzhanov, M.; Guseinov, N.; Sultanov, G.; Nadzafzade, M. Magnetic Heat Capacity and Susceptibility of the Pseudo-One-Dimensional Magnetic Systems TlFeS2 and TlFeSe2. Phys. Status Solidi B 1990, 159, K107–K110. [Google Scholar] [CrossRef]
- He, J.B.; Wang, D.M.; Shi, H.L.; Yang, H.X.; Li, J.Q.; Chen, G.F. Synthesis, structure, and magnetic properties of the layered iron oxychalcogenide Na2Fe2Se2O. Phys. Rev. B 2011, 84, 205212. [Google Scholar] [CrossRef]
- Lei, H.; Ryu, H.; Frenkel, A.I.; Petrovic, C. Anisotropy in BaFe2Se3 single crystals with double chains of FeSe tetrahedral. Phys. Rev. B 2011, 84, 214511. [Google Scholar] [CrossRef]
- Nambu, Y.; Ohgushi, K.; Suzuki, S.; Du, F.; Avdeev, M.; Uwatoko, Y.; Munakata, K.; Fukazawa, H.; Chi, S.; Ueda, Y.; et al. Block magnetism coupled with local distortion in the iron-based spin-ladder compound BaFe2Se3. Phys. Rev. B 2012, 85, 064413. [Google Scholar] [CrossRef]
- Lei, H.; Ryu, H.; Ivanovski, V.; Warren, J.B.; Frenkel, A.I.; Cekic, B.; Yin, W.-G.; Petrovic, C. Structure and physical properties of the layered iron oxychalcogenide BaFe2Se2O. Phys. Rev. B 2012, 85, 195133. [Google Scholar] [CrossRef]
- Du, F.; Ohgushi, K.; Nambu, Y.; Kawakami, T.; Avdeev, M.; Hirata, Y.; Watanabe, Y.; Sato, T.J.; Ueda, Y. Stripelike magnetism in a mixed-valence insulating state of the Fe-based ladder compound CsFe2Se3. Phys. Rev. B 2012, 85, 214436. [Google Scholar] [CrossRef]
- Seidov, Z.; Tsurkan, V.; Vagizov, F.; Krug von Nidda, H.-A.; Filipova, I.; Günther, A.; Gavrilova, T.; Kiiamov, A.G.; Tagirov, L.R.; Loidl, A. Magnetic properties of the covalent chain antiferromagnet RbFeSe2. Phys. Rev. B 2016, 94, 134414. [Google Scholar] [CrossRef]
- Nishioka, S.; Kuriyaki, H.; Hirakawa, K. Electric Conduction in Quasi-One-Dimensional Compound KFeS2. Synth. Met. 1995, 71, 1877–1878. [Google Scholar] [CrossRef]
- Kiiamov, A.G.; Lysogorskiy, Y.V.; Vagizov, F.G.; Tagirov, L.R.; Tayurskii, D.A.; Seidov, Z.; Krug von Nidda, H.-A.; Tsurkan, V.; Croitori, D.; Günther, A.; et al. Vibrational properties and magnetic specific heat of the covalent chain antiferromagnet RbFeSe2. Phys. Rev. B 2018, 98, 214411. [Google Scholar] [CrossRef]
- Kiiamov, A.; Seidov, Z.; Croitori, D.; Tsurkan, V.; Krug Von Nidda, H.-A.; Günther, A.; Tagirov, L.; Tayurskii, D. Magnetic and vibrational properties of the covalent chain antiferromagnet RbFeS2. J. Phys. Conf. Ser. 2022, 2164, 012026. [Google Scholar] [CrossRef]
- Dal Corso, A. Ab initio phonon dispersions of transition and noble metals: Effects of the exchange and correlation functional. J. Phys. Cond. Matter 2013, 25, 145401. [Google Scholar] [CrossRef] [PubMed]
- Rüffer, R.; Chumakov, A.I. Nuclear inelastic scattering. Hyperfine Interact. 2000, 128, 255–272. [Google Scholar] [CrossRef]
- Kohn, V.G.; Chumakov, A.I. DOS: Evaluation of phonon density of states from nuclear resonant inelastic absorption. Hyperfine Interact. 2000, 125, 205–221. [Google Scholar] [CrossRef]
- Kiiamov, A.; Tsurkan, V.; Croitori, D.; Krug von Nidda, H.-A.; Seidov, Z.; Wille, H.-C.; Sergueev, I.; Leupold, O.; Tayurskii, D.; Tagirov, L. Application of Nuclear Inelastic Scattering Spectroscopy to the Frequency Scale Calibration of Ab Initio Calculated Phonon Density of States of Quasi-One-Dimensional Ternary Iron Chalcogenide RbFeSe2. Appl. Sci. 2020, 10, 7212. [Google Scholar] [CrossRef]
- Rodriguez-Carvajal, J. Recent advances in magnetic structure determination by neutron powder diffraction + FullProf. Phys. B Condens. Matter 1993, 192, 55–56. [Google Scholar] [CrossRef]
- Jacobson, A.J.; McCandlish, L.E. In situ Mössbauer spectroscopy studies of the electrochemical reaction of lithium with KFeS2. J. Solid State Chem. 1979, 29, 355–365. [Google Scholar] [CrossRef]
- Zink, J.; Nagorny, P. Mössbauer spectroscopic investigations of the magnetic properties of potassium thioferrate-III. J. Phys. Chem. Solids 1988, 49, 1429–1434. [Google Scholar] [CrossRef]
- Wertheim, G.K. Mössbauer Effect: Principles and Applications; Academic Press: New York, NY, USA; London, UK, 1964. [Google Scholar]
- Wille, H.-C.; Franz, H.; Röhlsberger, R.; Caliebe, W.A.; Dill, F.-U. Nuclear resonant scattering at PETRA III: Brillant opportunities for nano- and extreme condition science. J. Phys. Conf. Ser. 2009, 217, 012008. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 1993, 47, 558. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initio molecular-dynamics simulation of the liquid-metal–amorphous-semiconductor transition in germanium. Phys. Rev. B 1994, 49, 14251. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmuller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comp. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmuller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169. [Google Scholar] [CrossRef] [PubMed]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188. [Google Scholar] [CrossRef]
- Parlinski, K.; Li, Z.Q.; Kawazoe, Y. First-Principles Determination of the Soft Mode in Cubic ZrO2. Phys. Rev. Lett. 1997, 78, 4063. [Google Scholar] [CrossRef]
- MedeA Version 2.20; MedeA Software: San Diego, CA, USA, 2018.
- Lysogorskiy, Y.V.; Eremina, R.M.; Gavrilova, T.P.; Nedopekin, O.V.; Tayurskii, D.A. Vibrational and magnetic properties of crystalline CuTe2O5. JETP Lett. 2015, 100, 652–656. [Google Scholar] [CrossRef]
- Kiiamov, A.; Lysogorskiy, Y.V.; Seidov, Z.; Krug von Nidda, H.-A.; Tsurkan, V.; Tayurskii, D.A.; Tagirov, L.R. Vibrational properties and lattice specific heat of KFeS2. AIP Conf. Proc. 2018, 2041, 040002. [Google Scholar] [CrossRef]
- Hubbard, J. Electron correlations in narrow energy bands. Proc. R. Soc. Lond. A 1963, 276, 238–257. [Google Scholar] [CrossRef]
- Kiiamov, A.G.; Kuznetsov, M.D.; Batulin, R.G.; Tayurskii, D.A. On the ab initio Calculations within DFT+ U Approach of Physical Properties of a Compound with Strong Electron-Electron Correlations by the Case of KFeS2. JETP Lett. 2022, 115, 98–101. [Google Scholar] [CrossRef]
- Carter, E.A. Challenges in modeling materials properties without experimental input. Science 2008, 321, 5890. [Google Scholar] [CrossRef] [PubMed]
- Finazzi, E.; Valentin, C.D.; Pacchioni, G.; Selloni, A. Excess electron states in reduced bulk anatase TiO2: Comparison of standard GGA, GGA+U, and hybrid DFT calculations. J. Chem. Phys. 2008, 129, 15. [Google Scholar] [CrossRef] [PubMed]
- Kittel, C. Introduction to Solid State Physics, 7th ed.; Wiley: New York, NY, USA, 1996; Chapter 4–5. [Google Scholar]
- Ziman, J.M. Electrons and Phonons, the International Series of Monographs on Physics; Clarendon Press: Oxford, UK, 1960; Chapter 1. [Google Scholar]
- Bonner, J.; Fischer, M. Linear magnetic chains with anisotropic coupling. Phys. Rev. 1964, 135, A640. [Google Scholar] [CrossRef]
- Heyd, J.; Scuseria, G.E.; Ernzerhof, M. Hybrid functionals based on a screened Coulomb potential. J. Chem. Phys. 2003, 118, 8207. [Google Scholar] [CrossRef]
- Krukau, A.V.; Vydrov, O.A.; Izmaylov, A.F.; Scuseria, G.E. Influence of the exchange screening parameter on the performance of screened hybrid functionals. J. Chem. Phys. 2006, 125, 224106. [Google Scholar] [CrossRef] [PubMed]
- Qin, L.; Duan, Y.; Shi, H.; Shi, L.; Tang, G. Hybrid density functional theory studies of AlN and GaN under uniaxial strain. J. Phys. Condens. Matter 2013, 25, 045801. [Google Scholar] [CrossRef]
- Lv, D.; Duan, Y.; Zhao, B.; Qin, L.; Shi, L.; Tang, G.; Shi, H. Comparing the effects of uniaxial and biaxial strains on the structural stability and electronic structure in wurtzite ZnS. J. Appl. Phys. 2013, 25, 045801. [Google Scholar] [CrossRef]
- Shinde, R.; Yamijala, S.S.R.K.C.; Wong, B.M. Improved band gaps and structural properties from Wannier–Fermi–Löwdin self-interaction corrections for periodic systems. J. Phys. Condens. Matter 2021, 33, 115501. [Google Scholar] [CrossRef]
- Tang, C.; Ostrikov, K.K.; Sanvito, S.; Du, A. Prediction of room-temperature ferromagnetism and large perpendicular magnetic anisotropy in a planar hypercoordinate FeB3 monolayer. Nanoscale Horiz. 2021, 6, 43–48. [Google Scholar] [CrossRef]
- Xinlei, Z.; Fengjie, M.; Zhing-Yi, L.; Tao, X. AFeSe2 (A=Tl, K, Rb, or Cs): Iron-based superconducting analog of the cuprates. Phys. Rev. B 2020, 101, 184504. [Google Scholar] [CrossRef]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).