
Combined Treatment with PI3K Inhibitors BYL-719 and CAL-101 Is a Promising Antiproliferative Strategy in Human Rhabdomyosarcoma Cells

 , , ,
, , , 
 , and
, and 
Abstract
:1. Introduction
2. Results
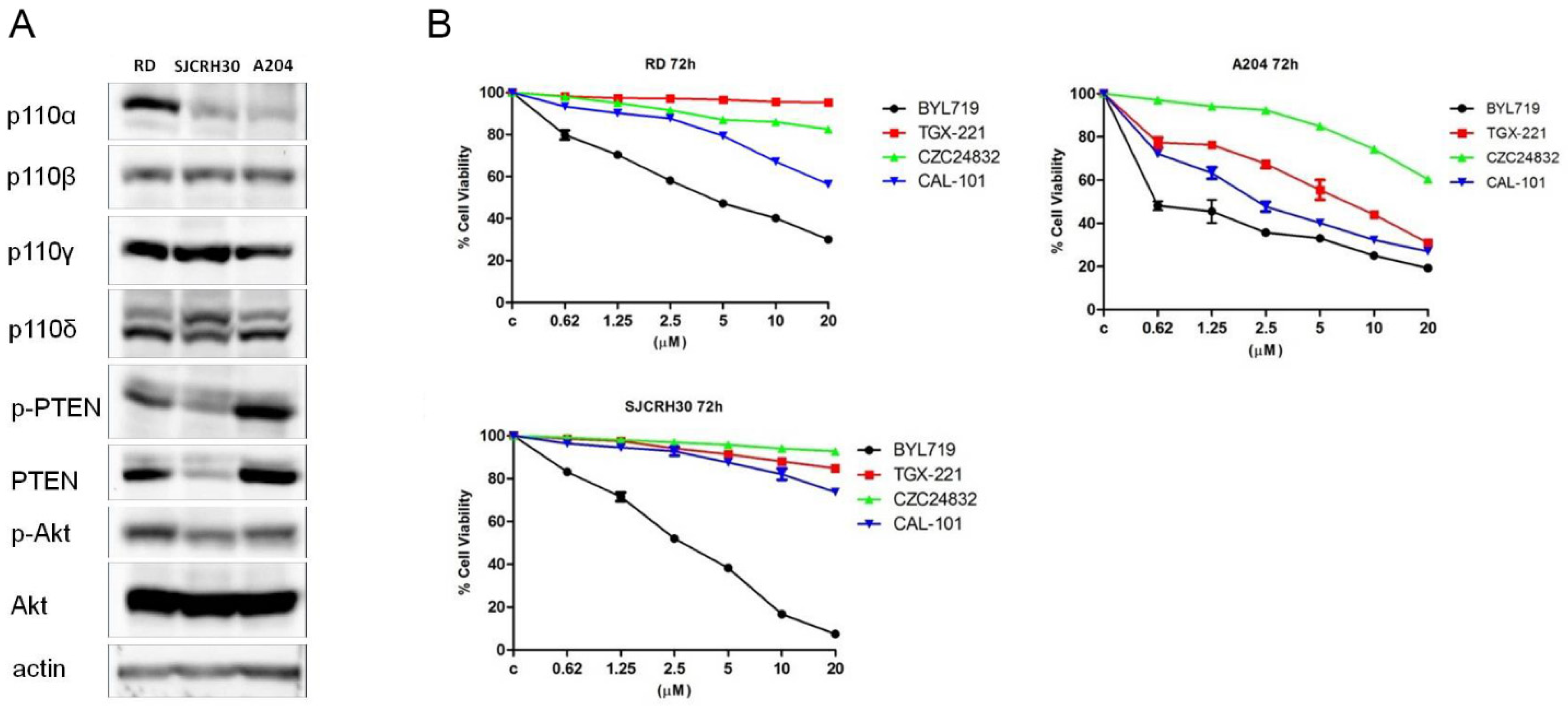
2.1. Single Treatment of Rhabdomyosarcoma Cell Lines with Alpelisib (BYL-719) and Idelalisib (CAL-101) Reduced Cell Proliferation
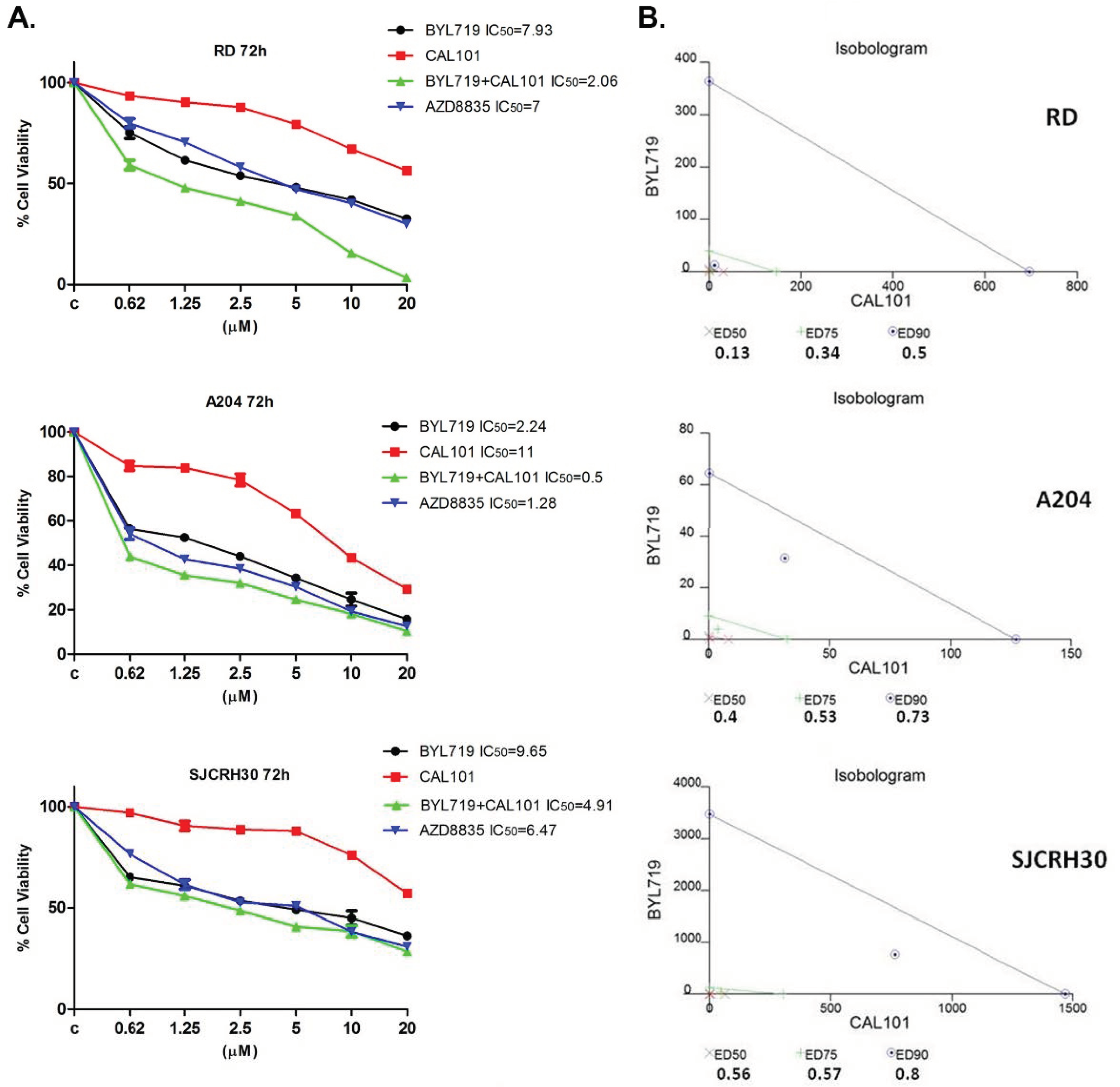
2.2. The Combined Treatment of Alpelisib (BYL-719) and Idelalisib (CAL-101) Reduce the Viability of RMS Cell Lines In Vitro in a Synergistic Manner
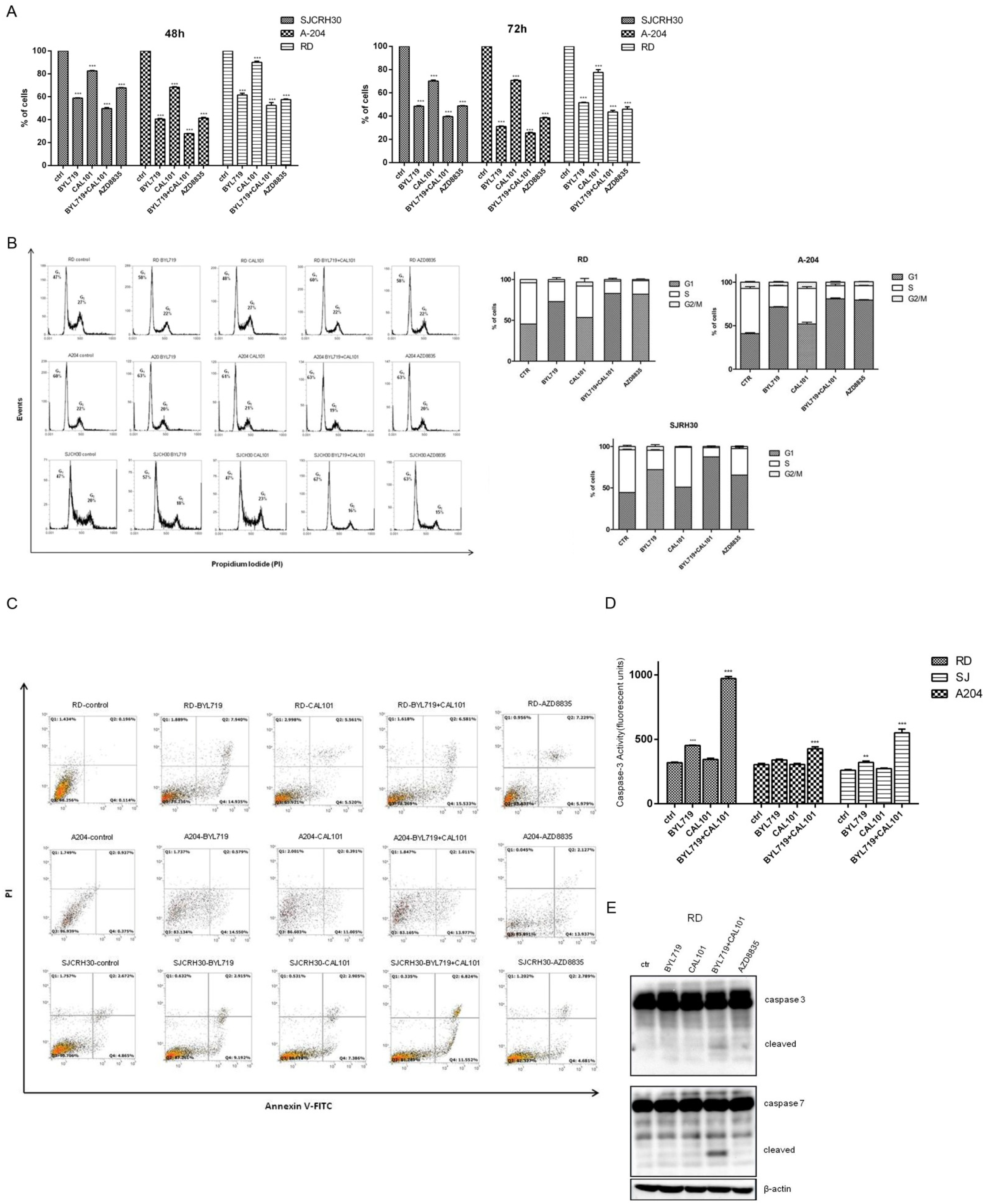
2.3. Effects of the Combined Treatment of Alpelisib (BYL-719) and Idelalisib (CAL-101) on Rhabdomyosarcoma Cell Homeostasis
2.3.1. BYL-719 + CAL-101 Induced Cell Cycle Arrest
2.3.2. BYL-719 + CAL-101 Sensitize Cells to Caspase-Dependent Apoptosis
2.4. The Combined Treatment of Alpelisib (BYL-719) and Idelalisib (CAL-101) Inhibited the Activity of Pi3k Downstream Signaling Effectors
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Reagent
4.2. MTT Assay
4.3. Protein Extraction and Western Blotting
4.4. Cell Viability
4.5. Cell Cycle Analysis
4.6. Apoptosis Assay
4.7. Fluorimetric Caspase-3 Enzyme Activity Assay
4.8. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Skapek, S.X.; Ferrari, A.; Gupta, A.A.; Lupo, P.J.; Butler, E.; Shipley, J.; Barr, F.G.; Hawkins, D.S. Rhabdomyosarcoma. Nat. Rev. Dis. Primers 2019, 5, 1–19. [Google Scholar] [CrossRef]
- Gallego Melcon, S.; Sanchez de Toledo Codina, J. Molecular biology of rhabdomyosarcoma. Clin. Transl. Oncol. 2007, 9, 415–419. [Google Scholar] [CrossRef]
- Scrable, H.; Witte, D.; Shimada, H.; Seemayer, T.; Sheng, W.W.; Soukup, S.; Koufos, A.; Houghton, P.; Lampkin, B.; Cavenee, W. Molecular differential pathology of rhabdomyosarcoma. Genes Chromosom. Cancer 1989, 1, 23–35. [Google Scholar] [CrossRef]
- Nguyen, T.H.; Barr, F.G. Therapeutic Approaches Targeting PAX3-FOXO1 and Its Regulatory and Transcriptional Pathways in Rhabdomyosarcoma. Molecules 2018, 23, 2798. [Google Scholar] [CrossRef] [Green Version]
- Davis, R.J.; Barr, F.G. Fusion genes resulting from alternative chromosomal translocations are overexpressed by gene-specific mechanisms in alveolar rhabdomyosarcoma. Proc. Natl. Acad. Sci. USA 1997, 94, 8047–8051. [Google Scholar] [CrossRef] [Green Version]
- Soleimani, V.D.; Rudnicki, M.A. New insights into the origin and the genetic basis of rhabdomyosarcomas. Cancer Cell 2011, 19, 157–159. [Google Scholar] [CrossRef] [Green Version]
- Aslam, M.I.; Hettmer, S.; Abraham, J.; Latocha, D.; Soundararajan, A.; Huang, E.T.; Goros, M.W.; Michalek, J.E.; Wang, S.; Mansoor, A.; et al. Dynamic and nuclear expression of PDGFRalpha and IGF-1R in alveolar Rhabdomyosarcoma. Mol. Cancer Res. 2013, 11, 1303–1313. [Google Scholar] [CrossRef] [Green Version]
- McKinnon, T.; Venier, R.; Yohe, M.; Sindiri, S.; Gryder, B.E.; Shern, J.F.; Kabaroff, L.; Dickson, B.; Schleicher, K.; Chouinard-Pelletier, G.; et al. Functional screening of FGFR4-driven tumorigenesis identifies PI3K/mTOR inhibition as a therapeutic strategy in rhabdomyosarcoma. Oncogene 2018, 37, 2630–2644. [Google Scholar] [CrossRef]
- Chelsky, Z.L.; Paulson, V.A.; Chen, E.Y. Molecular analysis of 10 pleomorphic rhabdomyosarcomas reveals potential prognostic markers and druggable targets. Genes Chromosom. Cancer 2022, 61, 138–147. [Google Scholar] [CrossRef]
- Crose, L.E.; Linardic, C.M. Receptor tyrosine kinases as therapeutic targets in rhabdomyosarcoma. Sarcoma 2011, 2011, 756982. [Google Scholar] [CrossRef] [Green Version]
- Ramadan, F.; Fahs, A.; Ghayad, S.E.; Saab, R. Signaling pathways in Rhabdomyosarcoma invasion and metastasis. Cancer Metastasis Rev. 2020, 39, 287–301. [Google Scholar] [CrossRef]
- Monti, E.; Fanzani, A. Uncovering metabolism in rhabdomyosarcoma. Cell Cycle 2016, 15, 184–195. [Google Scholar] [CrossRef] [Green Version]
- Crist, W.; Gehan, E.A.; Ragab, A.H.; Dickman, P.S.; Donaldson, S.S.; Fryer, C.; Hammond, D.; Hays, D.M.; Herrmann, J.; Heyn, R.; et al. The Third Intergroup Rhabdomyosarcoma Study. J. Clin. Oncol. 1995, 13, 610–630. [Google Scholar] [CrossRef]
- Breneman, J.C.; Lyden, E.; Pappo, A.S.; Link, M.P.; Anderson, J.R.; Parham, D.M.; Qualman, S.J.; Wharam, M.D.; Donaldson, S.S.; Maurer, H.M.; et al. Prognostic factors and clinical outcomes in children and adolescents with metastatic rhabdomyosarcoma--a report from the Intergroup Rhabdomyosarcoma Study IV. J. Clin. Oncol. 2003, 21, 78–84. [Google Scholar] [CrossRef]
- Thorpe, L.M.; Yuzugullu, H.; Zhao, J.J. PI3K in cancer: Divergent roles of isoforms, modes of activation and therapeutic targeting. Nat. Rev. Cancer 2015, 15, 7–24. [Google Scholar] [CrossRef]
- Kohsaka, S.; Shukla, N.; Ameur, N.; Ito, T.; Ng, C.K.; Wang, L.; Lim, D.; Marchetti, A.; Viale, A.; Pirun, M.; et al. A recurrent neomorphic mutation in MYOD1 defines a clinically aggressive subset of embryonal rhabdomyosarcoma associated with PI3K-AKT pathway mutations. Nat. Genet. 2014, 46, 595–600. [Google Scholar] [CrossRef] [Green Version]
- Liu, P.; Cheng, H.; Roberts, T.M.; Zhao, J.J. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat. Rev. Drug Discov. 2009, 8, 627–644. [Google Scholar] [CrossRef] [Green Version]
- Cantrell, D.A. Phosphoinositide 3-kinase signalling pathways. J. Cell Sci. 2001, 114, 1439–1445. [Google Scholar] [CrossRef]
- Thorpe, L.M.; Spangle, J.M.; Ohlson, C.E.; Cheng, H.; Roberts, T.M.; Cantley, L.C.; Zhao, J.J. PI3K-p110alpha mediates the oncogenic activity induced by loss of the novel tumor suppressor PI3K-p85alpha. Proc. Natl. Acad. Sci. USA 2017, 114, 7095–7100. [Google Scholar] [CrossRef] [Green Version]
- Janku, F.; Yap, T.A.; Meric-Bernstam, F. Targeting the PI3K pathway in cancer: Are we making headway? Nat. Rev. Clin. Oncol. 2018, 15, 273–291. [Google Scholar] [CrossRef]
- Alpelisib. In LiverTox: Clinical and Research Information on Drug-Induced Liver Injury; National Institute for Diabetes and Digestive and Kidney Diseases: Bethesda, MD, USA, 2012.
- Dienstmann, R.; Rodon, J.; Serra, V.; Tabernero, J. Picking the point of inhibition: A comparative review of PI3K/AKT/mTOR pathway inhibitors. Mol. Cancer Ther. 2014, 13, 1021–1031. [Google Scholar] [CrossRef] [Green Version]
- Vanhaesebroeck, B.; Perry, M.W.D.; Brown, J.R.; Andre, F.; Okkenhaug, K. PI3K inhibitors are finally coming of age. Nat. Rev. Drug Discov. 2021, 20, 741–769. [Google Scholar] [CrossRef]
- Massacesi, C.; Di Tomaso, E.; Urban, P.; Germa, C.; Quadt, C.; Trandafir, L.; Aimone, P.; Fretault, N.; Dharan, B.; Tavorath, R.; et al. PI3K inhibitors as new cancer therapeutics: Implications for clinical trial design. Onco. Targets Ther. 2016, 9, 203–210. [Google Scholar] [CrossRef] [Green Version]
- Akinleye, A.; Avvaru, P.; Furqan, M.; Song, Y.; Liu, D. Phosphatidylinositol 3-kinase (PI3K) inhibitors as cancer therapeutics. J. Hematol. Oncol. 2013, 6, 88. [Google Scholar] [CrossRef] [Green Version]
- Mishra, R.; Patel, H.; Alanazi, S.; Kilroy, M.K.; Garrett, J.T. PI3K Inhibitors in Cancer: Clinical Implications and Adverse Effects. Int. J. Mol. Sci 2021, 22, 3464. [Google Scholar] [CrossRef]
- Yang, J.; Nie, J.; Ma, X.; Wei, Y.; Peng, Y.; Wei, X. Targeting PI3K in cancer: Mechanisms and advances in clinical trials. Mol. Cancer 2019, 18, 26. [Google Scholar] [CrossRef] [Green Version]
- Gong, G.Q.; Kendall, J.D.; Dickson, J.M.J.; Rewcastle, G.W.; Buchanan, C.M.; Denny, W.A.; Shepherd, P.R.; Flanagan, J.U. Combining properties of different classes of PI3Kalpha inhibitors to understand the molecular features that confer selectivity. Biochem. J. 2017, 474, 2261–2276. [Google Scholar] [CrossRef]
- Markham, A. Alpelisib: First Global Approval. Drugs 2019, 79, 1249–1253. [Google Scholar] [CrossRef]
- Sidaway, P. Alpelisib effective in advanced-stage disease. Nat. Rev. Clin. Oncol. 2019, 16, 466. [Google Scholar] [CrossRef]
- Xu, H.; Chen, K.; Shang, R.; Chen, X.; Zhang, Y.; Song, X.; Evert, M.; Zhong, S.; Li, B.; Calvisi, D.F.; et al. Alpelisib combination treatment as novel targeted therapy against hepatocellular carcinoma. Cell Death Dis. 2021, 12, 920. [Google Scholar] [CrossRef]
- Yang, X.; Yang, J.A.; Liu, B.H.; Liao, J.M.; Yuan, F.E.; Tan, Y.Q.; Chen, Q.X. TGX-221 inhibits proliferation and induces apoptosis in human glioblastoma cells. Oncol. Rep. 2017, 38, 2836–2842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Markham, A. Idelalisib: First global approval. Drugs 2014, 74, 1701–1707. [Google Scholar] [CrossRef] [PubMed]
- Byrd, J.C.; Woyach, J.A.; Johnson, A.J. Translating PI3K-Delta Inhibitors to the Clinic in Chronic Lymphocytic Leukemia: The Story of CAL-101 (GS1101). Am. Soc. Clin. Oncol. Educ. Book 2012, 32, 691–694. [Google Scholar] [CrossRef] [PubMed]
- Herman, S.E.; Gordon, A.L.; Wagner, A.J.; Heerema, N.A.; Zhao, W.; Flynn, J.M.; Jones, J.; Andritsos, L.; Puri, K.D.; Lannutti, B.J.; et al. Phosphatidylinositol 3-kinase-delta inhibitor CAL-101 shows promising preclinical activity in chronic lymphocytic leukemia by antagonizing intrinsic and extrinsic cellular survival signals. Blood 2010, 116, 2078–2088. [Google Scholar] [CrossRef] [Green Version]
- Lannutti, B.J.; Meadows, S.A.; Herman, S.E.; Kashishian, A.; Steiner, B.; Johnson, A.J.; Byrd, J.C.; Tyner, J.W.; Loriaux, M.M.; Deininger, M.; et al. CAL-101, a p110delta selective phosphatidylinositol-3-kinase inhibitor for the treatment of B-cell malignancies, inhibits PI3K signaling and cellular viability. Blood 2011, 117, 591–594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barlaam, B.; Cosulich, S.; Delouvrie, B.; Ellston, R.; Fitzek, M.; Germain, H.; Green, S.; Hancox, U.; Harris, C.S.; Hudson, K.; et al. Discovery of 1-(4-(5-(5-amino-6-(5-tert-butyl-1,3,4-oxadiazol-2-yl)pyrazin-2-yl)-1-ethyl-1,2,4 -triazol-3-yl)piperidin-1-yl)-3-hydroxypropan-1-one (AZD8835): A potent and selective inhibitor of PI3Kalpha and PI3Kdelta for the treatment of cancers. Bioorg. Med. Chem. Lett. 2015, 25, 5155–5162. [Google Scholar] [CrossRef]
- Hudson, K.; Hancox, U.J.; Trigwell, C.; McEwen, R.; Polanska, U.M.; Nikolaou, M.; Morentin Gutierrez, P.; Avivar-Valderas, A.; Delpuech, O.; Dudley, P.; et al. Intermittent High-Dose Scheduling of AZD8835, a Novel Selective Inhibitor of PI3Kalpha and PI3Kdelta, Demonstrates Treatment Strategies for PIK3CA-Dependent Breast Cancers. Mol. Cancer Ther. 2016, 15, 877–889. [Google Scholar] [CrossRef] [Green Version]
- Anderson, J.L.; Park, A.; Akiyama, R.; Tap, W.D.; Denny, C.T.; Federman, N. Evaluation of In Vitro Activity of the Class I PI3K Inhibitor Buparlisib (BKM120) in Pediatric Bone and Soft Tissue Sarcomas. PLoS ONE 2015, 10, e0133610. [Google Scholar] [CrossRef] [Green Version]
- Ricker, C.A.; Crawford, K.; Matlock, K.; Lathara, M.; Seguin, B.; Rudzinski, E.R.; Berlow, N.E.; Keller, C. Defining an embryonal rhabdomyosarcoma endotype. Cold Spring Harb Mol. Case Stud. 2020, 6, a005066. [Google Scholar] [CrossRef]
- Kim, S.; Dodd, R.D.; Mito, J.K.; Ma, Y.; Kim, Y.; Riedel, R.F.; Kirsch, D.G. Efficacy of phosphatidylinositol-3 kinase inhibitors in a primary mouse model of undifferentiated pleomorphic sarcoma. Sarcoma 2012, 2012, 680708. [Google Scholar] [CrossRef] [Green Version]
- Brachmann, S.M.; Kleylein-Sohn, J.; Gaulis, S.; Kauffmann, A.; Blommers, M.J.; Kazic-Legueux, M.; Laborde, L.; Hattenberger, M.; Stauffer, F.; Vaxelaire, J.; et al. Characterization of the mechanism of action of the pan class I PI3K inhibitor NVP-BKM120 across a broad range of concentrations. Mol. Cancer Ther. 2012, 11, 1747–1757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bavelloni, A.; Focaccia, E.; Piazzi, M.; Orsini, A.; Ramazzotti, G.; Cocco, L.; Blalock, W.; Faenza, I. Therapeutic potential of nvp-bkm120 in human osteosarcomas cells. J. Cell Physiol. 2019, 234, 10907–10917. [Google Scholar] [CrossRef] [PubMed]
- Choo, F.; Odinstov, I.; Nusser, K.; Nicholson, K.S.; Davis, L.; Corless, C.L.; Stork, L.; Somwar, R.; Ladanyi, M.; Davis, J.L.; et al. Functional impact and targetability of PI3KCA, GNAS, and PTEN mutations in a spindle cell rhabdomyosarcoma with MYOD1 L122R mutation. Cold Spring Harb Mol. Case Stud. 2022, 8, a006140. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Pinillos, A.; Ferrari, A.C. mTOR signaling pathway and mTOR inhibitors in cancer therapy. Hematol. Oncol. Clin. North Am. 2012, 26, 483–505. [Google Scholar] [CrossRef]
- Rodon, J.; Dienstmann, R.; Serra, V.; Tabernero, J. Development of PI3K inhibitors: Lessons learned from early clinical trials. Nat. Rev. Clin. Oncol. 2013, 10, 143–153. [Google Scholar] [CrossRef]
- Drullinsky, P.R.; Hurvitz, S.A. Mechanistic basis for PI3K inhibitor antitumor activity and adverse reactions in advanced breast cancer. Breast Cancer Res. Treat. 2020, 181, 233–248. [Google Scholar] [CrossRef]
- Piazzi, M.; Kojic, S.; Capanni, C.; Stamenkovic, N.; Bavelloni, A.; Marin, O.; Lattanzi, G.; Blalock, W.; Cenni, V. Ectopic Expression of Ankrd2 Affects Proliferation, Motility and Clonogenic Potential of Human Osteosarcoma Cells. Cancers 2021, 13, 174. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Alternative Name | PI3K Isoform | Clinical Trials | Cancer | References | Clinical Application |
|---|---|---|---|---|---|---|
| BYL-719 | Alpelisib | P110α | Phase 3 Phase 1–2 | Breast Cancer Solid Tumors | Pubchem CID: 56649450 [21,29,30,31] | EMA authorized for breast neoplasms 27 July 2020 Commercial name: Piqray |
| TGX-221 | TGX221 | P110β | Pre-clinical | Pancreatic Cancer Glioblastoma | Pubchem CID: 9907093 [32] | |
| CZC24832 | P110γ | Pre-clinical | CLL | Pubchem CID: 42623951 | ||
| CAL-101 | Idelalisib, GS-1101 | P110δ | Phase 3 | CLL, ALL | Pubchem CID: 11625818 [33,34,35,36] | EMA authorized for lymphomas and leukemias 18 August 2014 Commercial name: Zydelig |
| AZD-8835 | AZD8835 | P110α P110δ | Phase1 | Solid Tumors and Breast Cancer | Pubchem CID: 76685059 [37,38] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Piazzi, M.; Bavelloni, A.; Cenni, V.; Salucci, S.; Bartoletti Stella, A.; Tomassini, E.; Scotlandi, K.; Blalock, W.L.; Faenza, I. Combined Treatment with PI3K Inhibitors BYL-719 and CAL-101 Is a Promising Antiproliferative Strategy in Human Rhabdomyosarcoma Cells. Molecules 2022, 27, 2742. https://doi.org/10.3390/molecules27092742
Piazzi M, Bavelloni A, Cenni V, Salucci S, Bartoletti Stella A, Tomassini E, Scotlandi K, Blalock WL, Faenza I. Combined Treatment with PI3K Inhibitors BYL-719 and CAL-101 Is a Promising Antiproliferative Strategy in Human Rhabdomyosarcoma Cells. Molecules. 2022; 27(9):2742. https://doi.org/10.3390/molecules27092742
Chicago/Turabian StylePiazzi, Manuela, Alberto Bavelloni, Vittoria Cenni, Sara Salucci, Anna Bartoletti Stella, Enrica Tomassini, Katia Scotlandi, William L. Blalock, and Irene Faenza. 2022. "Combined Treatment with PI3K Inhibitors BYL-719 and CAL-101 Is a Promising Antiproliferative Strategy in Human Rhabdomyosarcoma Cells" Molecules 27, no. 9: 2742. https://doi.org/10.3390/molecules27092742
APA StylePiazzi, M., Bavelloni, A., Cenni, V., Salucci, S., Bartoletti Stella, A., Tomassini, E., Scotlandi, K., Blalock, W. L., & Faenza, I. (2022). Combined Treatment with PI3K Inhibitors BYL-719 and CAL-101 Is a Promising Antiproliferative Strategy in Human Rhabdomyosarcoma Cells. Molecules, 27(9), 2742. https://doi.org/10.3390/molecules27092742