
Identification of Mtb GlmU Uridyltransferase Domain Inhibitors by Ligand-Based and Structure-Based Drug Design Approaches

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussions
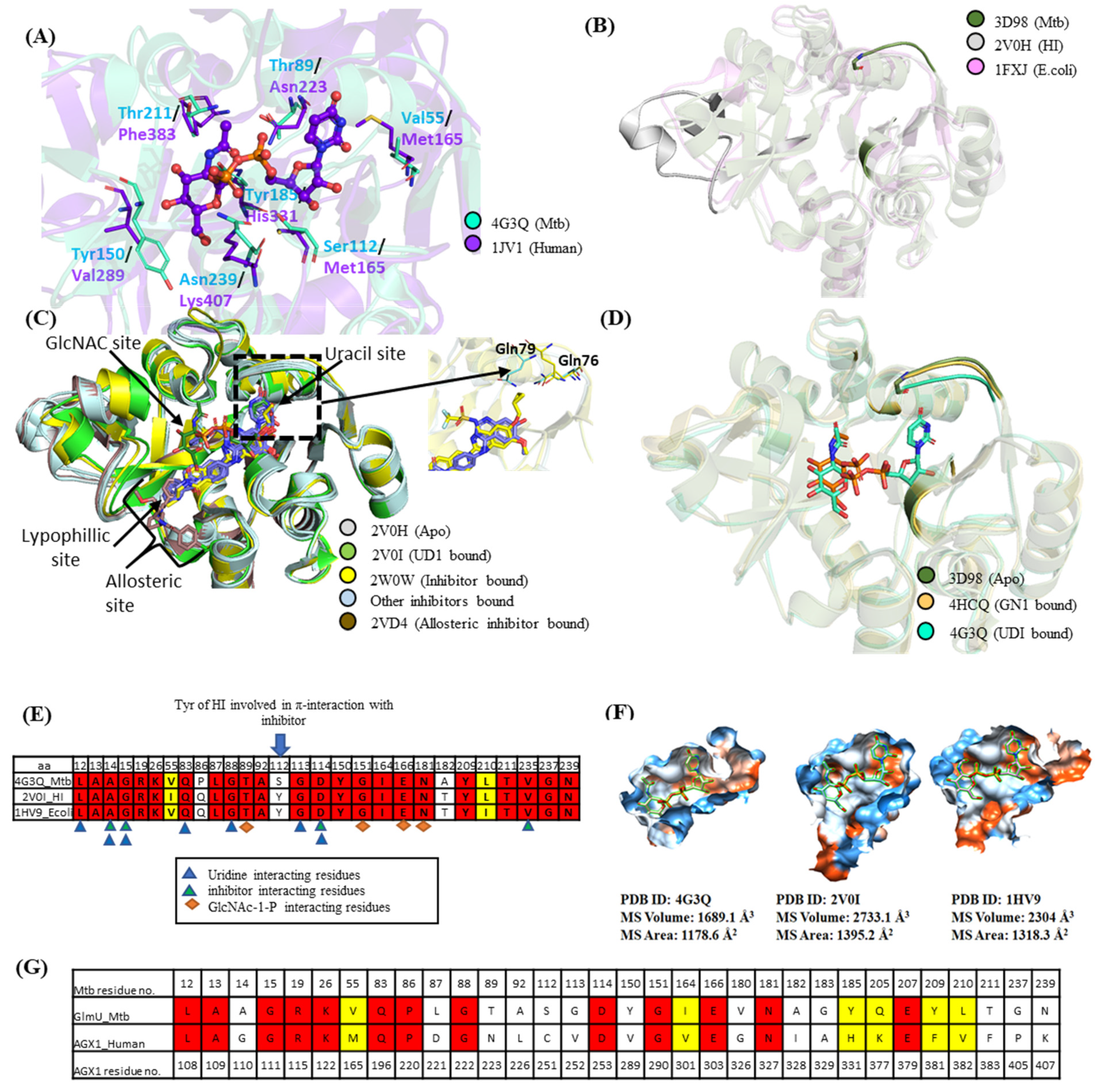
2.1. Pocket Comparison between Mtb GlmU Uridyltransferase and Human Homologs
2.2. Pocket Comparison among Bacterial GlmU Uridyltransferase Domain
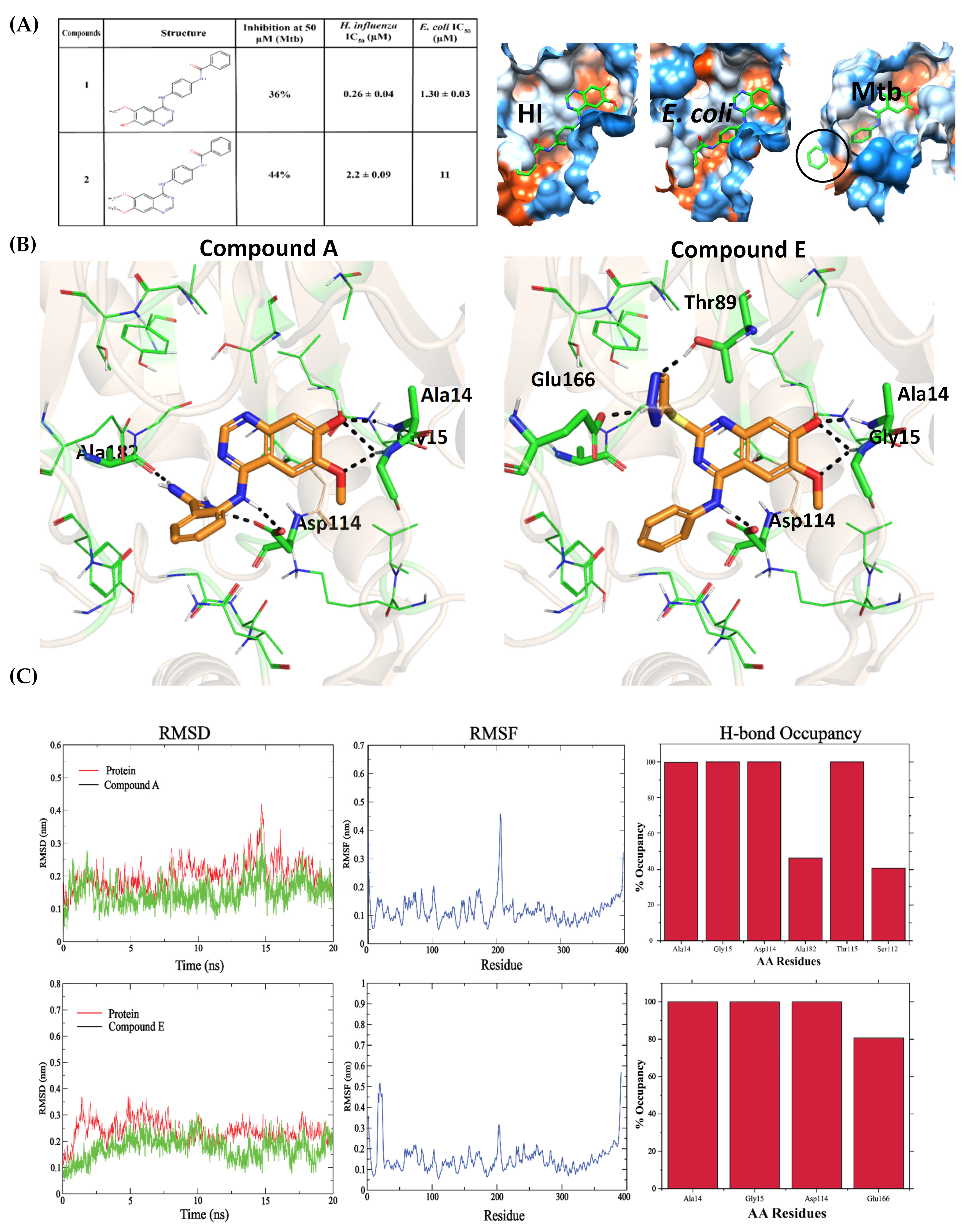
2.3. Optimization of Known Bacterial GlmU Inhibitors for Mtb GlmU
2.4. Ligand-Based Virtual Screening
2.5. Structure-Based Virtual Screening through Molecular Docking and E-pharmacophore Methods
2.6. Triaging Hits by MD Simulations of Top Poses
2.7. Selectivity against Human AGX1
3. Materials and Methods
3.1. Protein Preparation
3.2. Receptor Grid Generation
3.3. Molecular Docking
3.4. Ligand Growing
3.5. ZINC Database
3.6. Ligand Preparation
3.7. Conformational Sampling
3.8. Ligand-Based Screening
- (1)
- 2D-pharmacophore based screening using SMARTS pattern O=CNC=O of the uridine structure (which makes key interactions with residues Ala14, Gly88 and Gln88) against ~6.6 million ZINC compounds with 0.5 tanimoto coefficient (TC). Initially before the screen, known Mtb inhibitors were randomly seeded with the ZINC database and used for the 2D pharmacophore similarity search using MOE with both 0.5 and 0.4 TC. However, with 0.4, we obtained 27 Mtb inhibitors out of 37 (including active and inactive compounds) and with 0.5, none of the Mtb inhibitors were obtained in the screened hits. We chose 0.5 TC since all the Mtb inhibitors were obtained when we performed a similarity search with four known active Mtb inhibitors.
- (2)
- A similarity search was performed with a diverse set of known Mtb inhibitors and the substrate UTP against ~6.6 million ZINC compounds with 0.6 TC. In order to choose a diverse set of reference ligands, we clustered the known inhibitors and selected the one with the highest inhibition percentage from each cluster (201305, 201504, 201506, 201507; name is assigned based on the year of publication and the compound no. given in the respective article—for example, 201305: 2013 is year and 05 is compound no.). Additionally, we included UTP since we are targeting the UTP site. Hence, we selected five compounds for the similarity search.
3.9. Clustering
3.10. Virtual Screening
3.11. E-pharmacophore Generation
3.12. Molecular Dynamics Simulation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Fu, L.M.; Fu-Liu, C.S. Is Mycobacterium tuberculosis a closer relative to Gram-positive or Gram–negative bacterial pathogens? Tuberculosis 2002, 82, 85–90. [Google Scholar] [CrossRef] [PubMed]
- Tran, A.T.; Wen, D.; West, N.P.; Baker, E.N.; Britton, W.J.; Payne, R.J. Inhibition studies on Mycobacterium tuberculosis N-acetylglucosamine-1-phosphate uridyltransferase (GlmU). Org. Biomol. Chem. 2013, 11, 8113–8126. [Google Scholar] [CrossRef] [PubMed]
- Mengin-Lecreulx, D.; van Heijenoort, J. Copurification of glucosamine-1-phosphate acetyltransferase and N-acetylglucosamine-1-phosphate uridyltransferase activities of Escherichia coli: Characterization of the glmU gene product as a bifunctional enzyme catalyzing two subsequent steps in the pathway for UDP-N-acetylglucosamine synthesis. J. Bacteriol. 1994, 176, 5788–5795. [Google Scholar] [PubMed] [Green Version]
- Zhang, W.; Jones, V.C.; Scherman, M.S.; Mahapatra, S.; Crick, D.; Bhamidi, S.; Xin, Y.; McNeil, M.R.; Ma, Y. Expression, essentiality, and a microtiter plate assay for mycobacterial GlmU, the bifunctional glucosamine-1-phosphate acetyltransferase and N-acetylglucosamine-1-phosphate uridyltransferase. Int. J. Biochem. Cell Biol. 2008, 40, 2560–2571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gehring, A.M.; Lees, W.J.; Mindiola, D.J.; Walsh, C.T.; Brown, E.D. Acetyltransfer precedes uridylyltransfer in the formation of UDP-N-acetylglucosamine in separable active sites of the bifunctional GlmU protein of Escherichia coli. Biochemistry 1996, 35, 579–585. [Google Scholar] [CrossRef]
- Zhang, Z.; Bulloch, E.M.; Bunker, R.D.; Baker, E.N.; Squire, C.J. Structure and function of GlmU from Mycobacterium tuberculosis. Acta Crystallogr. Sect. D Biol. Crystallogr. 2009, 65 Pt 3, 275–283. [Google Scholar] [CrossRef] [Green Version]
- Mochalkin, I.; Lightle, S.; Zhu, Y.; Ohren, J.F.; Spessard, C.; Chirgadze, N.Y.; Banotai, C.; Melnick, M.; McDowell, L. Characterization of substrate binding and catalysis in the potential antibacterial target N-acetylglucosamine-1-phosphate uridyltransferase (GlmU). Protein Sci. A Publ. Protein Soc. 2007, 16, 2657–2666. [Google Scholar] [CrossRef]
- Jagtap, P.K.; Verma, S.K.; Vithani, N.; Bais, V.S.; Prakash, B. Crystal structures identify an atypical two-metal-ion mechanism for uridyltransfer in GlmU: Its significance to sugar nucleotidyl transferases. J. Mol. Biol. 2013, 425, 1745–1759. [Google Scholar] [CrossRef]
- Mio, T.; Yabe, T.; Arisawa, M.; Yamada-Okabe, H. The eukaryotic UDP-N-acetylglucosamine pyrophosphorylases. Gene cloning, protein expression, and catalytic mechanism. J. Biol. Chem. 1998, 273, 14392–14397. [Google Scholar] [CrossRef] [Green Version]
- Mio, T.; Yamada-Okabe, T.; Arisawa, M.; Yamada-Okabe, H. Saccharomyces cerevisiae GNA1, an essential gene encoding a novel acetyltransferase involved in UDP-N-acetylglucosamine synthesis. J. Biol. Chem. 1999, 274, 424–429. [Google Scholar] [CrossRef] [Green Version]
- Diekman, A.B.; Goldberg, E. Characterization of a human antigen with sera from infertile patients. Biol. Reprod. 1994, 50, 1087–1093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sassetti, C.M.; Boyd, D.H.; Rubin, E.J. Genes required for mycobacterial growth defined by high density mutagenesis. Mol. Microbiol. 2003, 48, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Soni, V.; Upadhayay, S.; Suryadevara, P.; Samla, G.; Singh, A.; Yogeeswari, P.; Sriram, D.; Nandicoori, V.K. Depletion of M. tuberculosis GlmU from Infected Murine Lungs Effects the Clearance of the Pathogen. PLoS Pathog. 2015, 11, e1005235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, V.K.; Das, K.; Seshadri, K. Kinetic modelling of GlmU reactions—Prioritization of reaction for therapeutic application. PLoS ONE 2012, 7, e43969. [Google Scholar] [CrossRef] [PubMed]
- Holm, L.; Sander, C. Dali: A network tool for protein structure comparison. Trends Biochem. Sci. 1995, 20, 478–480. [Google Scholar] [CrossRef]
- Yeturu, A.B.A.K. Site2Vec: A reference frame invariant algorithm for vector embedding of protein–ligand binding sites. Mach. Learn. Sci. Technol. 2021, 2, 015005. [Google Scholar]
- Tian, W.; Chen, C.; Lei, X.; Zhao, J.; Liang, J. CASTp 3.0: Computed atlas of surface topography of proteins. Nucleic Acids Res. 2018, 46, W363–W367. [Google Scholar] [CrossRef] [Green Version]
- Sanner, M.F.; Olson, A.J.; Spehner, J.C. Reduced surface: An efficient way to compute molecular surfaces. Biopolymers 1996, 38, 305–320. [Google Scholar] [CrossRef]
- Larsen, N.A.; Nash, T.J.; Morningstar, M.; Shapiro, A.B.; Joubran, C.; Blackett, C.J.; Patten, A.D.; Boriack-Sjodin, P.A.; Doig, P. An aminoquinazoline inhibitor of the essential bacterial cell wall synthetic enzyme GlmU has a unique non-protein-kinase-like binding mode. Biochem. J. 2012, 446, 405–413. [Google Scholar] [CrossRef] [Green Version]
- Doig, P.; Boriack-Sjodin, P.A.; Dumas, J.; Hu, J.; Itoh, K.; Johnson, K.; Kazmirski, S.; Kinoshita, T.; Kuroda, S.; Sato, T.O.; et al. Rational design of inhibitors of the bacterial cell wall synthetic enzyme GlmU using virtual screening and lead-hopping. Bioorg. Med. Chem. 2014, 22, 6256–6269. [Google Scholar] [CrossRef]
- Min, J.; Lin, D.; Zhang, Q.; Zhang, J.; Yu, Z. Structure-based virtual screening of novel inhibitors of the uridyltransferase activity of Xanthomonas oryzae pv. oryzae GlmU. Eur. J. Med. Chem. 2012, 53, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.; Deng, W.; Gao, M.; Mao, B.; Xu, S.; Chen, C.; Zhang, Q. Novel lead compound optimization and synthesized based on the target structure of Xanthomonas oryzae pv. oryzae GlmU. Pestic. Biochem. Physiol. 2015, 122, 22–28. [Google Scholar] [CrossRef] [PubMed]
- Parikh, A.; Verma, S.K.; Khan, S.; Prakash, B.; Nandicoori, V.K. PknB-mediated phosphorylation of a novel substrate, N-acetylglucosamine-1-phosphate uridyltransferase, modulates its acetyltransferase activity. J. Mol. Biol. 2009, 386, 451–464. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.K.; Jaiswal, M.; Kumar, N.; Parikh, A.; Nandicoori, V.K.; Prakash, B. Structure of N-acetylglucosamine-1-phosphate uridyltransferase (GlmU) from Mycobacterium tuberculosis in a cubic space group. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2009, 65 Pt 5, 435–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schrödinger Release 2018: Protein Preparation Wizard; Epik, Schrödinger, LLC: New York, NY, USA, 2020; Impact, Schrödinger, LLC: New York, NY, USA, 2020; Prime, Schrödinger, LLC: New York, NY, USA, 2020, 2018.
- Sastry, G.M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef]
- Cheeseright, T.; Mackey, M.; Rose, S.; Vinter, A. Molecular field extrema as descriptors of biological activity: Definition and validation. J. Chem. Inf. Model. 2006, 46, 665–676. [Google Scholar] [CrossRef]
- Sterling, T.; Irwin, J.J. ZINC 15—Ligand Discovery for Everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337. [Google Scholar] [CrossRef]
- Schrödinger Release 2018: Canvas; Schrödinger, LLC: New York, NY, USA, 2020.
- Duan, J.; Dixon, S.L.; Lowrie, J.F.; Sherman, W. Analysis and comparison of 2D fingerprints: Insights into database screening performance using eight fingerprint methods. J. Mol. Graph. Model. 2010, 29, 157–170. [Google Scholar] [CrossRef] [PubMed]
- Sastry, M.; Lowrie, J.F.; Dixon, S.L.; Sherman, W. Large-scale systematic analysis of 2D fingerprint methods and parameters to improve virtual screening enrichments. J. Chem. Inf. Model. 2010, 50, 771–784. [Google Scholar] [CrossRef] [PubMed]
- Watts, K.S.; Dalal, P.; Murphy, R.B.; Sherman, W.; Friesner, R.A.; Shelley, J.C. ConfGen: A conformational search method for efficient generation of bioactive conformers. J. Chem. Inf. Model. 2010, 50, 534–546. [Google Scholar] [CrossRef] [PubMed]
- Molecular Operating Environment (MOE); Chemical Computing Group ULC: Montreal, QC, Canada, 2018.
- Salam, N.K.; Nuti, R.; Sherman, W. Novel method for generating structure-based pharmacophores using energetic analysis. J. Chem. Inf. Model. 2009, 49, 2356–2368. [Google Scholar] [CrossRef]
- Loving, K.; Salam, N.K.; Sherman, W. Energetic analysis of fragment docking and application to structure-based pharmacophore hypothesis generation. J. Comput. Aided Mol. Des. 2009, 23, 541–554. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Hornak, V.; Abel, R.; Okur, A.; Strockbine, B.; Roitberg, A.; Simmerling, C. Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins 2006, 65, 712–725. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Wang, W.; Kollman, P.A.; Case, D.A. Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graph. Model. 2006, 25, 247–260. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N⋅ log (N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef] [Green Version]
- Hess, B.; Beker, H.; Berendsen, H.J.; Fraaije, J.G. LINCS: A linear constraint solver for molecular simulations. J. Comp. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Singh, M.; Kempanna, P.; Bharatham, K. Identification of Mtb GlmU Uridyltransferase Domain Inhibitors by Ligand-Based and Structure-Based Drug Design Approaches. Molecules 2022, 27, 2805. https://doi.org/10.3390/molecules27092805
Singh M, Kempanna P, Bharatham K. Identification of Mtb GlmU Uridyltransferase Domain Inhibitors by Ligand-Based and Structure-Based Drug Design Approaches. Molecules. 2022; 27(9):2805. https://doi.org/10.3390/molecules27092805
Chicago/Turabian StyleSingh, Manvi, Priya Kempanna, and Kavitha Bharatham. 2022. "Identification of Mtb GlmU Uridyltransferase Domain Inhibitors by Ligand-Based and Structure-Based Drug Design Approaches" Molecules 27, no. 9: 2805. https://doi.org/10.3390/molecules27092805
APA StyleSingh, M., Kempanna, P., & Bharatham, K. (2022). Identification of Mtb GlmU Uridyltransferase Domain Inhibitors by Ligand-Based and Structure-Based Drug Design Approaches. Molecules, 27(9), 2805. https://doi.org/10.3390/molecules27092805