
Interactions among Endothelial Nitric Oxide Synthase, Cardiovascular System, and Nociception during Physiological and Pathophysiological States


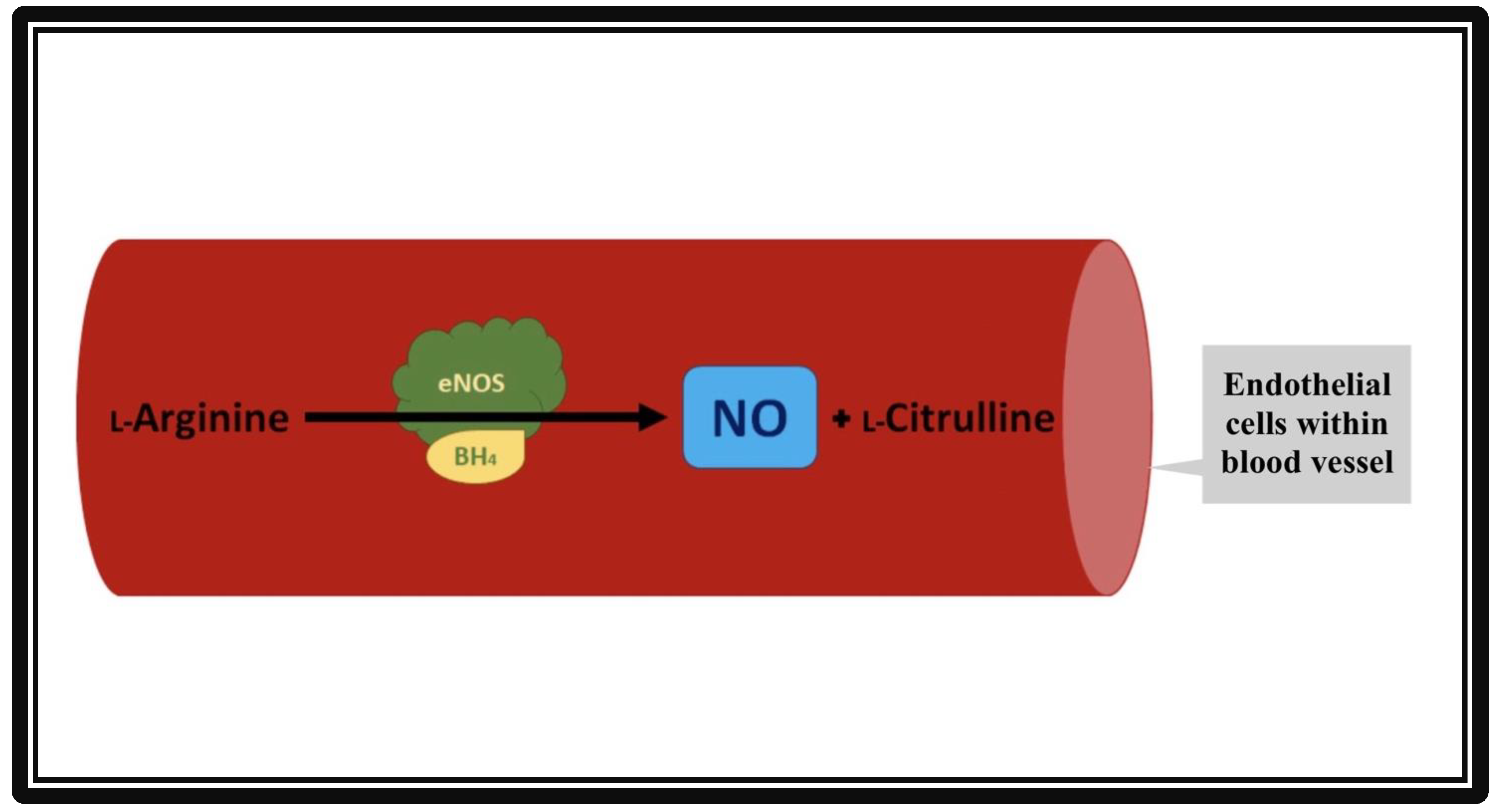{kind=link}
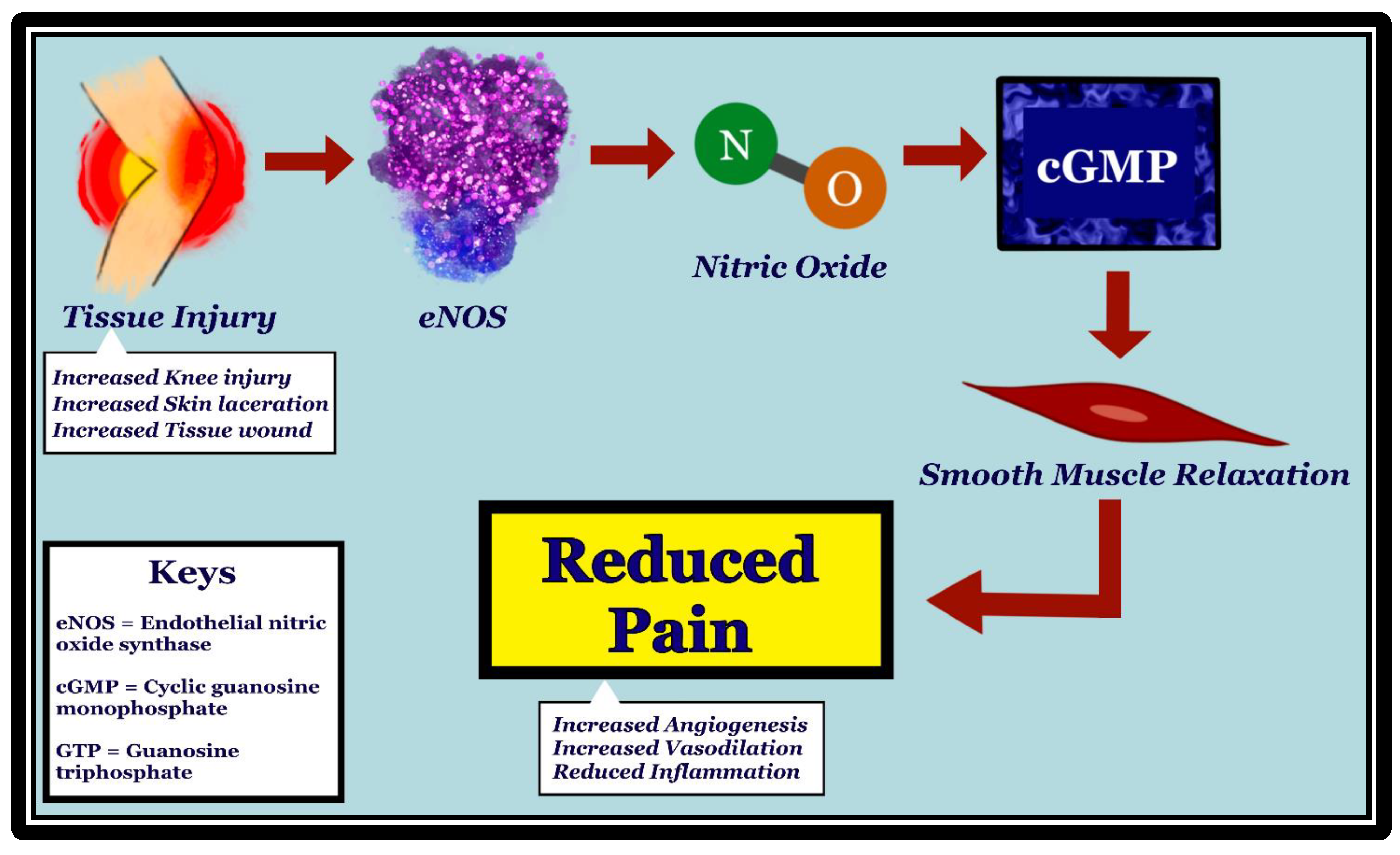{kind=link}
{kind=link}
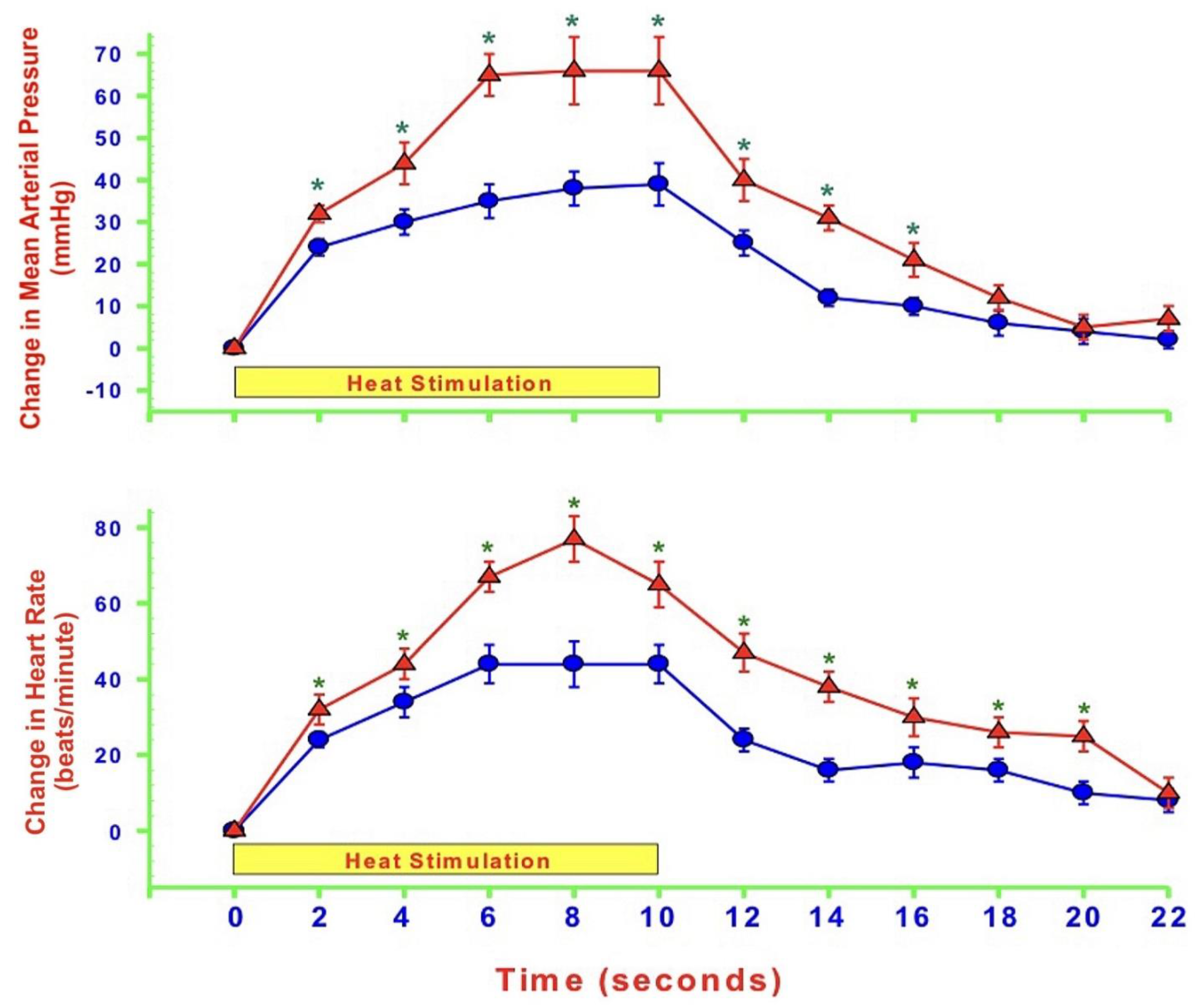{kind=link}
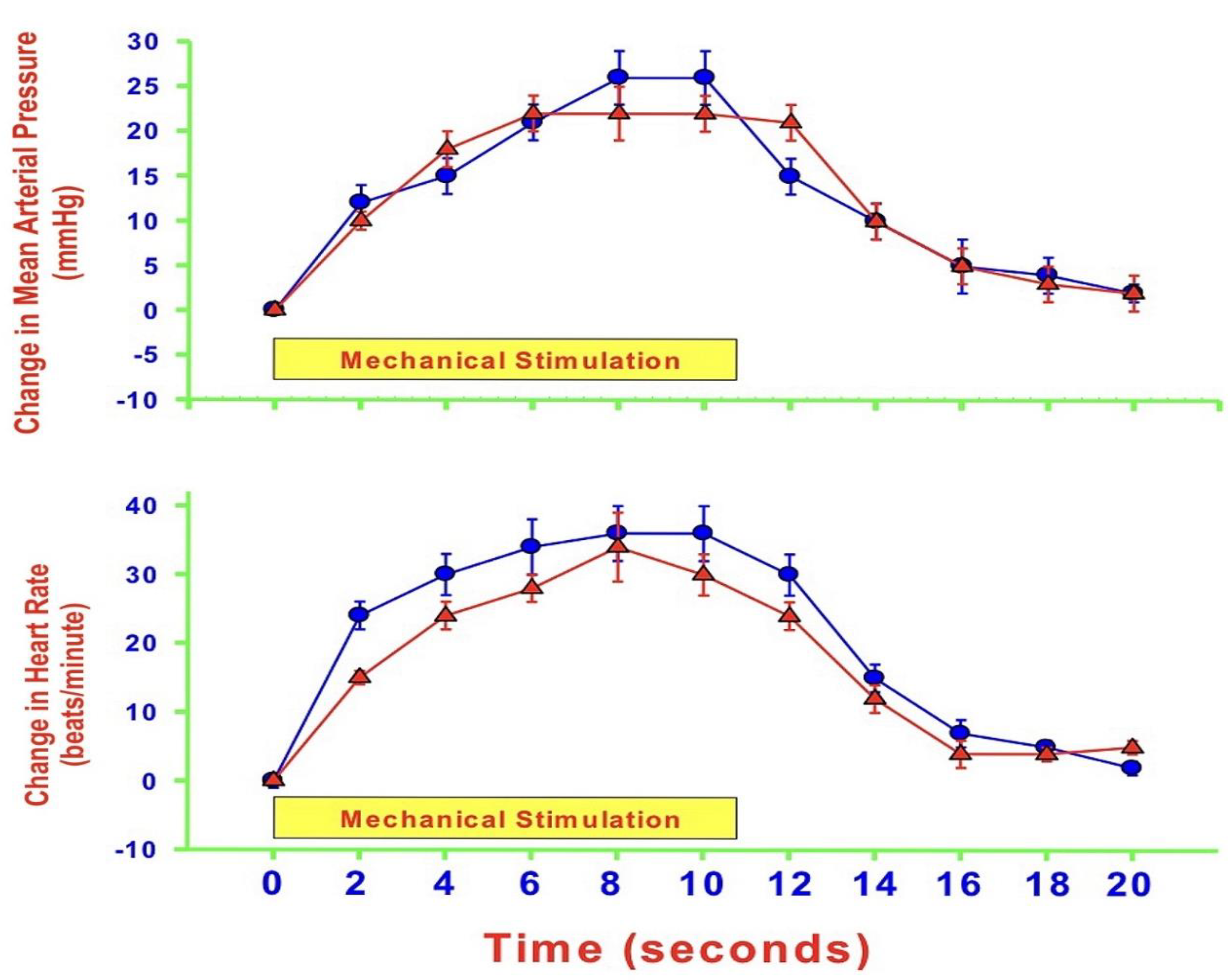
{kind=link}
Abstract
:1. Introduction

1.1. Nociception
1.2. Cardiovascular System
2. Endothelial Nitric Oxide Synthase and Nociception
3. Endothelial Nitric Oxide Synthase and the Cardiovascular System

4. Interaction between Endothelial Nitric Oxide Synthase, Nociception, and the Cardiovascular System
- The presence of eNOS in dorsal root ganglia and other nervous tissues has been shown to play a role in nociception.
- The eNOS pathway can be activated by the stimulation of nociceptors.
- Through its vasodilatory effect, eNOS may affect nociception by regulating the movement of important mediators across the blood vessel walls.
- Endothelial NOS have pro- or anti-nociceptive effects.
- Endothelial NOS can also affect nociception through its formation of peroxynitrite and its interaction with TRPV1 as well as with pro- and anti-inflammatory mediators.
- MAP and HR are both altered during mechanically, heat- and cold-induced nociception. Inhibition of eNOS in PAG affects MAP and HR responses to heat- and cold- but not to mechanical-induced nociception.
5. Conclusions and Future Direction
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pollock, J.S.; Forstermann, U.; Mitchell, J.A.; Warner, T.D.; Schmidt, H.H.; Nakane, M.; Murad, F. Purification and characterization of particulate endothelium-derived relaxing factor synthase from cultured and native bovine aortic endothelial cells. Proc. Natl. Acad. Sci. USA 1991, 88, 10480–10484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, T.; Zhang, Z.; Chen, Y.; Su, H.; Deng, X.; Liu, X.; Fan, Y. Delivery of Nitric Oxide in the Cardiovascular System: Implications for Clinical Diagnosis and Therapy. Int. J. Mol. Sci. 2021, 22, 12166. [Google Scholar] [CrossRef] [PubMed]
- Borsani, E.; Giovannozzi, S.; Cocchi, M.A.; Boninsegna, R.; Rezzani, R.; Rodella, L.F. Endothelial nitric oxide synthase in dorsal root ganglia during chronic inflammatory nociception. Cells Tissues Organs 2013, 197, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Forstermann, U.; Munzel, T. Endothelial nitric oxide synthase in vascular disease: From marvel to menace. Circulation 2006, 113, 1708–1714. [Google Scholar] [CrossRef] [Green Version]
- Dreno, B.; Bettoli, V.; Perez, M.; Bouloc, A.; Ochsendorf, F. Cutaneous lesions caused by mechanical injury. Eur. J. Dermatol. 2015, 25, 114–121. [Google Scholar] [CrossRef]
- Frias, B.; Merighi, A. Capsaicin, Nociception and Pain. Molecules 2016, 21, 797. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Kandle, P.F.; Murray, I.; Fitzgerald, L.A.; Sehdev, J.S. Physiology, Pain; StatPearls Publishing: Treasure Island, FL, USA, 2022. Available online: https://www.ncbi.nlm.nih.gov/books/NBK539789/ (accessed on 25 April 2022).
- Mills, E.P.; Keay, K.A.; Henderson, L.A. Brainstem Pain-Modulation Circuitry and Its Plasticity in Neuropathic Pain: Insights From Human Brain Imaging Investigations. Front Pain Res. (Lausanne) 2021, 2, 705345. [Google Scholar] [CrossRef]
- Ally, A.; Powell, I.; Ally, M.M.; Chaitoff, K.; Nauli, S.M. Role of neuronal nitric oxide synthase on cardiovascular functions in physiological and pathophysiological states. Nitric Oxide 2020, 102, 52–73. [Google Scholar] [CrossRef]
- Jain, V.; Bordes, S.; Bhardwaj, A. Physiology, Pulmonary Circulatory System. In StatPearls [Internet]; Treasure Island, FL, USA, 2022. Available online: https://www.ncbi.nlm.nih.gov/books/NBK525948/ (accessed on 26 February 2022).
- Albrecht, E.W.; Stegeman, C.A.; Heeringa, P.; Henning, R.H.; van Goor, H. Protective role of endothelial nitric oxide synthase. J. Pathol. 2003, 199, 8–17. [Google Scholar] [CrossRef]
- Ozaki, M.; Kawashima, S.; Yamashita, T.; Hirase, T.; Namiki, M.; Inoue, N.; Hirata, K.; Yasui, H.; Sakurai, H.; Yoshida, Y.; et al. Overexpression of endothelial nitric oxide synthase accelerates atherosclerotic lesion formation in apoE-deficient mice. J. Clin. Investig. 2002, 110, 331–340. [Google Scholar] [CrossRef]
- Tran, N.; Garcia, T.; Aniqa, M.; Ali, S.; Ally, A.; Nauli, S.M. Endothelial Nitric Oxide Synthase (eNOS) and the Cardiovascular System: In Physiology and in Disease States. Am. J. Biomed. Sci. Res. 2022, 15, 153–177. [Google Scholar] [PubMed]
- Raja, S.N.; Carr, D.B.; Cohen, M.; Finnerup, N.B.; Flor, H.; Gibson, S.; Keefe, F.J.; Mogil, J.S.; Ringkamp, M.; Sluka, K.A.; et al. The revised International Association for the Study of Pain definition of pain: Concepts, challenges, and compromises. Pain 2020, 161, 1976–1982. [Google Scholar] [CrossRef] [PubMed]
- Yam, M.F.; Loh, Y.C.; Tan, C.S.; Khadijah Adam, S.; Abdul Manan, N.; Basir, R. General Pathways of Pain Sensation and the Major Neurotransmitters Involved in Pain Regulation. Int. J. Mol. Sci. 2018, 19, 2164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karlsson, G.A.; Chaitoff, K.A.; Hossain, S.; Bohlke, M.; Maher, T.J.; Ally, A. Modulation of cardiovascular responses and neurotransmission during peripheral nociception following nNOS antagonism within the periaqueductal gray. Brain Res. 2007, 1143, 150–160. [Google Scholar] [CrossRef] [PubMed]
- Douki, T.; Cadet, J. Peroxynitrite mediated oxidation of purine bases of nucleosides and isolated DNA. Free Radic Res. 1996, 24, 369–380. [Google Scholar] [CrossRef] [PubMed]
- McMahon, S.B. Wall and Melzack’s Textbook of Pain; Elsevier/Saunders Publishing: Philadelphia, PA, USA, 2013. [Google Scholar]
- Bakshi, N.; Morris, C.R. The role of the arginine metabolome in pain: Implications for sickle cell disease. J. Pain Res. 2016, 9, 167–175. [Google Scholar]
- Xanthos, D.N.; Bennett, G.J.; Coderre, T.J. Norepinephrine-induced nociception and vasoconstrictor hypersensitivity in rats with chronic post-ischemia pain. Pain 2008, 137, 640–651. [Google Scholar] [CrossRef] [Green Version]
- Da Silva-Azevedo, L.; Baum, O.; Zakrzewicz, A.; Pries, A.R. Vascular endothelial growth factor is expressed in endothelial cells isolated from skeletal muscles of nitric oxide synthase knockout mice during prazosin-induced angiogenesis. Biochem. Biophys. Res. Commun. 2002, 297, 1270–1276. [Google Scholar] [CrossRef]
- Bhattacharya, D.; Singh, M.K.; Chaudhuri, S.; Acharya, S.; Basu, A.K.; Chaudhuri, S. T11TS impedes glioma angiogenesis by inhibiting VEGF signaling and pro-survival PI3K/Akt/eNOS pathway with concomitant upregulation of PTEN in brain endothelial cells. J. Neurooncol. 2013, 113, 13–25. [Google Scholar] [CrossRef]
- Chu, L.W.; Chen, J.Y.; Yu, K.L.; Cheng, K.I.; Wu, P.C.; Wu, B.N. Neuroprotective and anti-inflammatory activities of atorvastatin in a rat chronic constriction injury model. Int. J. Immunopathol. Pharmacol. 2012, 25, 219–230. [Google Scholar] [CrossRef] [Green Version]
- Li, S.T.; Pan, J.; Hua, X.M.; Liu, H.; Shen, S.; Liu, J.F.; Li, B.; Tao, B.B.; Ge, X.L.; Wang, X.H.; et al. Endothelial nitric oxide synthase protects neurons against ischemic injury through regulation of brain-derived neurotrophic factor expression. CNS Neurosci. Ther. 2014, 20, 154–164. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Jiao, B.; Zhu, B.; Xiong, B.; Lu, P.; Ai, L.; Yang, N.; Zhao, Y.; Xu, H. Nitric Oxide in the Spinal Cord Is Involved in the Hyperalgesia Induced by Tetrahydrobiopterin in Chronic Restraint Stress Rats. Front Neurosci. 2021, 15, 593654. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Niu, X.; Wang, G.; Huang, J.; Liu, M.; Peng, B. Chronic prostatitis/chronic pelvic pain syndrome impairs erectile function through increased endothelial dysfunction, oxidative stress, apoptosis, and corporal fibrosis in a rat model. Andrology 2016, 4, 1209–1216. [Google Scholar] [CrossRef] [PubMed]
- Han, T.H.; Hyduke, D.R.; Vaughn, M.W.; Fukuto, J.M.; Liao, J.C. Nitric oxide reaction with red blood cells and hemoglobin under heterogeneous conditions. Proc. Natl. Acad. Sci. USA 2002, 99, 7763–7768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helms, C.; Kim-Shapiro, D.B. Hemoglobin-mediated nitric oxide signaling. Free Radic. Biol. Med. 2013, 61, 464–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boger, R.H. L-Arginine therapy in cardiovascular pathologies: Beneficial or dangerous? Curr. Opin. Clin. Nutr. Metab. Care 2008, 11, 55–61. [Google Scholar] [CrossRef]
- Grimble, G.K. Adverse gastrointestinal effects of arginine and related amino acids. J. Nutr. 2007, 137 (Suppl. 2), 1693S–1701S. [Google Scholar] [CrossRef] [Green Version]
- Rysz, J.; Franczyk, B.; Rysz-Gorzynska, M.; Gluba-Brzozka, A. Pharmacogenomics of Hypertension Treatment. Int. J. Mol. Sci. 2020, 21, 4709. [Google Scholar] [CrossRef]
- Zhao, Q.; Zhang, J.; Wang, H. PGC-1alpha overexpression suppresses blood pressure elevation in DOCA-salt hypertensive mice. Biosci. Rep. 2015, 35, e00217. [Google Scholar] [CrossRef] [Green Version]
- Du, Q.; Liao, Q.; Chen, C.; Yang, X.; Xie, R.; Xu, J. The Role of Transient Receptor Potential Vanilloid 1 in Common Diseases of the Digestive Tract and the Cardiovascular and Respiratory System. Front Physiol. 2019, 10, 1064. [Google Scholar] [CrossRef]
- Randhawa, P.K.; Jaggi, A.S. TRPV1 channels in cardiovascular system: A double edged sword? Int. J. Cardiol. 2017, 228, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Ching, L.C.; Kou, Y.R.; Shyue, S.K.; Su, K.H.; Wei, J.; Cheng, L.C.; Yu, Y.B.; Pan, C.C.; Lee, T.S. Molecular mechanisms of activation of endothelial nitric oxide synthase mediated by transient receptor potential vanilloid type 1. Cardiovasc. Res. 2011, 91, 492–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, L.; Wang, S.; Li, B.; Sun, A.; Zou, Y.; Ge, J. A protective role of ciglitazone in ox-LDL-induced rat microvascular endothelial cells via modulating PPARgamma-dependent AMPK/eNOS pathway. J. Cell Mol. Med. 2015, 19, 92–102. [Google Scholar] [CrossRef]
- Nunes, A.K.; Raposo, C.; Rocha, S.W.; Barbosa, K.P.; Luna, R.L.; da Cruz-Hofling, M.A.; Peixoto, C.A. Involvement of AMPK, IKbetaalpha-NFkappaB and eNOS in the sildenafil anti-inflammatory mechanism in a demyelination model. Brain Res. 2015, 1627, 119–133. [Google Scholar] [CrossRef]
- Buchwalow, I.B.; Cacanyiova, S.; Neumann, J.; Samoilova, V.E.; Boecker, W.; Kristek, F. The role of arterial smooth muscle in vasorelaxation. Biochem. Biophys. Res. Commun. 2008, 377, 504–507. [Google Scholar] [CrossRef] [PubMed]
- Furchgott, R.F.; Zawadzki, J.V. The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature 1980, 288, 373–376. [Google Scholar] [CrossRef]
- Mayhan, W.G. Role of nitric oxide in modulating permeability of hamster cheek pouch in response to adenosine 5′-diphosphate and bradykinin. Inflammation 1992, 16, 295–305. [Google Scholar] [CrossRef]
- Yuan, Y.; Granger, H.J.; Zawieja, D.C.; Chilian, W.M. Flow modulates coronary venular permeability by a nitric oxide-related mechanism. Am. J. Physiol. 1992, 263 Pt 2, H641–H646. [Google Scholar] [CrossRef]
- Hatakeyama, T.; Pappas, P.J.; Hobson, R.W., 2nd; Boric, M.P.; Sessa, W.C.; Duran, W.N. Endothelial nitric oxide synthase regulates microvascular hyperpermeability in vivo. J. Physiol. 2006, 574 Pt 1, 275–281. [Google Scholar] [CrossRef]
- Fukumura, D.; Gohongi, T.; Kadambi, A.; Izumi, Y.; Ang, J.; Yun, C.O.; Buerk, D.G.; Huang, P.L.; Jain, R.K. Predominant role of endothelial nitric oxide synthase in vascular endothelial growth factor-induced angiogenesis and vascular permeability. Proc. Natl. Acad. Sci. USA 2001, 98, 2604–2609. [Google Scholar] [CrossRef] [Green Version]
- Lal, B.K.; Varma, S.; Pappas, P.J.; Hobson, R.W., 2nd; Duran, W.N. VEGF increases permeability of the endothelial cell monolayer by activation of PKB/akt, endothelial nitric-oxide synthase, and MAP kinase pathways. Microvasc. Res. 2001, 62, 252–262. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Granger, H.J.; Zawieja, D.C.; DeFily, D.V.; Chilian, W.M. Histamine increases venular permeability via a phospholipase C-NO synthase-guanylate cyclase cascade. Am. J. Physiol. 1993, 264 Pt 2, H1734–H1739. [Google Scholar] [CrossRef] [PubMed]
- Varma, S.; Breslin, J.W.; Lal, B.K.; Pappas, P.J.; Hobson, R.W., 2nd; Duran, W.N. p42/44MAPK regulates baseline permeability and cGMP-induced hyperpermeability in endothelial cells. Microvasc. Res. 2002, 63, 172–178. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.L. eNOS, metabolic syndrome and cardiovascular disease. Trends Endocrinol. Metab. 2009, 20, 295–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deanfield, J.E.; Halcox, J.P.; Rabelink, T.J. Endothelial function and dysfunction: Testing and clinical relevance. Circulation 2007, 115, 1285–1295. [Google Scholar] [CrossRef]
- Widlansky, M.E.; Gokce, N.; Keaney, J.F., Jr.; Vita, J.A. The clinical implications of endothelial dysfunction. J. Am. Coll. Cardiol. 2003, 42, 1149–1160. [Google Scholar] [CrossRef] [Green Version]
- Jonk, A.M.; Houben, A.J.; de Jongh, R.T.; Serne, E.H.; Schaper, N.C.; Stehouwer, C.D. Microvascular dysfunction in obesity: A potential mechanism in the pathogenesis of obesity-associated insulin resistance and hypertension. Physiology 2007, 22, 252–260. [Google Scholar] [CrossRef]
- Semenkovich, C.F. Insulin resistance and atherosclerosis. J. Clin. Investig. 2006, 116, 1813–1822. [Google Scholar] [CrossRef]
- Daiber, A.; Di Lisa, F.; Oelze, M.; Kroller-Schon, S.; Steven, S.; Schulz, E.; Munzel, T. Crosstalk of mitochondria with NADPH oxidase via reactive oxygen and nitrogen species signalling and its role for vascular function. Br. J. Pharmacol. 2017, 174, 1670–1689. [Google Scholar] [CrossRef] [Green Version]
- Schulz, E.; Wenzel, P.; Munzel, T.; Daiber, A. Mitochondrial redox signaling: Interaction of mitochondrial reactive oxygen species with other sources of oxidative stress. Antioxid. Redox. Signal. 2014, 20, 308–324. [Google Scholar] [CrossRef]
- Munzel, T.; Daiber, A.; Gori, T. Nitrate therapy: New aspects concerning molecular action and tolerance. Circulation 2011, 123, 2132–2144. [Google Scholar] [CrossRef] [PubMed]
- Munzel, T.; Daiber, A.; Ullrich, V.; Mulsch, A. Vascular consequences of endothelial nitric oxide synthase uncoupling for the activity and expression of the soluble guanylyl cyclase and the cGMP-dependent protein kinase. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1551–1557. [Google Scholar] [CrossRef] [PubMed]
- Chaitoff, K.A.; Toner, F.; Tedesco, A.; Maher, T.J.; Ally, A. Effects of inducible nitric oxide synthase blockade within the periaqueductal gray on cardiovascular responses during mechanical, heat, and cold nociception. Neurol. Sci. 2012, 33, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, G.A.; Preuss, C.V.; Chaitoff, K.A.; Maher, T.J.; Ally, A. Medullary monoamines and NMDA-receptor regulation of cardiovascular responses during peripheral nociceptive stimuli. Neurosci. Res. 2006, 55, 316–326. [Google Scholar] [CrossRef] [PubMed]
- Ishide, T.; Maher, T.J.; Ally, A. Role of nitric oxide in the ventrolateral medulla on cardiovascular responses and glutamate neurotransmission during mechanical and thermal stimuli. Pharmacol. Res. 2003, 47, 59–68. [Google Scholar] [CrossRef]
- Gray, T.K.; Lewis, E., 3rd; Maher, T.J.; Ally, A. AMPA-receptor blockade within the RVLM modulates cardiovascular responses via glutamate during peripheral stimuli. Pharmacol. Res. 2001, 43, 47–54. [Google Scholar] [CrossRef]
- Ahlawat, A.; Rana, A.; Goyal, N.; Sharma, S. Potential role of nitric oxide synthase isoforms in pathophysiology of neuropathic pain. Inflammopharmacology 2014, 22, 269–278. [Google Scholar] [CrossRef]
- Boettger, M.K.; Uceyler, N.; Zelenka, M.; Schmitt, A.; Reif, A.; Chen, Y.; Sommer, C. Differences in inflammatory pain in nNOS-, iNOS- and eNOS-deficient mice. Eur. J. Pain 2007, 11, 810–818. [Google Scholar] [CrossRef] [Green Version]
- Lassen, L.H.; Ashina, M.; Christiansen, I.; Ulrich, V.; Grover, R.; Donaldson, J.; Olesen, J. Nitric oxide synthase inhibition: A new principle in the treatment of migraine attacks. Cephalalgia 1998, 18, 27–32. [Google Scholar] [CrossRef]
- Ashina, M.; Lassen, L.H.; Bendtsen, L.; Jensen, R.; Olesen, J. Effect of inhibition of nitric oxide synthase on chronic tension-type headache: A randomised crossover trial. Lancet 1999, 353, 287–289. [Google Scholar] [CrossRef]
- Chaitoff, K.A.; Patel, D.; Ally, A. Effects of endothelial NOS antagonism within the periaqueductal gray on cardiovascular responses and neurotransmission during mechanical, heat, and cold nociception. Brain Res. 2008, 1236, 93–104. [Google Scholar] [CrossRef] [PubMed]
- Miclescu, A.; Gordh, T. Nitric oxide and pain: ‘Something old, something new’. Acta Anaesthesiol. Scand. 2009, 53, 1107–1120. [Google Scholar] [CrossRef] [PubMed]
- Miclescu, A. Chronic pain patient and anaesthesia. Rom. J. Anaesth. Intensive Care 2019, 26, 59–66. [Google Scholar] [PubMed]
- Lyden, J.; Binswanger, I.A. The United States opioid epidemic. Semin. Perinatol. 2019, 43, 123–131. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sarmah, N.; Nauli, A.M.; Ally, A.; Nauli, S.M. Interactions among Endothelial Nitric Oxide Synthase, Cardiovascular System, and Nociception during Physiological and Pathophysiological States. Molecules 2022, 27, 2835. https://doi.org/10.3390/molecules27092835
Sarmah N, Nauli AM, Ally A, Nauli SM. Interactions among Endothelial Nitric Oxide Synthase, Cardiovascular System, and Nociception during Physiological and Pathophysiological States. Molecules. 2022; 27(9):2835. https://doi.org/10.3390/molecules27092835
Chicago/Turabian StyleSarmah, Niribili, Andromeda M. Nauli, Ahmmed Ally, and Surya M. Nauli. 2022. "Interactions among Endothelial Nitric Oxide Synthase, Cardiovascular System, and Nociception during Physiological and Pathophysiological States" Molecules 27, no. 9: 2835. https://doi.org/10.3390/molecules27092835
APA StyleSarmah, N., Nauli, A. M., Ally, A., & Nauli, S. M. (2022). Interactions among Endothelial Nitric Oxide Synthase, Cardiovascular System, and Nociception during Physiological and Pathophysiological States. Molecules, 27(9), 2835. https://doi.org/10.3390/molecules27092835