
Identifying Protein Features and Pathways Responsible for Toxicity Using Machine Learning and Tox21: Implications for Predictive Toxicology

 , ,
, ,  , and
, and 
Abstract
1. Introduction
2. Results and Discussion
2.1. Preprocessed Data
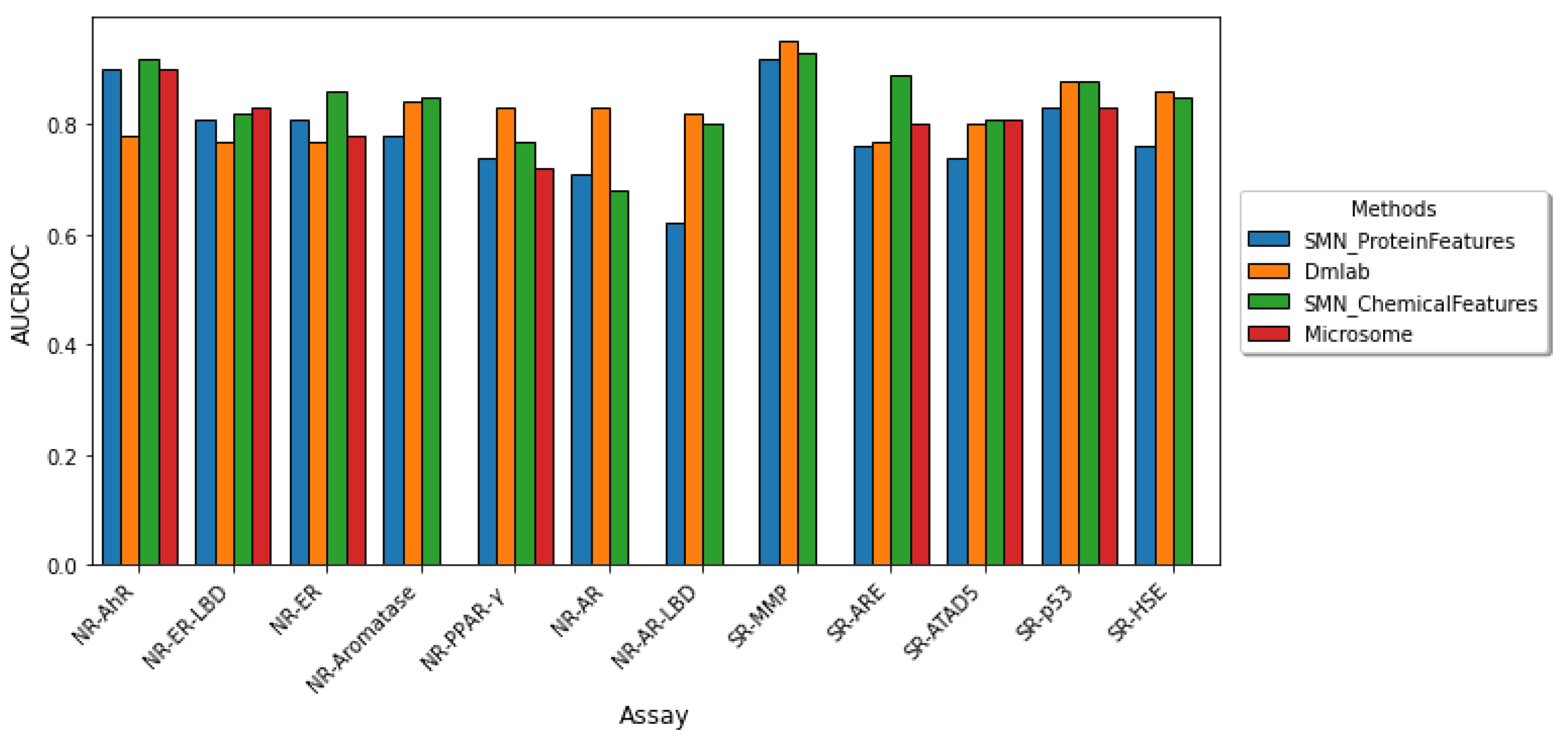
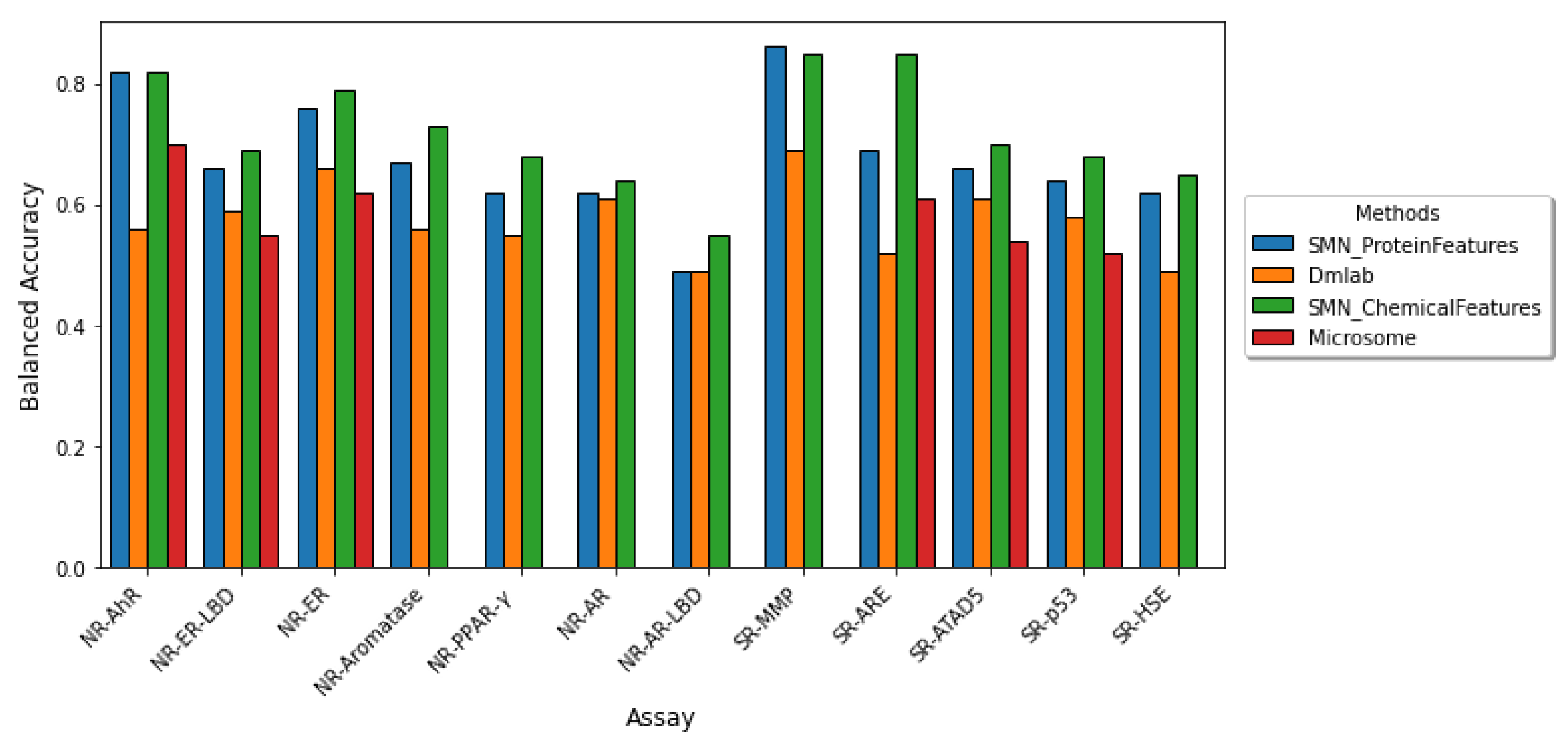
2.2. Comparison to Other Tox21 Studies
2.3. Enrichment Analysis
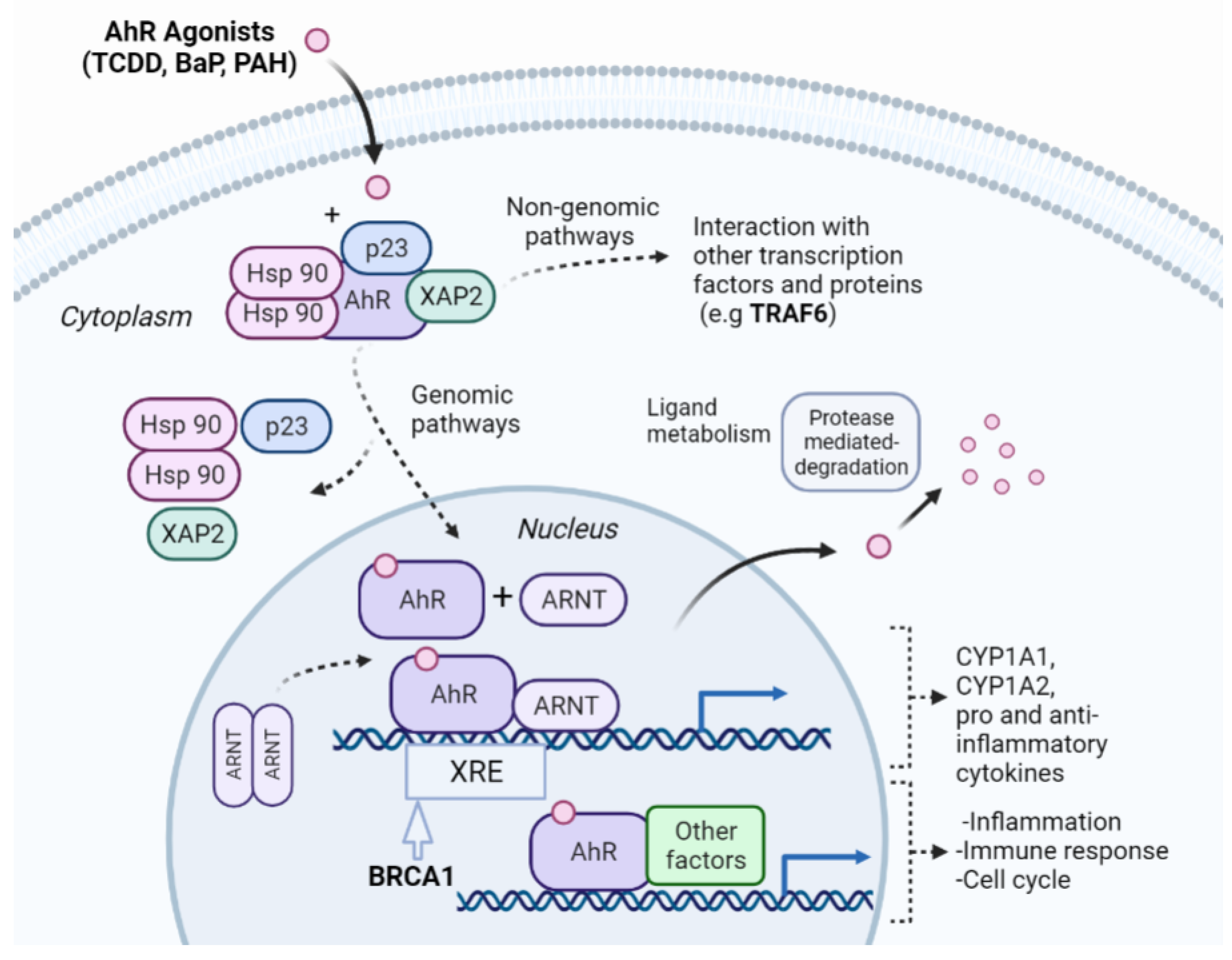
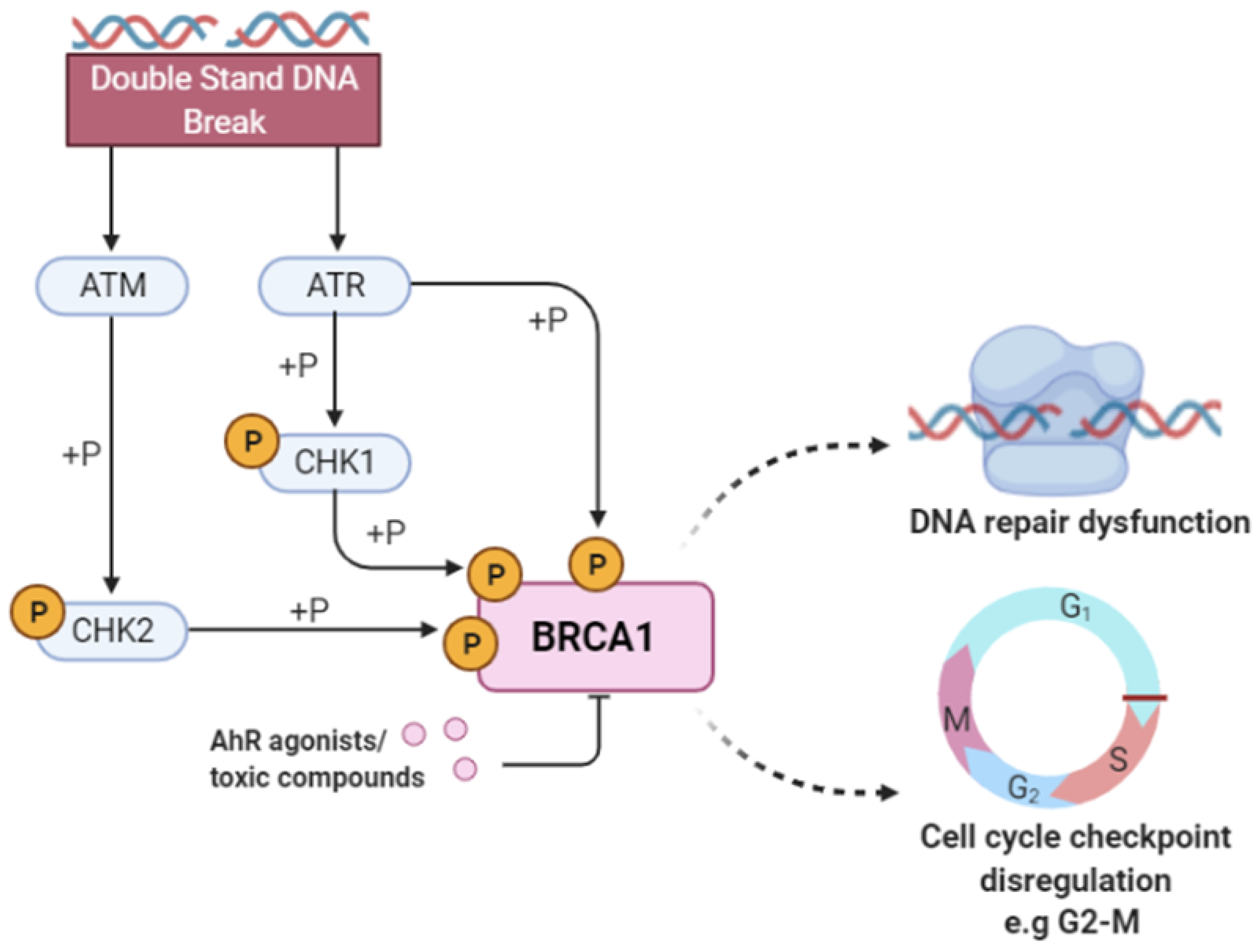
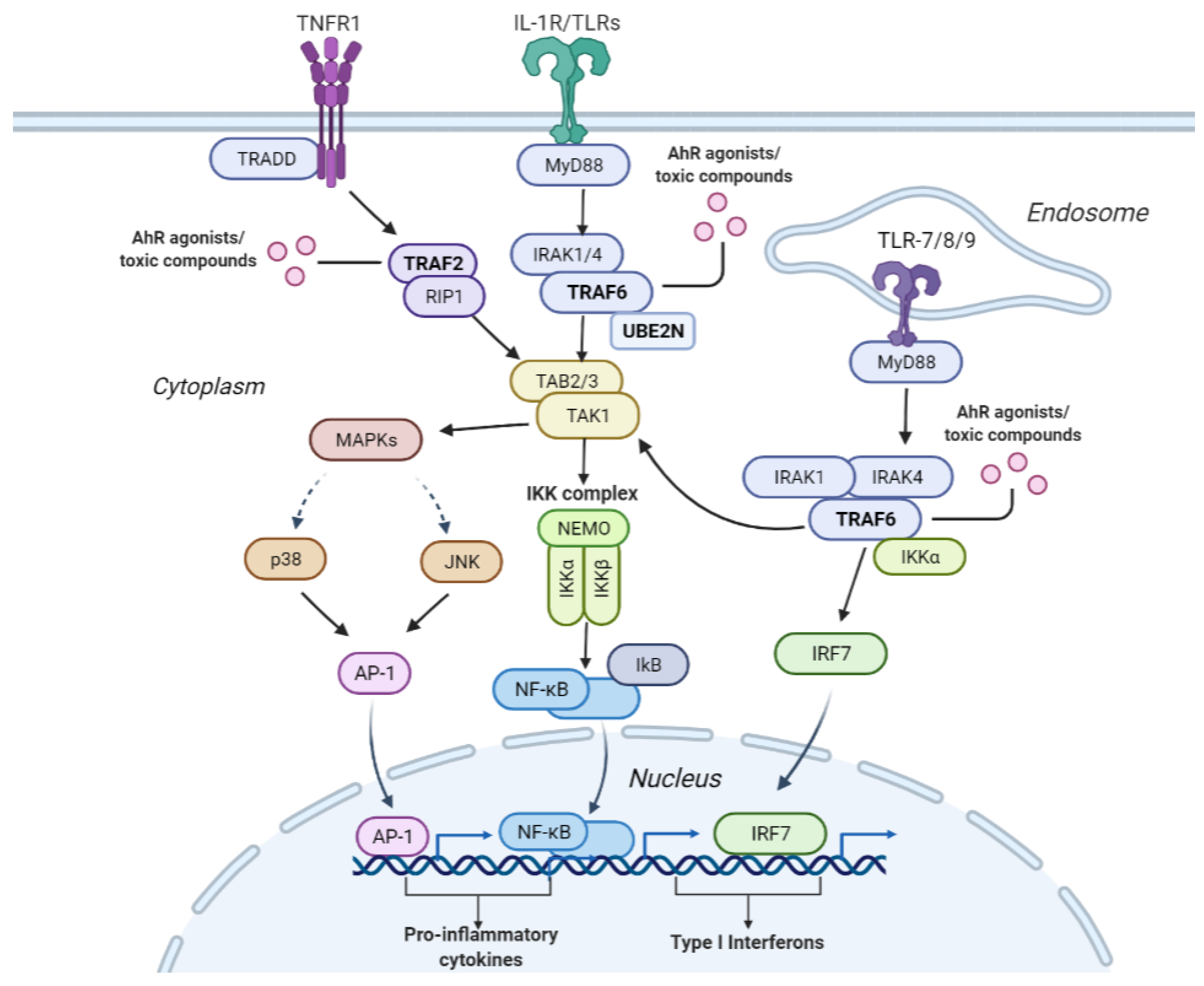
2.4. Case Study for NR-AhR
3. Limitations and Future Work
4. Materials and Methods
4.1. Tox21 Datasets
4.2. UniProt
4.3. Protein-Compound Interaction Scoring Protocol
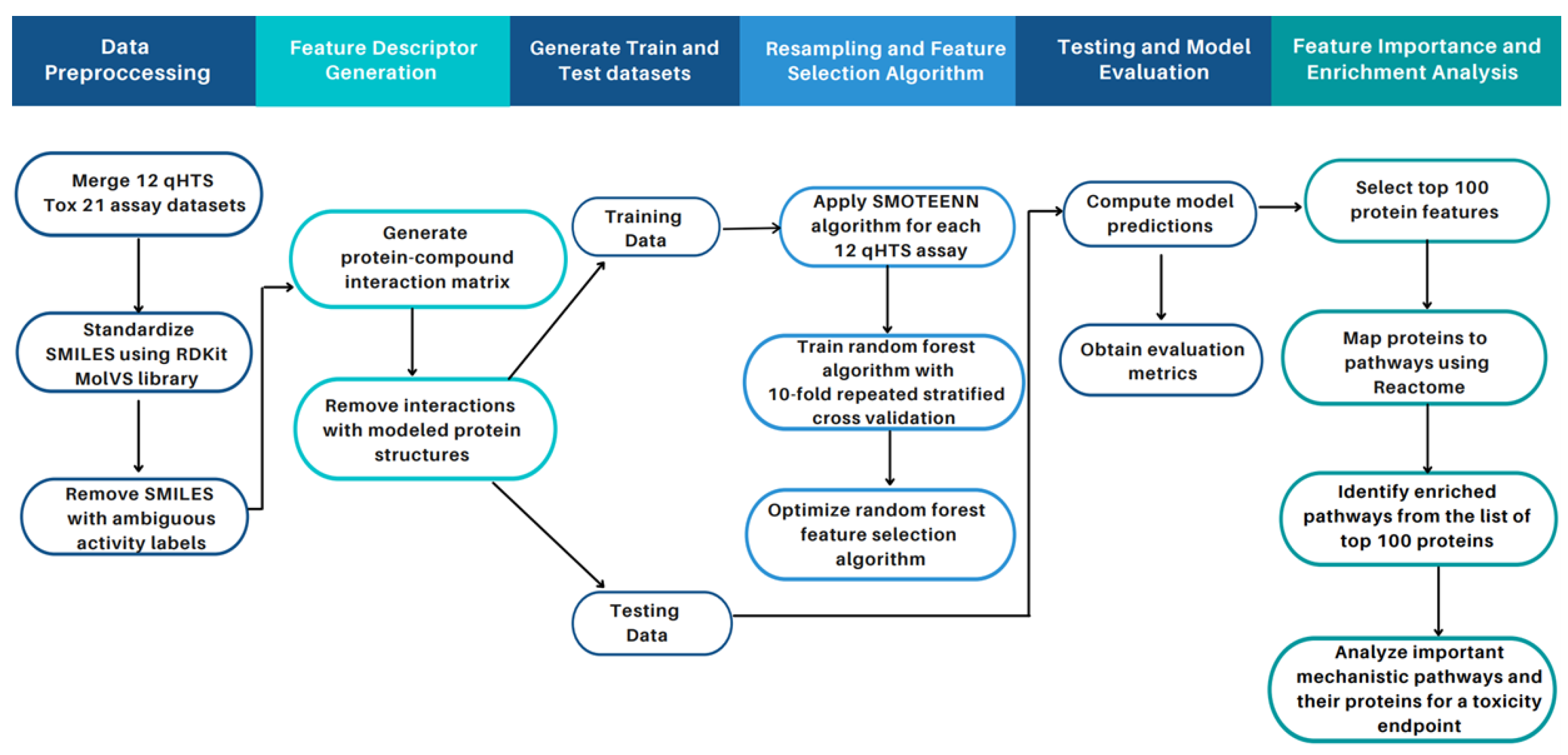
4.4. Study Design
4.4.1. Data Pre-Processing and Feature Generation
4.4.2. Data Resampling for Predictive Modeling
4.4.3. Random Forest
- (1)
- Draw a bootstrap sample: we randomly sample N compounds with replacement from the original dataset;
- (2)
- Create maximum decision trees: we construct a decision tree for each bootstrap sample by randomly sampling a subset of features at each node and choosing the best split among those features;
- (3)
- Construct a forest by repeating steps 1 and 2 for N trees;
- (4)
- Predict the outcome: from the built forest, the prediction is obtained by aggregating the predictions of the N trees (i.e., majority votes for classification and average for regression tasks).
4.4.4. Random Forest for Feature Selection
4.4.5. Model Training and Testing
4.4.6. Performance Evaluation Metrics
4.4.7. Enrichment Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Binetti, R.; Costamagna, F.M.; Marcello, I. Exponential growth of new chemicals and evolution of information relevant to risk control. Ann.-Ist. Super. Sanità 2008, 44, 13. [Google Scholar]
- Ekins, S.; Nikolsky, Y.; Nikolskaya, T. Techniques: Application of systems biology to absorption, distribution, metabolism, excretion and toxicity. Trends Pharmacol. Sci. 2005, 26, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Kavlock, R.; Dix, D. Computational toxicology as implemented by the US EPA: Providing high throughput decision support tools for screening and assessing chemical exposure, hazard and risk. J. Toxicol. Environ. Health Part B 2010, 13, 197–217. [Google Scholar] [CrossRef] [PubMed]
- Inglese, J.; Auld, D.S.; Jadhav, A.; Johnson, R.L.; Simeonov, A.; Yasgar, A.; Zheng, W.; Austin, C.P. Quantitative high-throughput screening: A titration-based approach that efficiently identifies biological activities in large chemical libraries. Proc. Natl. Acad. Sci. USA 2006, 103, 11473–11478. [Google Scholar] [CrossRef]
- Shukla, S.J.; Huang, R.; Austin, C.P.; Xia, M. The future of toxicity testing: A focus on in vitro methods using a quantitative high-throughput screening platform. Drug Discov. Today 2010, 15, 997–1007. [Google Scholar] [CrossRef]
- Sun, H.; Xia, M.; Austin, C.P.; Huang, R. Paradigm shift in toxicity testing and modeling. AAPS J. 2012, 14, 473–480. [Google Scholar] [CrossRef]
- Krewski, D.; Acosta, D., Jr.; Andersen, M.; Anderson, H.; Bailar, J.C., III; Boekelheide, K.; Brent, R.; Charnley, G.; Cheung, V.G.; Green, S., Jr.; et al. Toxicity testing in the 21st century: A vision and a strategy. J. Toxicol. Environ. Health Part B 2010, 13, 51–138. [Google Scholar] [CrossRef] [PubMed]
- Kavlock, R.J.; Austin, C.P.; Tice, R. Toxicity testing in the 21st century: Implications for human health risk assessment. Risk Anal. Off. Publ. Soc. Risk Anal. 2009, 29, 485. [Google Scholar] [CrossRef]
- Tice, R.R.; Austin, C.P.; Kavlock, R.J.; Bucher, J.R. Improving the human hazard characterization of chemicals: A Tox21 update. Environ. Health Perspect. 2013, 121, 756–765. [Google Scholar] [CrossRef]
- Collins, F.S.; Gray, G.M.; Bucher, J.R. Transforming environmental health protection. Science 2008, 319, 906. [Google Scholar] [CrossRef]
- Centre, I.I. The Tox21 Data Challenge. 2014. Available online: https://tripod.nih.gov/tox21/challenge/ (accessed on 1 June 2021).
- Chen, S.; Hsieh, J.H.; Huang, R.; Sakamuru, S.; Hsin, L.Y.; Xia, M.; Shockley, K.R.; Auerbach, S.; Kanaya, N.; Lu, H.; et al. Cell-based high-throughput screening for aromatase inhibitors in the Tox21 10K library. Toxicol. Sci. 2015, 147, 446–457. [Google Scholar] [CrossRef] [PubMed]
- Attene-Ramos, M.S.; Huang, R.; Michael, S.; Witt, K.L.; Richard, A.; Tice, R.R.; Simeonov, A.; Austin, C.P.; Xia, M. Profiling of the Tox21 chemical collection for mitochondrial function to identify compounds that acutely decrease mitochondrial membrane potential. Environ. Health Perspect. 2015, 123, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.; Sakamuru, S.; Martin, M.T.; Reif, D.M.; Judson, R.S.; Houck, K.A.; Casey, W.; Hsieh, J.H.; Shockley, K.R.; Ceger, P.; et al. Profiling of the Tox21 10K compound library for agonists and antagonists of the estrogen receptor alpha signaling pathway. Sci. Rep. 2014, 4, 5664. [Google Scholar] [CrossRef] [PubMed]
- Paul-Friedman, K.; Martin, M.; Crofton, K.M.; Hsu, C.W.; Sakamuru, S.; Zhao, J.; Xia, M.; Huang, R.; Stavreva, D.A.; Soni, V.; et al. Limited chemical structural diversity found to modulate thyroid hormone receptor in the Tox21 chemical library. Environ. Health Perspect. 2019, 127, 097009. [Google Scholar] [CrossRef]
- Huang, R.; Xia, M.; Nguyen, D.T.; Zhao, T.; Sakamuru, S.; Zhao, J.; Shahane, S.A.; Rossoshek, A.; Simeonov, A. Tox21Challenge to build predictive models of nuclear receptor and stress response pathways as mediated by exposure to environmental chemicals and drugs. Front. Environ. Sci. 2016, 3, 85. [Google Scholar] [CrossRef]
- Stefaniak, F. Prediction of compounds activity in nuclear receptor signaling and stress pathway assays using machine learning algorithms and low-dimensional molecular descriptors. Front. Environ. Sci. 2015, 3, 77. [Google Scholar] [CrossRef]
- Judson, R.S.; Magpantay, F.M.; Chickarmane, V.; Haskell, C.; Tania, N.; Taylor, J.; Xia, M.; Huang, R.; Rotroff, D.M.; Filer, D.L.; et al. Integrated model of chemical perturbations of a biological pathway using 18 in vitro high-throughput screening assays for the estrogen receptor. Toxicol. Sci. 2015, 148, 137–154. [Google Scholar] [CrossRef]
- Uesawa, Y. Rigorous selection of random forest models for identifying compounds that activate toxicity-related pathways. Front. Environ. Sci. 2016, 4, 9. [Google Scholar] [CrossRef]
- Barta, G. Identifying biological pathway interrupting toxins using multi-tree ensembles. Front. Environ. Sci. 2016, 4, 52. [Google Scholar] [CrossRef]
- Koutsoukas, A.; St Amand, J.; Mishra, M.; Huan, J. Predictive toxicology: Modeling chemical induced toxicological response combining circular fingerprints with random forest and support vector machine. Front. Environ. Sci. 2016, 4, 11. [Google Scholar] [CrossRef]
- Mayr, A.; Klambauer, G.; Unterthiner, T.; Hochreiter, S. DeepTox: Toxicity prediction using deep learning. Front. Environ. Sci. 2016, 3, 80. [Google Scholar] [CrossRef]
- Guyon, I.; Gunn, S.; Nikravesh, M.; Zadeh, L. Feature Extraction: Foundations and Applications; Studies in Fuzziness and Soft Computing; Springer: Berlin/Heidelberg, Germany, 2008. [Google Scholar]
- Chandrashekar, G.; Sahin, F. A survey on feature selection methods. Comput. Electr. Eng. 2014, 40, 16–28. [Google Scholar] [CrossRef]
- Jaiswal, J.K.; Samikannu, R. Application of random forest algorithm on feature subset selection and classification and regression. In Proceedings of the 2017 World Congress on Computing and Communication Technologies (WCCCT), Tiruchirappalli, India, 2–4 February 2017; pp. 65–68. [Google Scholar]
- Remeseiro, B.; Bolon-Canedo, V. A review of feature selection methods in medical applications. Comput. Biol. Med. 2019, 112, 103375. [Google Scholar] [CrossRef] [PubMed]
- Breiman, L. Random Forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef]
- Chen, R.C.; Dewi, C.; Huang, S.W.; Caraka, R.E. Selecting critical features for data classification based on machine learning methods. J. Big Data 2020, 7, 52. [Google Scholar] [CrossRef]
- Díaz-Uriarte, R.; De Andres, S.A. Gene selection and classification of microarray data using random forest. BMC Bioinform. 2006, 7, 3. [Google Scholar] [CrossRef]
- Rogers, J.; Gunn, S. Identifying feature relevance using a random forest. In International Statistical and Optimization Perspectives Workshop” Subspace, Latent Structure and Feature Selection”; Springer: Berlin/Heidelberg, Germany, 2005; pp. 173–184. [Google Scholar]
- Idakwo, G.; Thangapandian, S.; Luttrell, J.; Li, Y.; Wang, N.; Zhou, Z.; Hong, H.; Yang, B.; Zhang, C.; Gong, P. Structure–activity relationship-based chemical classification of highly imbalanced Tox21 datasets. J. Cheminform. 2020, 12, 66. [Google Scholar] [CrossRef]
- Banerjee, P.; Dehnbostel, F.O.; Preissner, R. Prediction is a balancing act: Importance of sampling methods to balance sensitivity and specificity of predictive models based on imbalanced chemical data sets. Front. Chem. 2018, 6, 362. [Google Scholar] [CrossRef]
- Ring, C.; Sipes, N.S.; Hsieh, J.H.; Carberry, C.; Koval, L.E.; Klaren, W.D.; Harris, M.A.; Auerbach, S.S.; Rager, J.E. Predictive modeling of biological responses in the rat liver using in vitro Tox21 bioactivity: Benefits from high-throughput toxicokinetics. Comput. Toxicol. 2021, 18, 100166. [Google Scholar] [CrossRef]
- Zhang, J.; Mucs, D.; Norinder, U.; Svensson, F. LightGBM: An effective and scalable algorithm for prediction of chemical toxicity–application to the Tox21 and mutagenicity data sets. J. Chem. Inf. Model. 2019, 59, 4150–4158. [Google Scholar] [CrossRef]
- LI, J.C. Imbalanced Toxicity Prediction Using Multi-Task Learning and Over-Sampling. In Proceedings of the 2020 International Conference on Machine Learning and Cybernetics (ICMLC), Adelaide, Australia, 2 December 2020; pp. 1–7. [Google Scholar]
- Jenwitheesuk, E.; Samudrala, R. Prediction of HIV-1 protease inhibitor resistance using a protein-inhibitor flexible docking approach. Antivir. Ther. 2005, 10, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Jenwitheesuk, E.; Samudrala, R. New paradigms for drug discovery: Computational multitarget screening. Trends Pharmacol. Sci. 2008, 29, 62–71. [Google Scholar] [CrossRef] [PubMed]
- Minie, M.; Chopra, G.; Sethi, G.; Horst, J.; White, G.; Roy, A.; Hatti, K.; Samudrala, R. CANDO and the infinite drug discovery frontier. Drug Discov. Today 2014, 19, 1353–1363. [Google Scholar] [CrossRef] [PubMed]
- Sethi, G.; Chopra, G.; Samudrala, R. Multiscale modelling of relationships between protein classes and drug behavior across all diseases using the CANDO platform. Mini Rev. Med. Chem. 2015, 15, 705–717. [Google Scholar] [CrossRef] [PubMed]
- Chopra, G.; Samudrala, R. Exploring polypharmacology in drug discovery and repurposing using the CANDO platform. Curr. Pharm. Des. 2016, 22, 3109–3123. [Google Scholar] [CrossRef] [PubMed]
- Chopra, G.; Kaushik, S.; Elkin, P.; Samudrala, R. Combating Ebola with repurposed therapeutics using the CANDO platform. Molecules 2016, 21, 1537. [Google Scholar] [CrossRef]
- Mangione, W.; Samudrala, R. Identifying protein features responsible for improved drug repurposing accuracies using the CANDO platform: Implications for drug design. Molecules 2019, 24, 167. [Google Scholar] [CrossRef]
- Falls, Z.; Mangione, W.; Schuler, J.; Samudrala, R. Exploration of interaction scoring criteria in the CANDO platform. BMC Res. Notes 2019, 12, 318. [Google Scholar] [CrossRef]
- Schuler, J.; Samudrala, R. Fingerprinting CANDO: Increased Accuracy with Structure-and Ligand-Based Shotgun Drug Repurposing. ACS Omega 2019, 4, 17393–17403. [Google Scholar] [CrossRef]
- Fine, J.; Lacker, R.; Samudrala, R.; Chopra, G. Computational chemoproteomics to understand the role of selected psychoactives in treating mental health disorders. Sci. Rep. 2019, 9, 13155. [Google Scholar] [CrossRef]
- Mangione, W.; Falls, Z.; Melendy, T.; Chopra, G.; Samudrala, R. Shotgun drug repurposing biotechnology to tackle epidemics and pandemics. Drug Discov. Today 2020, 25, 1126–1128. [Google Scholar] [CrossRef] [PubMed]
- Mangione, W.; Falls, Z.; Chopra, G.; Samudrala, R. cando. py: Open Source Software for Predictive Bioanalytics of Large Scale Drug–Protein–Disease Data. J. Chem. Inf. Model. 2020, 60, 4131–4136. [Google Scholar] [CrossRef] [PubMed]
- Hudson, M.L.; Samudrala, R. Multiscale virtual screening optimization for shotgun drug repurposing using the CANDO platform. Molecules 2021, 26, 2581. [Google Scholar] [CrossRef] [PubMed]
- Schuler, J.; Falls, Z.; Mangione, W.; Hudson, M.L.; Bruggemann, L.; Samudrala, R. Evaluating the performance of drug-repurposing technologies. Drug Discov. Today 2021, 27, 49–64. [Google Scholar] [CrossRef] [PubMed]
- Deric; Sforna, G.; Landrum, G.; Winter, H.D. RDKit MolVS Python Package. 2016. Available online: https://molvs.readthedocs.io/en/latest/ (accessed on 1 September 2021).
- Swanson, H.I.; Bradfield, C.A. The AH-receptor: Genetics, structure and function. Pharmacogenetics 1993, 3, 213–230. [Google Scholar] [CrossRef] [PubMed]
- Mimura, J.; Fujii-Kuriyama, Y. Functional role of AhR in the expression of toxic effects by TCDD. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2003, 1619, 263–268. [Google Scholar] [CrossRef]
- Vorderstrasse, B.A.; Steppan, L.B.; Silverstone, A.E.; Kerkvliet, N.I. Aryl hydrocarbon receptor-deficient mice generate normal immune responses to model antigens and are resistant to TCDD-induced immune suppression. Toxicol. Appl. Pharmacol. 2001, 171, 157–164. [Google Scholar] [CrossRef]
- Huff, J.; Lucier, G.; Tritscher, A. Carcinogenicity of TCDD: Experimental, mechanistic, and epidemiologic evidence. Annu. Rev. Pharmacol. Toxicol. 1994, 34, 343–372. [Google Scholar] [CrossRef]
- Marlowe, J.L.; Puga, A. Aryl hydrocarbon receptor, cell cycle regulation, toxicity, and tumorigenesis. J. Cell. Biochem. 2005, 96, 1174–1184. [Google Scholar] [CrossRef]
- Dietrich, C.; Kaina, B. The aryl hydrocarbon receptor (AhR) in the regulation of cell–cell contact and tumor growth. Carcinogenesis 2010, 31, 1319–1328. [Google Scholar] [CrossRef]
- Quintana, F.J.; Sherr, D.H. Aryl hydrocarbon receptor control of adaptive immunity. Pharmacol. Rev. 2013, 65, 1148–1161. [Google Scholar] [CrossRef] [PubMed]
- Winans, B.; Humble, M.C.; Lawrence, B.P. Environmental toxicants and the developing immune system: A missing link in the global battle against infectious disease? Reprod. Toxicol. 2011, 31, 327–336. [Google Scholar] [CrossRef] [PubMed]
- Vezina, C.M.; Walker, N.J.; Olson, J.R. Subchronic exposure to TCDD, PeCDF, PCB126, and PCB153: Effect on hepatic gene expression. Environ. Health Perspect. 2004, 112, 1636–1644. [Google Scholar] [CrossRef] [PubMed]
- Tijet, N.; Boutros, P.C.; Moffat, I.D.; Okey, A.B.; Tuomisto, J.; Pohjanvirta, R. Aryl hydrocarbon receptor regulates distinct dioxin-dependent and dioxin-independent gene batteries. Mol. Pharmacol. 2006, 69, 140–153. [Google Scholar] [CrossRef]
- Matsumura, F. The significance of the nongenomic pathway in mediating inflammatory signaling of the dioxin-activated Ah receptor to cause toxic effects. Biochem. Pharmacol. 2009, 77, 608–626. [Google Scholar] [CrossRef]
- Kang, H.J.; Kim, H.J.; Kim, S.K.; Barouki, R.; Cho, C.H.; Khanna, K.K.; Rosen, E.M.; Bae, I. BRCA1 modulates xenobiotic stress-inducible gene expression by interacting with ARNT in human breast cancer cells. J. Biol. Chem. 2006, 281, 14654–14662. [Google Scholar] [CrossRef]
- Chan, C.Y.; Kim, P.M.; Winn, L.M. TCDD affects DNA double strand-break repair. Toxicol. Sci. 2004, 81, 133–138. [Google Scholar] [CrossRef][Green Version]
- Rattenborg, T.; Gjermandsen, I.; Bonefeld-Jørgensen, E.C. Inhibition of E2-induced expression of BRCA1 by persistent organochlorines. Breast Cancer Res. 2002, 4, R12. [Google Scholar] [CrossRef]
- MacLachlan, T.K.; Somasundaram, K.; Sgagias, M.; Shifman, Y.; Muschel, R.J.; Cowan, K.H.; El-Deiry, W.S. BRCA1 effects on the cell cycle and the DNA damage response are linked to altered gene expression. J. Biol. Chem. 2000, 275, 2777–2785. [Google Scholar] [CrossRef]
- Gowen, L.C.; Avrutskaya, A.V.; Latour, A.M.; Koller, B.H.; Leadon, S.A. BRCA1 required for transcription-coupled repair of oxidative DNA damage. Science 1998, 281, 1009–1012. [Google Scholar] [CrossRef]
- Rosen, E.M.; Fan, S.; Pestell, R.G.; Goldberg, I.D. BRCA1 gene in breast cancer. J. Cell. Physiol. 2003, 196, 19–41. [Google Scholar] [CrossRef] [PubMed]
- Jeffy, B.D.; Schultz, E.U.; Selmin, O.; Gudas, J.M.; Bowden, G.T.; Romagnolo, D. Inhibition of BRCA-1 expression by benzo [a] pyrene and its diol epoxide. Mol. Carcinog. Publ. Coop. Univ. Tex. MD Anderson Cancer Cent. 1999, 26, 100–118. [Google Scholar] [CrossRef]
- Foo, T.K.; Vincelli, G.; Huselid, E.; Her, J.; Zheng, H.; Simhadri, S.; Wang, M.; Huo, Y.; Li, T.; Yu, X.; et al. ATR/ATM-Mediated Phosphorylation of BRCA1 T1394 Promotes Homologous Recombinational Repair and G2–M Checkpoint Maintenance. Cancer Res. 2021, 81, 4676–4684. [Google Scholar] [CrossRef] [PubMed]
- Hankinson, O. Role of coactivators in transcriptional activation by the aryl hydrocarbon receptor. Arch. Biochem. Biophys. 2005, 433, 379–386. [Google Scholar] [CrossRef]
- Voronov, I.; Li, K.; Tenenbaum, H.; Manolson, M. Benzo [a] pyrene inhibits osteoclastogenesis by affecting RANKL-induced activation of NF-κB. Biochem. Pharmacol. 2008, 75, 2034–2044. [Google Scholar] [CrossRef]
- Hayden, M.S.; Ghosh, S. Signaling to NF-κB. Genes Dev. 2004, 18, 2195–2224. [Google Scholar] [CrossRef]
- Zheng, W.; Li, R.; Pan, H.; He, D.; Xu, R.; Guo, T.B.; Guo, Y.; Zhang, J.Z. Role of osteopontin in induction of monocyte chemoattractant protein 1 and macrophage inflammatory protein 1β through the NF-κB and MAPK pathways in rheumatoid arthritis. Arthritis Rheum. Off. J. Am. Coll. Rheumatol. 2009, 60, 1957–1965. [Google Scholar] [CrossRef]
- Tanaka, S.; Nakamura, K.; Takahasi, N.; Suda, T. Role of RANKL in physiological and pathological bone resorption and therapeutics targeting the RANKL–RANK signaling system. Immunol. Rev. 2005, 208, 30–49. [Google Scholar] [CrossRef]
- Fu, J.; Nogueira, S.V.; van Drongelen, V.; Coit, P.; Ling, S.; Rosloniec, E.F.; Sawalha, A.H.; Holoshitz, J. Shared epitope–aryl hydrocarbon receptor crosstalk underlies the mechanism of gene–environment interaction in autoimmune arthritis. Proc. Natl. Acad. Sci. USA 2018, 115, 4755–4760. [Google Scholar] [CrossRef]
- Weiss, C.; Faust, D.; Dürk, H.; Kolluri, S.K.; Pelzer, A.; Schneider, S.; Dietrich, C.; Oesch, F.; Göttlicher, M. TCDD induces c-jun expression via a novel Ah (dioxin) receptor-mediated p38–MAPK-dependent pathway. Oncogene 2005, 24, 4975–4983. [Google Scholar] [CrossRef]
- Deng, L.; Wang, C.; Spencer, E.; Yang, L.; Braun, A.; You, J.; Slaughter, C.; Pickart, C.; Chen, Z.J. Activation of the IκB kinase complex by TRAF6 requires a dimeric ubiquitin-conjugating enzyme complex and a unique polyubiquitin chain. Cell 2000, 103, 351–361. [Google Scholar] [CrossRef]
- Biswas, R.; Bagchi, A. Inhibition of TRAF6-Ubc13 interaction in NFkB inflammatory pathway by analyzing the hotspot amino acid residues and protein–protein interactions using molecular docking simulations. Comput. Biol. Chem. 2017, 70, 116–124. [Google Scholar] [CrossRef] [PubMed]
- Consortium, T.U. UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2020, 49, D480–D489. [Google Scholar] [CrossRef] [PubMed]
- Protein Data Bank: The single global archive for 3D macromolecular structure data. Nucleic Acids Res. 2019, 47, D520–D528. [CrossRef]
- Jassal, B.; Matthews, L.; Viteri, G.; Gong, C.; Lorente, P.; Fabregat, A.; Sidiropoulos, K.; Cook, J.; Gillespie, M.; Haw, R.; et al. The reactome pathway knowledgebase. Nucleic Acids Res. 2020, 48, D498–D503. [Google Scholar] [CrossRef]
- Yang, J.; Roy, A.; Zhang, Y. Protein–ligand binding site recognition using complementary binding-specific substructure comparison and sequence profile alignment. Bioinformatics 2013, 29, 2588–2595. [Google Scholar] [CrossRef]
- Sorensen, T.A. A method of establishing groups of equal amplitude in plant sociology based on similarity of species content and its application to analyses of the vegetation on Danish commons. Biol. Skar. 1948, 5, 1–34. [Google Scholar]
- Deric; Sforna, G.; Landrum, G.; Winter, H.D. Rdkit Github Page. 2012. Available online: https://github.com/rdkit (accessed on 1 September 2021).
- Bellman, R. Dynamic programming. Science 1966, 153, 34–37. [Google Scholar] [CrossRef]
- Chawla, N.V.; Bowyer, K.W.; Hall, L.O.; Kegelmeyer, W.P. SMOTE: Synthetic minority over-sampling technique. J. Artif. Intell. Res. 2002, 16, 321–357. [Google Scholar] [CrossRef]
- Beckmann, M.; Ebecken, N.F.; de Lima, B.S.P. A KNN undersampling approach for data balancing. J. Intell. Learn. Syst. Appl. 2015, 7, 104. [Google Scholar] [CrossRef]
- Batista, G.E.; Prati, R.C.; Monard, M.C. A Study of The Behavior of Several Methods for Balancing Machine Learning Training Data. ACM SIGKDD Explor. Newsl. 2004, 6, 20–29. [Google Scholar] [CrossRef]
- Svetnik, V.; Liaw, A.; Tong, C.; Culberson, J.C.; Sheridan, R.P.; Feuston, B.P. Random forest: A classification and regression tool for compound classification and QSAR modeling. J. Chem. Inf. Comput. Sci. 2003, 43, 1947–1958. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y. Random forest for bioinformatics. In Ensemble Machine Learning; Springer: Berlin/Heidelberg, Germany, 2012; pp. 307–323. [Google Scholar]
- Dietterich, T.G. An experimental comparison of three methods for constructing ensembles of decision trees: Bagging, boosting, and randomization. Mach. Learn. 2000, 40, 139–157. [Google Scholar] [CrossRef]
- Saeys, Y.; Inza, I.; Larranaga, P. A review of feature selection techniques in bioinformatics. Bioinformatics 2007, 23, 2507–2517. [Google Scholar] [CrossRef] [PubMed]
- Nembrini, S.; König, I.R.; Wright, M.N. The revival of the Gini importance? Bioinformatics 2018, 34, 3711–3718. [Google Scholar] [CrossRef]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-learn: Machine learning in python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Lemaıtre, G.; Nogueira, F.; Christos, K. Aridas. Imbalanced-Learn: A Python Toolbox to Tackle the Curse of Imbalanced Datasets in Machine Learning. J. Mach. Learn. Res. 2017, 18, 559–563. [Google Scholar]
- Davis, J.; Goadrich, M. The Relationship between Precision-Recall and ROC Curves. 2006, pp. 233–240. Available online: https://www.biostat.wisc.edu/~page/rocpr.pdf (accessed on 30 October 2021).
- Huang, J.; Ling, C.X. Using AUC and accuracy in evaluating learning algorithms. IEEE Trans. Knowl. Data Eng. 2005, 17, 299–310. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| In Vitro qHTS Assay Identifier | Target/Assay | Number of Compounds | Training Set | Test Set | ||||
|---|---|---|---|---|---|---|---|---|
| Inactive | Active | IR | Inactive | Active | IR | |||
| NR-AhR | Aryl hydrocarbon receptor | 7103 | 5777 | 734 | 7.87 | 521 | 71 | 7.34 |
| NR-ER-LBD | Estrogen receptor (luciferase assay) | 7509 | 6643 | 282 | 23.56 | 564 | 20 | 28.20 |
| NR-ER | Estrogen receptor | 6630 | 5474 | 651 | 8.41 | 456 | 49 | 9.31 |
| NR-Aromatase | Aromatase | 6286 | 5496 | 274 | 20.06 | 479 | 37 | 12.94 |
| NR-PPAR- | Peroxisome proliferator-activated receptor | 7039 | 6283 | 167 | 37.62 | 559 | 30 | 18.63 |
| NR-AR | Androgen receptor | 7783 | 6958 | 252 | 27.61 | 561 | 12 | 46.75 |
| NR-AR-LBD | Androgen receptor (luciferase assay) | 7298 | 6521 | 211 | 30.90 | 558 | 8 | 69.75 |
| SR-MMP | Mitochondrial membrane potential | 6316 | 4899 | 888 | 5.52 | 474 | 55 | 8.62 |
| SR-ARE | Nuclear factor (erythroid-derived 2)-like 2 antioxidant responsive element | 6339 | 4919 | 881 | 5.58 | 450 | 89 | 5.06 |
| SR-ATAD5 | Genotoxicity indicated by ATAD5 | 7646 | 6787 | 256 | 26.51 | 569 | 34 | 16.73 |
| SR-p53 | DNA damage p53-pathway | 7358 | 6351 | 409 | 15.53 | 560 | 38 | 14.74 |
| SR-HSE | Heat shock factor response element | 7040 | 6144 | 305 | 20.14 | 574 | 17 | 33.76 |
| Assays | F1 | Precision | Recall | AUCROC | AUPRC | BA | MCC | Specificity | Accuracy |
|---|---|---|---|---|---|---|---|---|---|
| NR-AhR | 0.471 | 0.318 | 0.901 | 0.896 | 0.560 | 0.819 | 0.438 | 0.737 | 0.757 |
| NR-ER-LBD | 0.342 | 0.333 | 0.350 | 0.810 | 0.272 | 0.663 | 0.318 | 0.975 | 0.954 |
| NR-ER | 0.420 | 0.301 | 0.694 | 0.806 | 0.414 | 0.760 | 0.370 | 0.827 | 0.814 |
| NR-Aromatase | 0.317 | 0.250 | 0.432 | 0.795 | 0.282 | 0.666 | 0.260 | 0.900 | 0.866 |
| NR-PPAR- | 0.286 | 0.308 | 0.267 | 0.745 | 0.241 | 0.617 | 0.251 | 0.968 | 0.932 |
| NR-AR | 0.261 | 0.273 | 0.250 | 0.706 | 0.196 | 0.618 | 0.178 | 0.988 | 0.970 |
| NR-AR-LBD | 0.000 | 0.000 | 0.000 | 0.618 | 0.036 | 0.493 | −0.014 | 0.986 | 0.972 |
| SR-MMP | 0.488 | 0.331 | 0.927 | 0.916 | 0.597 | 0.855 | 0.478 | 0.783 | 0.798 |
| SR-ARE | 0.425 | 0.305 | 0.697 | 0.757 | 0.403 | 0.692 | 0.294 | 0.687 | 0.688 |
| SR-ATAD5 | 0.325 | 0.283 | 0.382 | 0.744 | 0.230 | 0.662 | 0.282 | 0.942 | 0.910 |
| SR-p53 | 0.235 | 0.159 | 0.447 | 0.830 | 0.198 | 0.643 | 0.182 | 0.839 | 0.814 |
| SR-HSE | 0.286 | 0.308 | 0.267 | 0.759 | 0.240 | 0.617 | 0.251 | 0.968 | 0.932 |
| Pathway | Total Proteins | Selected Proteins (Gene IDs) | p-Value |
|---|---|---|---|
| Nonhomologous End-Joining (NHEJ) | 52 | RNF8,UBE2N, BRCA1,NSD2 | 1.36 × 10 |
| Recruitment and ATM-mediated phosphorylation of repair and signaling proteins at DNA double strand breaks | 59 | RNF8,UBE2N, BRCA1,NSD2 | 2.22 × 10 |
| TRAF6 mediated NF-B activation | 24 | TRAF2,TRAF6 | 2.35 × 10 |
| DNA Double Strand Break Response | 60 | RNF8,UBE2N, BRCA1,NSD2 | 2.36 × 10 |
| TRAF6 mediated IRF7 activation | 28 | TRAF2,TRAF6 | 3.73 × 10 |
| Neurofascin interactions | 7 | NRCAM,CNTN1 | 5.28 × 10 |
| DDX58/IFIH1-mediated induction of interferon-alpha/beta | 77 | TRAF2,RNF125, TRAF6,DDX58 | 6.04 × 10 |
| RUNX3 regulates YAP1-mediated transcription | 8 | TEAD1,TEAD4 | 7.01 × 10 |
| SUMOylation of transcription cofactors | 42 | RNF2,UHRF2,PIAS3 | 1.22 × 10 |
| IRAK1 recruits IKK complex | 14 | TRAF6,UBE2N | 2.21 × 10 |
| IRAK1 recruits IKK complex upon TLR7/8 or 9 stimulation | 14 | TRAF6,UBE2N | 2.21 × 10 |
| YAP1- and WWTR1 (TAZ)-stimulated gene expression | 14 | TEAD1,TEAD4 | 2.21 × 10 |
| TRAF6 mediated IRF7 activation in TLR7/8 or 9 signaling | 14 | TRAF6,UBE2N | 2.21 × 10 |
| TICAM1, RIP1-mediated IKK complex recruitment | 19 | TRAF6,UBE2N | 4.05 × 10 |
| Signal transduction by L1 | 20 | NRP1,NCAM1 | 4.48 × 10 |
| G2/M DNA damage checkpoint | 78 | RNF8,UBE2N,BRCA1, NSD2,RPA1 | 4.64 × 10 |
| Regulation of FZD by ubiquitination | 21 | LRP6,LGR5 | 4.92 × 10 |
| IKK complex recruitment mediated by RIP1 | 22 | TRAF6,UBE2N | 5.39 × 10 |
| JNK (c-Jun kinases) phosphorylation and activation mediated by activated human TAK1 | 22 | TRAF6,UBE2N | 5.39 × 10 |
| Processing of DNA double-strand break ends | 81 | RNF8,UBE2N,BRCA1, NSD2,RPA1 | 5.55 × 10 |
| Activated TAK1 mediates p38 MAPK activation | 23 | TRAF6,UBE2N | 5.87 × 10 |
| Formation of Incision Complex in GG-NER | 43 | UBE2N,PIAS3, RBX1,RPA1 | 6.52 × 10 |
| Recognition of DNA damage by PCNA-containing replication complex | 31 | RBX1,RPA1 | 1.03 × 10 |
| TAK1 activates NFkB by phosphorylation and activation of IKKs complex | 32 | TRAF6,UBE2N | 1.10 × 10 |
| DNA strand elongation | 32 | GINS2,RPA1 | 1.10 × 10 |
| Sialic acid metabolism | 33 | GLB1,NANP | 1.16 × 10 |
| Transcriptional Regulation by E2F6 | 34 | RNF2,BRCA1 | 1.23 × 10 |
| Negative regulators of DDX58/IFIH1 signaling | 34 | RNF125,DDX58 | 1.23 × 10 |
| NOD1/2 Signaling Pathway | 35 | TRAF6,UBE2N | 1.30 × 10 |
| RUNX1 interacts with co-factors whose precise effect on RUNX1 targets is not known | 36 | RNF2,PCGF5 | 1.37 × 10 |
| HDR through Single Strand Annealing (SSA) | 37 | BRCA1,RPA1 | 1.44 × 10 |
| Ovarian tumor domain proteases | 38 | TRAF6,DDX58 | 1.51 × 10 |
| Presynaptic phase of homologous DNA pairing and strand exchange | 39 | BRCA1,RPA1 | 1.58 × 10 |
| Formation of Fibrin Clot (Clotting Cascade) | 39 | PROCR,GP1BB | 1.58 × 10 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moukheiber, L.; Mangione, W.; Moukheiber, M.; Maleki, S.; Falls, Z.; Gao, M.; Samudrala, R. Identifying Protein Features and Pathways Responsible for Toxicity Using Machine Learning and Tox21: Implications for Predictive Toxicology. Molecules 2022, 27, 3021. https://doi.org/10.3390/molecules27093021
Moukheiber L, Mangione W, Moukheiber M, Maleki S, Falls Z, Gao M, Samudrala R. Identifying Protein Features and Pathways Responsible for Toxicity Using Machine Learning and Tox21: Implications for Predictive Toxicology. Molecules. 2022; 27(9):3021. https://doi.org/10.3390/molecules27093021
Chicago/Turabian StyleMoukheiber, Lama, William Mangione, Mira Moukheiber, Saeed Maleki, Zackary Falls, Mingchen Gao, and Ram Samudrala. 2022. "Identifying Protein Features and Pathways Responsible for Toxicity Using Machine Learning and Tox21: Implications for Predictive Toxicology" Molecules 27, no. 9: 3021. https://doi.org/10.3390/molecules27093021
APA StyleMoukheiber, L., Mangione, W., Moukheiber, M., Maleki, S., Falls, Z., Gao, M., & Samudrala, R. (2022). Identifying Protein Features and Pathways Responsible for Toxicity Using Machine Learning and Tox21: Implications for Predictive Toxicology. Molecules, 27(9), 3021. https://doi.org/10.3390/molecules27093021