
Intermolecular Interactions and Charge Resonance Contributions to Triplet and Singlet Exciton States of Oligoacene Aggregates
 , , , and
, , , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
2.1. Orbital Space and Diabatic States for the Analysis of Exciton States
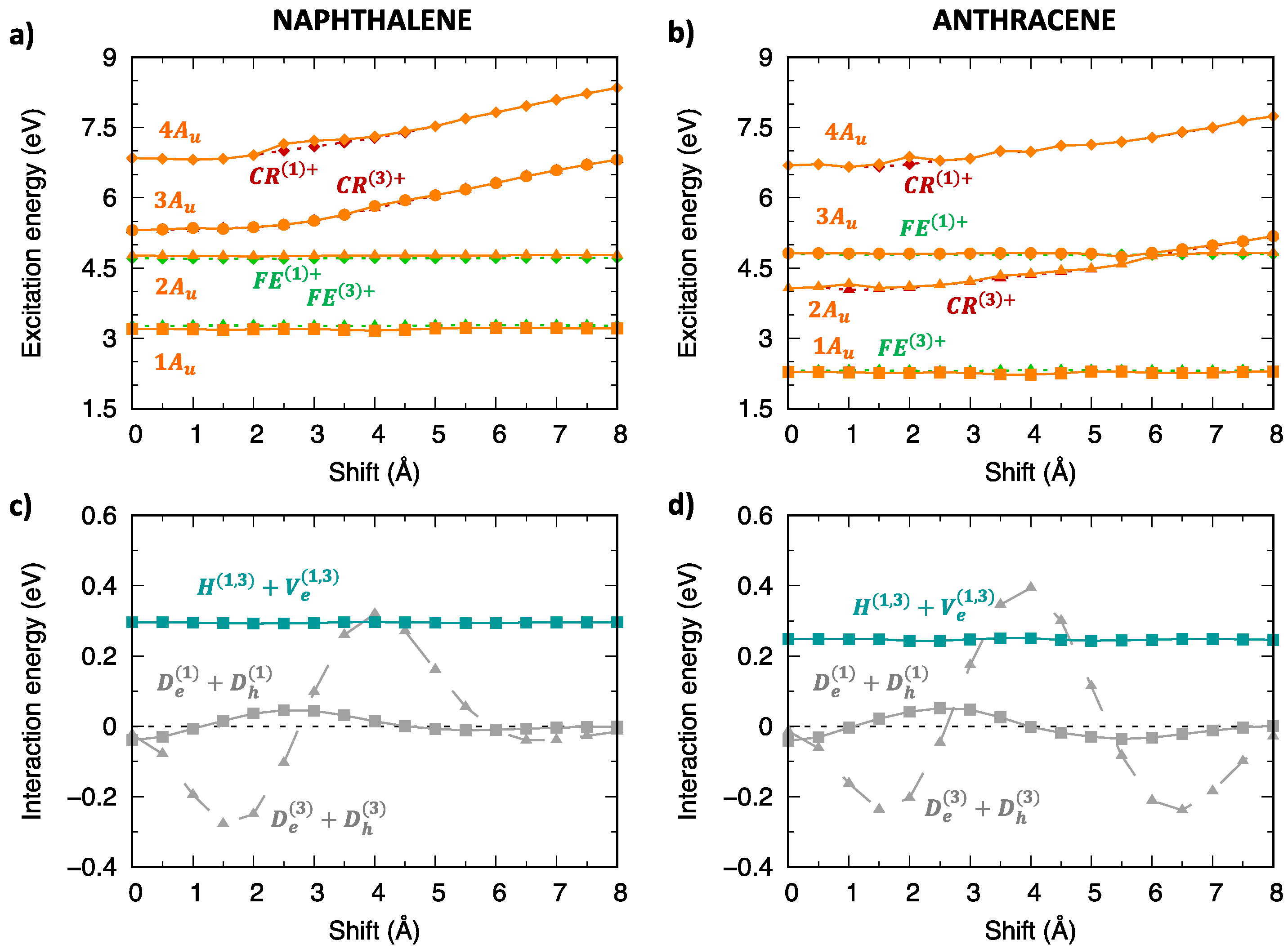
2.2. Singlet Exciton States of Naphthalene and Anthracene Dimers
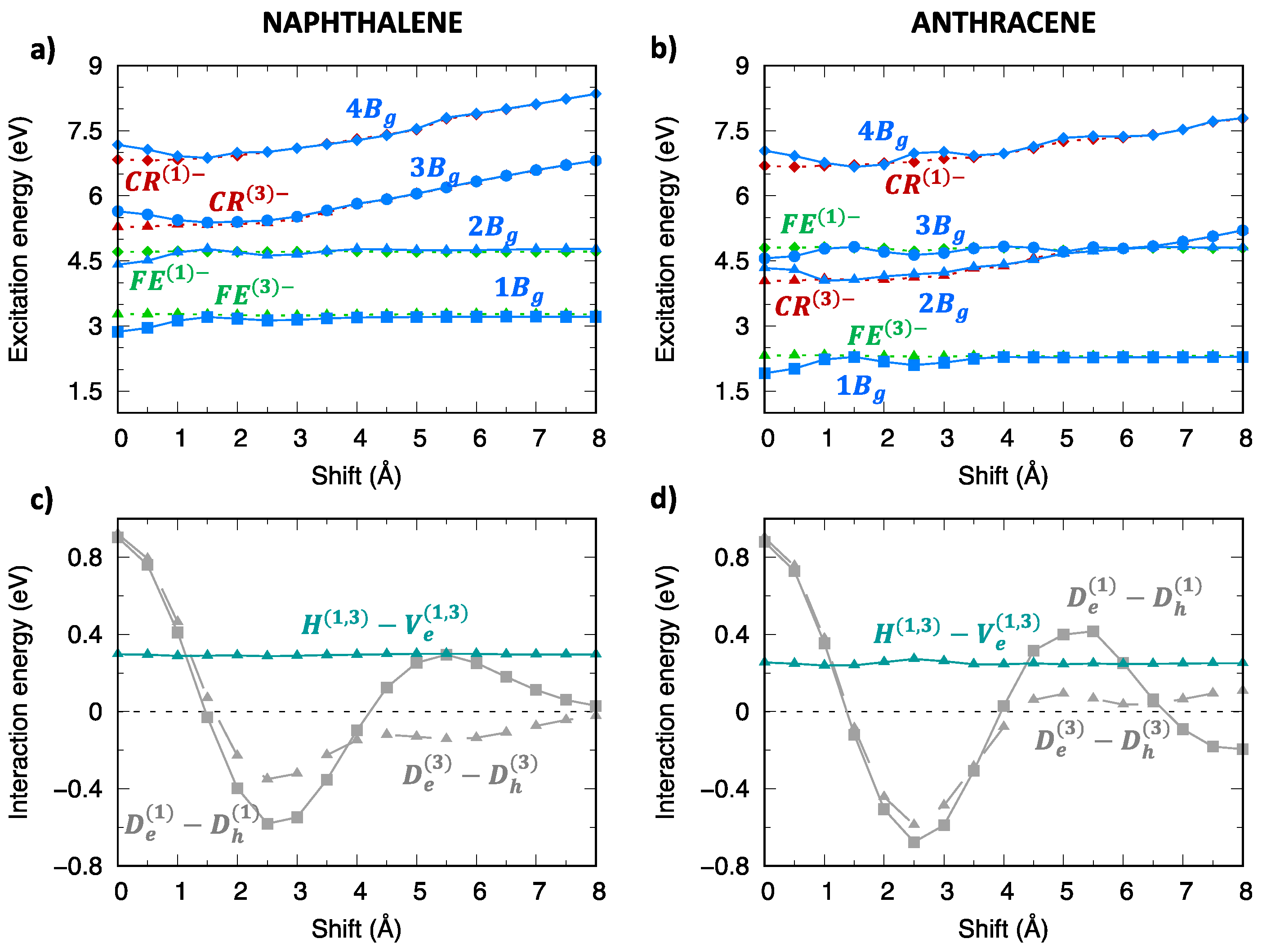
2.3. Triplet Exciton States of Naphthalene and Anthracene Dimers
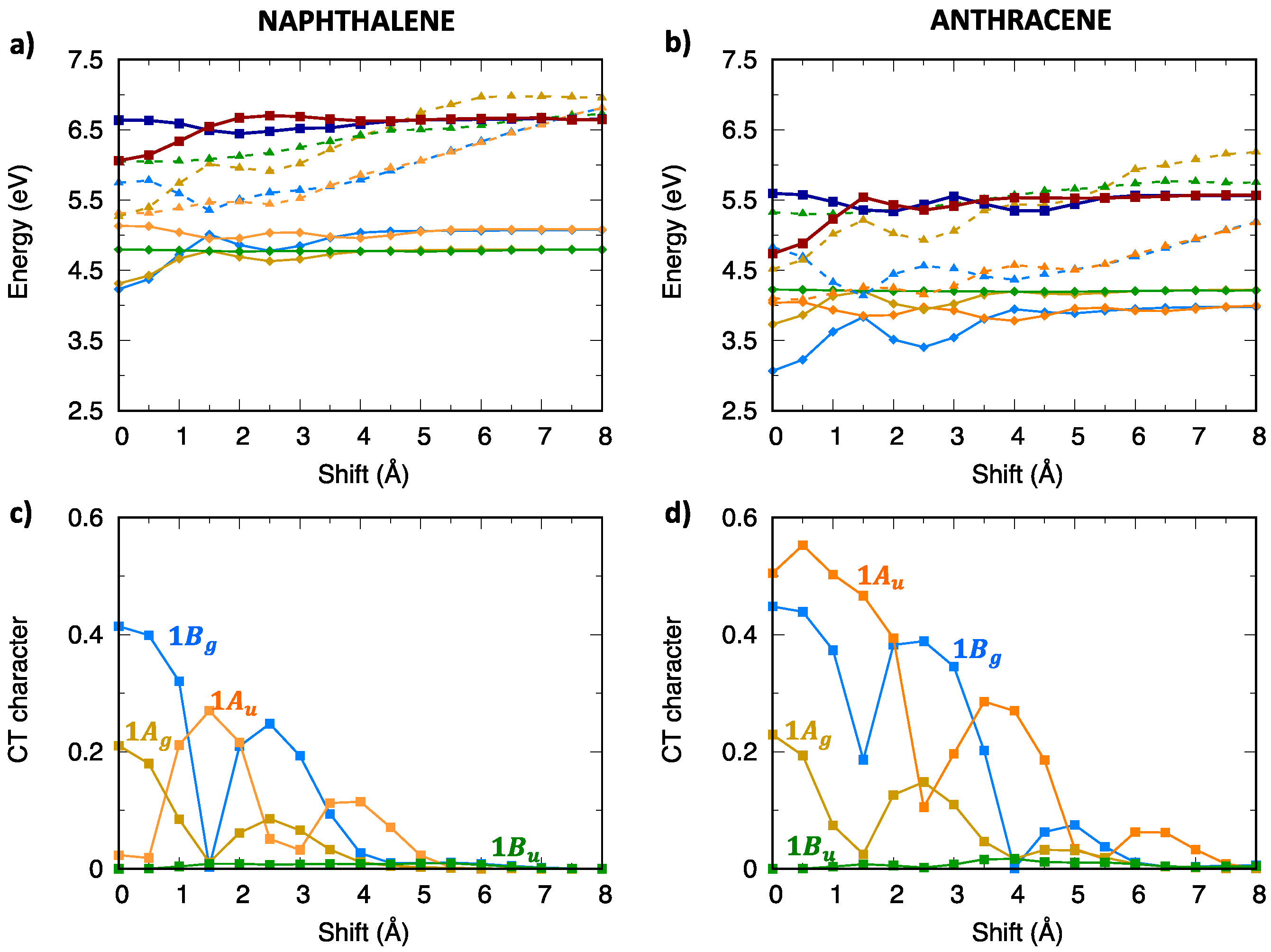
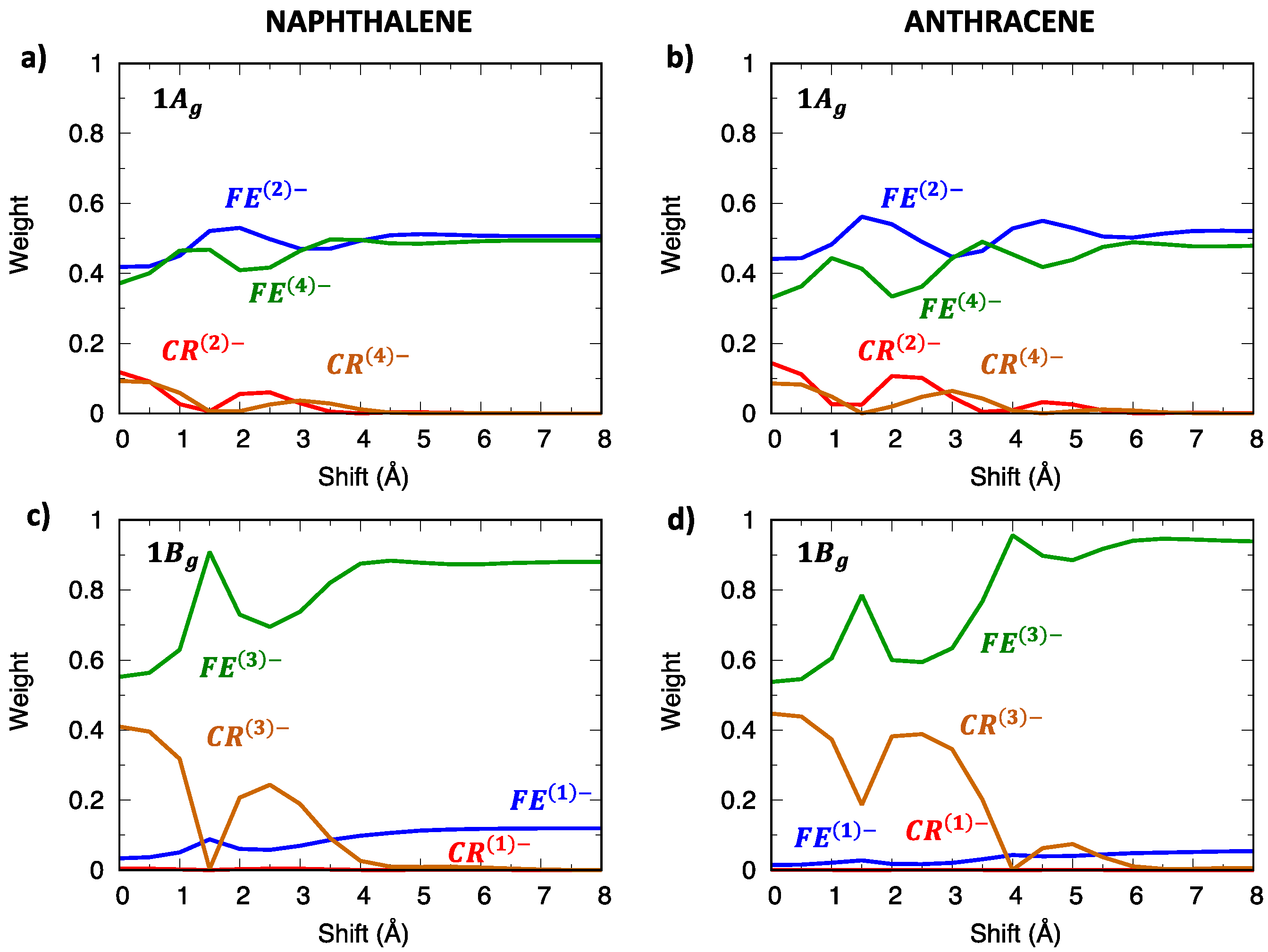
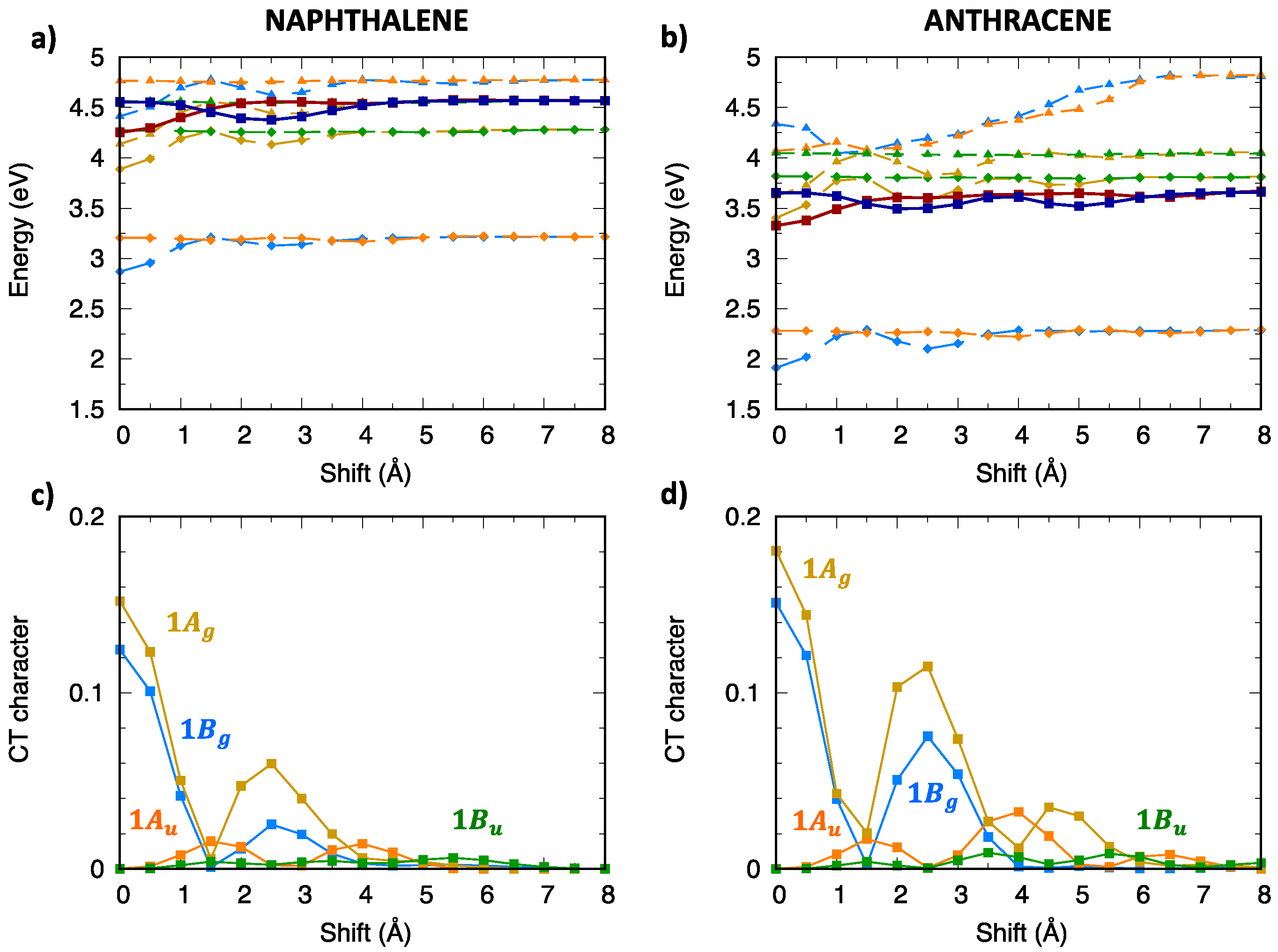
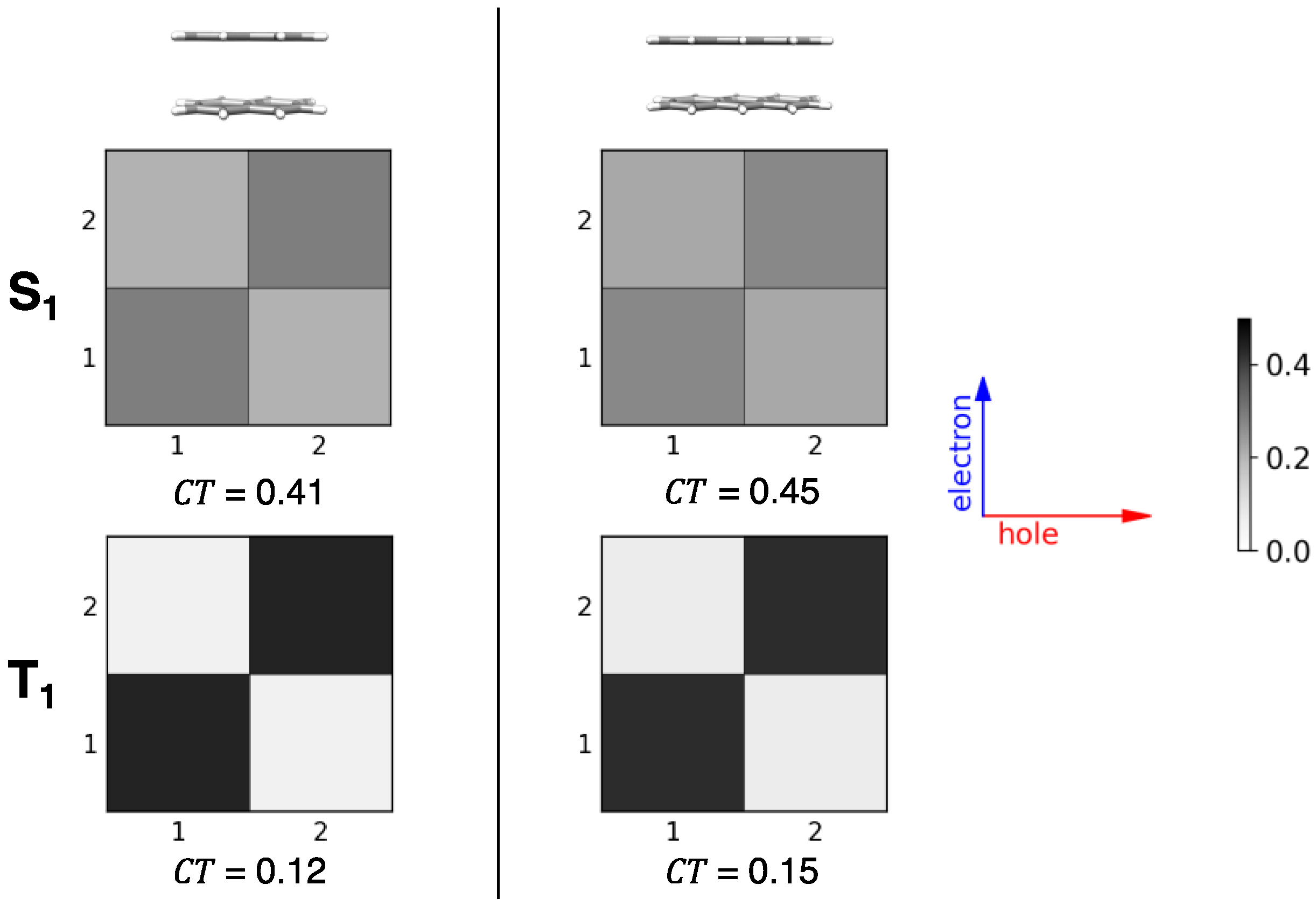
2.4. CT Character of Singlet and Triplet Excitons of Oligoacenes
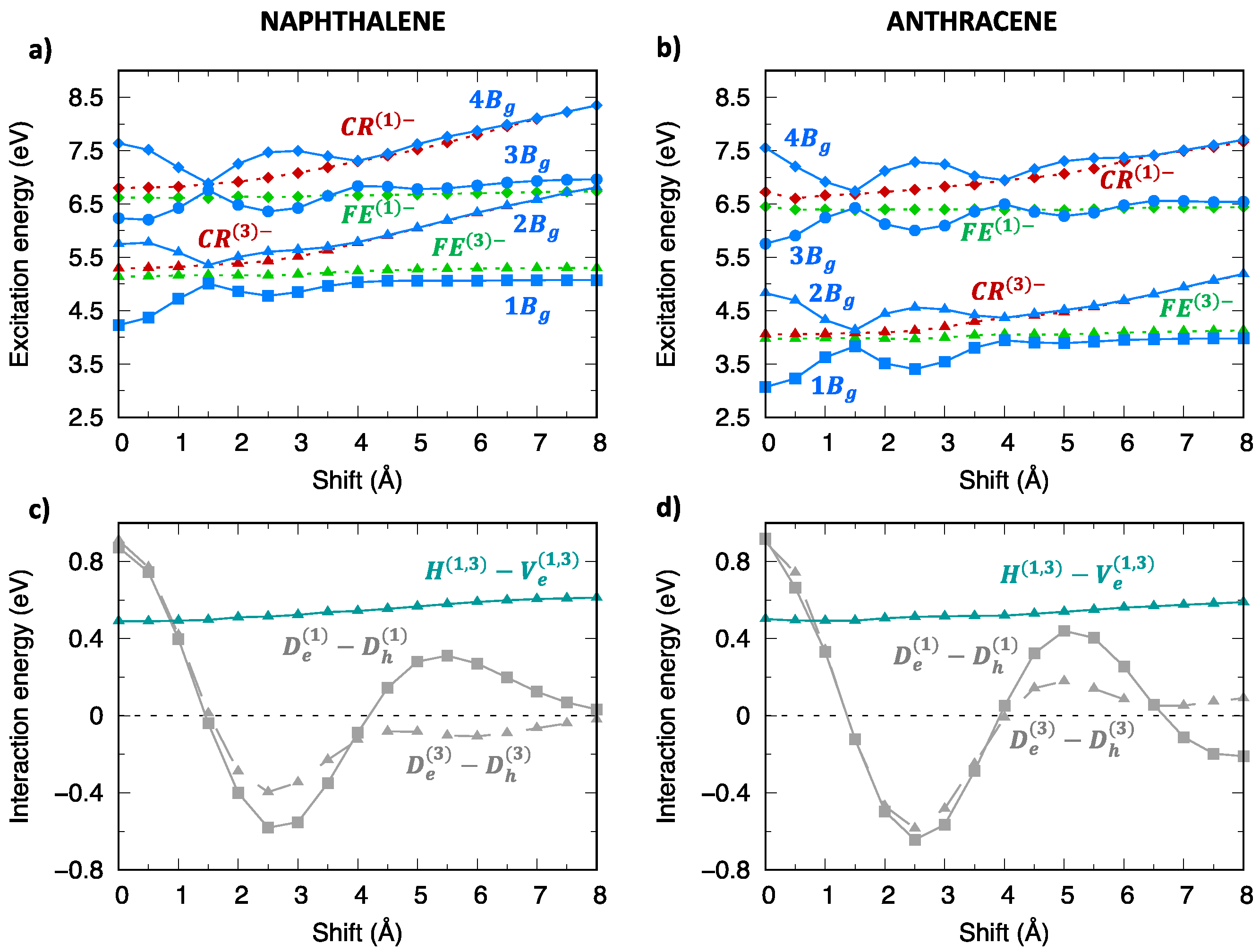
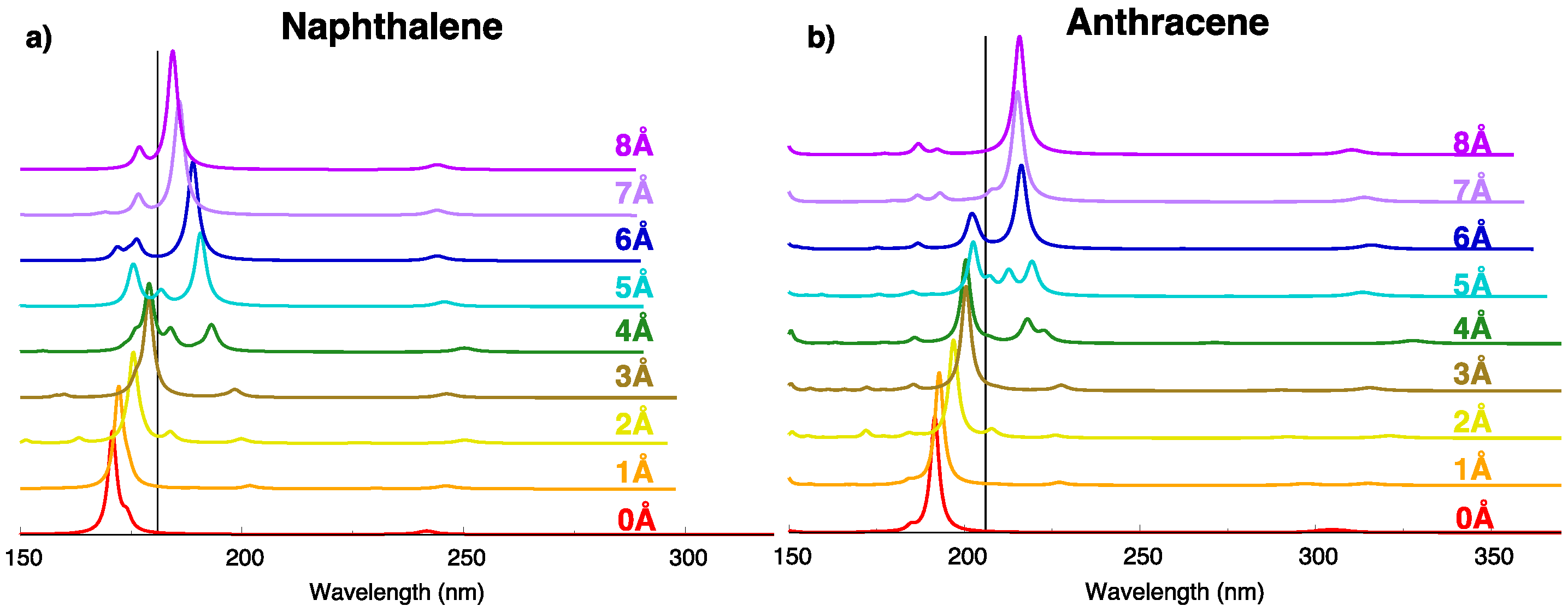
2.5. Absorption Spectrum and H-/J- Character Switch along the Longitudinal Translation Coordinate
3. Computational Models
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Ahmad, S. Organic semiconductors for device applications: Current trends and future prospects. J. Polym. Eng. 2014, 34, 279–338. [Google Scholar] [CrossRef]
- Shirota, Y.; Kageyama, H. Charge carrier transporting molecular materials and their applications in devices. Chem. Rev. 2007, 107, 953–1010. [Google Scholar] [CrossRef] [PubMed]
- Friend, R. Organic Materials for Large Area Electronics. Mater. Sci. Forum 2008, 608, 159–179. [Google Scholar] [CrossRef]
- Lee, N.E.; Zhou, J.J.; Agapito, L.A.; Bernardi, M. Charge transport in organic molecular semiconductors from first principles: The bandlike hole mobility in a naphthalene crystal. Phys. Rev. B 2018, 97, 115203. [Google Scholar] [CrossRef] [Green Version]
- Mohajeri, A.; Farmani, M. Toward More Efficient Organic Semiconductors: The Relationship between Morphology, Charge Transport, and Photophysical Properties. ACS Appl. Electron. Mater. 2022, 4, 246–258. [Google Scholar] [CrossRef]
- Ostroverkhova, O. Organic Optoelectronic Materials: Mechanisms and Applications. Chem. Rev. 2016, 116, 13279–13412. [Google Scholar] [CrossRef]
- Xu, Y.; Xu, P.; Hu, D.; Ma, Y. Recent progress in hot exciton materials for organic light-emitting diodes. Chem. Soc. Rev. 2021, 50, 1030–1069. [Google Scholar] [CrossRef]
- Yu, P.; Zhen, Y.; Dong, H.; Hu, W. Crystal Engineering of Organic Optoelectronic Materials. Chem 2019, 5, 2814–2853. [Google Scholar] [CrossRef]
- Würthner, F.; Saha-Möller, C.R.; Fimmel, B.; Ogi, S.; Leowanawat, P.; Schmidt, D. Perylene Bisimide Dye Assemblies as Archetype Functional Supramolecular Materials. Chem. Rev. 2016, 116, 962–1052. [Google Scholar] [CrossRef]
- Hecht, M.; Würthner, F. Supramolecularly Engineered J-Aggregates Based on Perylene Bisimide Dyes. Acc. Chem. Res. 2021, 54, 642–653. [Google Scholar] [CrossRef]
- Hoeben, F.J.M.; Jonkheijm, P.; Meijer, E.W.; Schenning, A.P.H.J. About supramolecular assemblies of pi-conjugated systems. Chem. Rev. 2005, 105, 1491–1546. [Google Scholar] [CrossRef] [PubMed]
- Su, F.; Chen, G.; Korevaar, P.A.; Pan, F.; Liu, H.; Guo, Z.; Schenning, A.P.H.J.; Zhang, H.; Lin, J.; Jiang, Y. Discrete π-Stacks from Self-Assembled Perylenediimide Analogues. Angew. Chem. Int. Ed. Engl. 2019, 58, 15273–15277. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Langer, P.; Davies, E.S.; Baldoni, M.; Wickham, K.; Besley, N.A.; Besley, E.; Champness, N.R. Synthesis and characterisation of rylene diimide dimers using molecular handcuffs. Chem. Sci. 2019, 10, 3723–3732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaufmann, C.; Bialas, D.; Stolte, M.; Würthner, F. Discrete π-Stacks of Perylene Bisimide Dyes within Folda-Dimers: Insight into Long- and Short-Range Exciton Coupling. J. Am. Chem. Soc. 2018, 140, 9986–9995. [Google Scholar] [CrossRef]
- Kaufmann, C.; Kim, W.; Nowak-Król, A.; Hong, Y.; Kim, D.; Würthner, F. Ultrafast Exciton Delocalization, Localization, and Excimer Formation Dynamics in a Highly Defined Perylene Bisimide Quadruple π-Stack. J. Am. Chem. Soc. 2018, 140, 4253–4258. [Google Scholar] [CrossRef] [PubMed]
- Samanta, S.; Chaudhuri, D. Suppressing Excimers in H-Aggregates of Perylene Bisimide Folda-Dimer: Role of Dimer Conformation and Competing Assembly Pathways. J. Phys. Chem. Lett. 2017, 8, 3427–3432. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.; Kim, J.; Kim, W.; Kaufmann, C.; Kim, H.; Würthner, F.; Kim, D. Efficient Multiexciton State Generation in Charge-Transfer-Coupled Perylene Bisimide Dimers via Structural Control. J. Am. Chem. Soc. 2020, 142, 7845–7857. [Google Scholar] [CrossRef]
- Haldar, R.; Mazel, A.; Krstić, M.; Zhang, Q.; Jakoby, M.; Howard, I.A.; Richards, B.S.; Jung, N.; Jacquemin, D.; Diring, S.; et al. A de novo strategy for predictive crystal engineering to tune excitonic coupling. Nat. Commun. 2019, 10, 2048. [Google Scholar] [CrossRef] [Green Version]
- Haldar, R.; Mazel, A.; Joseph, R.; Adams, M.; Howard, I.A.; Richards, B.S.; Tsotsalas, M.; Redel, E.; Diring, S.; Odobel, F.; et al. Excitonically Coupled States in Crystalline Coordination Networks. Chem. A Eur. J. 2017, 23, 14316–14322. [Google Scholar] [CrossRef]
- Spano, F.C. The Spectral Signatures of Frenkel Polarons in H- and J-Aggregates. Acc. Chem. Res. 2010, 43, 429–439. [Google Scholar] [CrossRef]
- Hestand, N.J.; Spano, F.C. Molecular Aggregate Photophysics beyond the Kasha Model: Novel Design Principles for Organic Materials. Acc. Chem. Res. 2017, 50, 341–350. [Google Scholar] [CrossRef] [PubMed]
- Hestand, N.J.; Spano, F.C. Expanded Theory of H- and J-Molecular Aggregates: The Effects of Vibronic Coupling and Intermolecular Charge Transfer. Chem. Rev. 2018, 118, 7069–7163. [Google Scholar] [CrossRef] [PubMed]
- Yamagata, H.; Norton, J.; Hontz, E.; Olivier, Y.; Beljonne, D.; Brédas, J.L.; Silbey, R.J.; Spano, F.C. The nature of singlet excitons in oligoacene molecular crystals. J. Chem. Phys. 2011, 134, 204703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hestand, N.J.; Yamagata, H.; Xu, B.; Sun, D.; Zhong, Y.; Harutyunyan, A.R.; Chen, G.; Dai, H.-L.; Rao, Y.; Spano, F.C. Polarized Absorption in Crystalline Pentacene: Theory vs Experiment. J. Phys. Chem. C 2015, 119, 22137–22147. [Google Scholar] [CrossRef]
- Kim, D. A Theoretical Analysis of the Excited State of Oligoacene Aggregates: Local Excitation vs. Charge-Transfer Transition. Bull. Korean Chem. Soc. 2015, 36, 2284–2289. [Google Scholar] [CrossRef]
- Shirai, S.; Iwata, S.; Tani, T.; Inagaki, S. Ab Initio Studies of Aromatic Excimers Using Multiconfiguration Quasi-Degenerate Perturbation Theory. J. Phys. Chem. A 2011, 115, 7687–7699. [Google Scholar] [CrossRef]
- Fliegl, H.; You, Z.; Hsu, C.; Sundholm, D. The Excitation Spectra of Naphthalene Dimers: Frenkel and Charge-Transfer Excitons. J. Chinese Chem. Soc. 2016, 63, 20–32. [Google Scholar] [CrossRef]
- Puschnig, P.; Ambrosch-Draxl, C. Excitons in organic semiconductors. Comptes Rendus Phys. 2009, 10, 504–513. [Google Scholar] [CrossRef]
- Liu, W.; Canola, S.; Köhn, A.; Engels, B.; Negri, F.; Fink, R.F. A model hamiltonian tuned toward high level ab initio calculations to describe the character of excitonic states in perylenebisimide aggregates. J. Comput. Chem. 2018, 39, 1979–1989. [Google Scholar] [CrossRef]
- Canola, S.; Bagnara, G.; Dai, Y.; Ricci, G.; Calzolari, A.; Negri, F. Addressing the Frenkel and charge transfer character of exciton states with a model Hamiltonian based on dimer calculations: Application to large aggregates of perylene bisimide. J. Chem. Phys. 2021, 154, 124101. [Google Scholar] [CrossRef]
- Liu, W.; Settels, V.; Harbach, P.H.P.; Dreuw, A.; Fink, R.F.; Engels, B. Assessment of TD-DFT- and TD-HF-based approaches for the prediction of exciton coupling parameters, potential energy curves, and electronic characters of electronically excited aggregates. J. Comput. Chem. 2011, 32, 1971–1981. [Google Scholar] [CrossRef] [PubMed]
- Stehr, V.; Engels, B.; Deibel, C.; Fink, R.F. Anisotropy of singlet exciton diffusion in organic semiconductor crystals from ab initio approaches. J. Chem. Phys. 2014, 140, 024503. [Google Scholar] [CrossRef] [PubMed]
- Engels, B.; Engel, V. The dimer-approach to characterize opto-electronic properties of and exciton trapping and diffusion in organic semiconductor aggregates and crystals. Phys. Chem. Chem. Phys. 2017, 19, 12604–12619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walter, C.; Krämer, V.; Engels, B. On the applicability of time-dependent density functional theory (TDDFT) and semiempirical methods to the computation of excited-state potential energy surfaces of perylene-based dye-aggregates. Int. J. Quantum Chem. 2017, 117, e25337. [Google Scholar] [CrossRef]
- Jortner, J.; Rice, S.A.; Katz, J.L.; Choi, S. Triplet Excitons in Crystals of Aromatic Molecules. J. Chem. Phys. 1965, 42, 309–323. [Google Scholar] [CrossRef]
- Zubiria-Ulacia, M.; Matxain, J.M.; Casanova, D. The role of CT excitations in PDI aggregates. Phys. Chem. Chem. Phys. 2020, 22, 15908–15918. [Google Scholar] [CrossRef]
- Diaz-Andres, A.; Casanova, D. Benzene Excimer and Excited Multimers: Electronic Character, Interaction Nature, and Aromaticity. J. Phys. Chem. Lett. 2021, 12, 7400–7408. [Google Scholar] [CrossRef]
- Dai, Y.; Zubiria-Ulacia, M.; Casanova, D.; Negri, F. Impact of Charge-Resonance Excitations on CT-Mediated J-Type Aggregation in Singlet and Triplet Exciton States of Perylene Di-Imide Aggregates: A TDDFT Investigation. Computation 2022, 10, 18. [Google Scholar] [CrossRef]
- Krishnan, A.; Diaz-Andres, A.; Sudhakaran, K.P.; John, A.T.; Hariharan, M.; Casanova, D. Deciphering the role of (anti)aromaticity in cofacial excimers of linear acenes. J. Phys. Org. Chem. 2022, 36, e4438. [Google Scholar] [CrossRef]
- Hartzler, D.A.; Slipchenko, L.V.; Savikhin, S. Triplet-Triplet Coupling in Chromophore Dimers: Theory and Experiment. J. Phys. Chem. A 2018, 122, 6713–6723. [Google Scholar] [CrossRef]
- You, Z.-Q.; Hsu, C.-P.; Fleming, G.R. Triplet-triplet energy-transfer coupling: Theory and calculation. J. Chem. Phys. 2006, 124, 044506. [Google Scholar] [CrossRef]
- East, A.L.L.; Lim, E.C. Naphthalene dimer: Electronic states, excimers, and triplet decay. J. Chem. Phys. 2000, 113, 8981–8994. [Google Scholar] [CrossRef] [Green Version]
- Pabst, M.; Lunkenheimer, B.; Köhn, A. The Triplet Excimer of Naphthalene: A Model System for Triplet−Triplet Interactions and Its Spectral Properties. J. Phys. Chem. C 2011, 115, 8335–8344. [Google Scholar] [CrossRef]
- Gillett, A.J.; Privitera, A.; Dilmurat, R.; Karki, A.; Qian, D.; Pershin, A.; Londi, G.; Myers, W.K.; Lee, J.; Yuan, J.; et al. The role of charge recombination to triplet excitons in organic solar cells. Nature 2021, 597, 666–671. [Google Scholar] [CrossRef]
- Cakmak, Y.; Kolemen, S.; Duman, S.; Dede, Y.; Dolen, Y.; Kilic, B.; Kostereli, Z.; Yildirim, L.T.; Dogan, A.L.; Guc, D.; et al. Designing excited states: Theory-guided access to efficient photosensitizers for photodynamic action. Angew. Chem. Int. Ed. 2011, 50, 11937–11941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bassan, E.; Gualandi, A.; Cozzi, P.G.; Ceroni, P. Design of BODIPY dyes as triplet photosensitizers: Electronic properties tailored for solar energy conversion, photoredox catalysis and photodynamic therapy. Chem. Sci. 2021, 12, 6607–6628. [Google Scholar] [CrossRef] [PubMed]
- Gibbons, D.J.; Farawar, A.; Mazzella, P.; Leroy-Lhez, S.; Williams, R.M. Making triplets from photo-generated charges: Observations, mechanisms and theory. Photochem. Photobiol. Sci. 2020, 19, 136–158. [Google Scholar] [CrossRef] [Green Version]
- Bardeen, C.J. The Structure and Dynamics of Molecular Excitons. Annu. Rev. Phys. Chem. 2014, 65, 127–148. [Google Scholar] [CrossRef] [PubMed]
- Mewes, S.A.; Dreuw, A. Density-based descriptors and exciton analyses for visualizing and understanding the electronic structure of excited states. Phys. Chem. Chem. Phys. 2019, 21, 2843–2856. [Google Scholar] [CrossRef]
- Mewes, S.A.; Plasser, F.; Krylov, A.; Dreuw, A. Benchmarking Excited-State Calculations Using Exciton Properties. J. Chem. Theory Comput. 2018, 14, 710–725. [Google Scholar] [CrossRef] [PubMed]
- Jurinovich, S.; Cupellini, L.; Guido, C.A.; Mennucci, B. EXAT: EXcitonic analysis tool. J. Comput. Chem. 2018, 39, 279–286. [Google Scholar] [CrossRef] [PubMed]
- Accomasso, D.; Persico, M.; Granucci, G. Diabatization by Localization in the Framework of Configuration Interaction Based on Floating Occupation Molecular Orbitals (FOMO−CI). ChemPhotoChem 2019, 3, 933–944. [Google Scholar] [CrossRef]
- Tamura, H. Diabatization for Time-Dependent Density Functional Theory: Exciton Transfers and Related Conical Intersections. J. Phys. Chem. A 2016, 120, 9341–9347. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Lunkenheimer, B.; Settels, V.; Engels, B.; Fink, R.F.; Köhn, A. A general ansatz for constructing quasi-diabatic states in electronically excited aggregated systems. J. Chem. Phys. 2015, 143, 084106. [Google Scholar] [CrossRef]
- Carreras, A.; Uranga-Barandiaran, O.; Castet, F.; Casanova, D. Photophysics of Molecular Aggregates from Excited State Diabatization. J. Chem. Theory Comput. 2019, 15, 2320–2330. [Google Scholar] [CrossRef]
- Darghouth, A.A.M.H.M.; Correa, G.C.; Juillard, S.; Casida, M.E.; Humeniuk, A.; Mitrić, R. Davydov-type excitonic effects on the absorption spectra of parallel-stacked and herringbone aggregates of pentacene: Time-dependent density-functional theory and time-dependent density-functional tight binding. J. Chem. Phys. 2018, 149, 134111. [Google Scholar] [CrossRef] [Green Version]
- Casanova, D. Theoretical investigations of the perylene electronic structure: Monomer, dimers, and excimers. Int. J. Quantum Chem. 2015, 115, 442–452. [Google Scholar] [CrossRef]
- Mao, Y.; Montoya-Castillo, A.; Markland, T.E. Accurate and efficient DFT-based diabatization for hole and electron transfer using absolutely localized molecular orbitals. J. Chem. Phys. 2019, 151, 164114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, Y.; Montoya-Castillo, A.; Markland, T.E. Excited state diabatization on the cheap using DFT: Photoinduced electron and hole transfer. J. Chem. Phys. 2020, 153, 244111. [Google Scholar] [CrossRef] [PubMed]
- Lv, M.; Lu, X.; Jiang, Y.; Sandoval-Salinas, M.E.; Casanova, D.; Sun, H.; Sun, Z.; Xu, J.; Yang, Y.; Chen, J. Near-Unity Triplet Generation Promoted via Spiro-Conjugation. Angew. Chem. Int. Ed. 2021, 61, e202113190. [Google Scholar] [CrossRef]
- Margulies, E.A.; Logsdon, J.L.; Miller, C.E.; Ma, L.; Simonoff, E.; Young, R.M.; Schatz, G.C.; Wasielewski, M.R. Direct Observation of a Charge-Transfer State Preceding High-Yield Singlet Fission in Terrylenediimide Thin Films. J. Am. Chem. Soc. 2017, 139, 663–671. [Google Scholar] [CrossRef] [PubMed]
- Margulies, E.A.; Miller, C.E.; Wu, Y.; Ma, L.; Schatz, G.C.; Young, R.M.; Wasielewski, M.R. Enabling singlet fission by controlling intramolecular charge transfer in π-stacked covalent terrylenediimide dimers. Nat. Chem. 2016, 8, 1120–1125. [Google Scholar] [CrossRef]
- Young, R.M.; Wasielewski, M.R. Mixed Electronic States in Molecular Dimers: Connecting Singlet Fission, Excimer Formation, and Symmetry-Breaking Charge Transfer. Acc. Chem. Res. 2020, 53, 1957–1968. [Google Scholar] [CrossRef] [PubMed]
- Beljonne, D.; Yamagata, H.; Brédas, J.L.; Spano, F.C.; Olivier, Y. Charge-Transfer Excitations Steer the Davydov Splitting and Mediate Singlet Exciton Fission in Pentacene. Phys. Rev. Lett. 2013, 110, 226402. [Google Scholar] [CrossRef] [Green Version]
- Berkelbach, T.C.; Hybertsen, M.S.; Reichman, D.R. Microscopic theory of singlet exciton fission. II. Application to pentacene dimers and the role of superexchange. J. Chem. Phys. 2013, 138, 114103. [Google Scholar] [CrossRef] [Green Version]
- Zeng, T.; Hoffmann, R.; Ananth, N. The Low-Lying Electronic States of Pentacene and Their Roles in Singlet Fission. J. Am. Chem. Soc. 2014, 136, 5755–5764. [Google Scholar] [CrossRef] [PubMed]
- Casanova, D. Theoretical Modeling of Singlet Fission. Chem. Rev. 2018, 118, 7164–7207. [Google Scholar] [CrossRef] [PubMed]
- Han, G.; Yi, Y. Local Excitation/Charge-Transfer Hybridization Simultaneously Promotes Charge Generation and Reduces Nonradiative Voltage Loss in Nonfullerene Organic Solar Cells. J. Phys. Chem. Lett. 2019, 10, 2911–2918. [Google Scholar] [CrossRef]
- Coropceanu, V.; Chen, X.-K.; Wang, T.; Zheng, Z.; Brédas, J.-L. Charge-transfer electronic states in organic solar cells. Nat. Rev. Mater. 2019, 4, 689–707. [Google Scholar] [CrossRef]
- Dong, Y.; Nikolis, V.C.; Talnack, F.; Chin, Y.-C.; Benduhn, J.; Londi, G.; Kublitski, J.; Zheng, X.; Mannsfeld, S.C.B.; Spoltore, D.; et al. Orientation dependent molecular electrostatics drives efficient charge generation in homojunction organic solar cells. Nat. Commun. 2020, 11, 4617. [Google Scholar] [CrossRef]
- Plasser, F. TheoDORE: A toolbox for a detailed and automated analysis of electronic excited state computations. J. Chem. Phys. 2020, 152, 084108. [Google Scholar] [CrossRef] [Green Version]
- Hirata, S.; Head-Gordon, M. Time-dependent density functional theory within the Tamm–Dancoff approximation. Chem. Phys. Lett. 1999, 314, 291–299. [Google Scholar] [CrossRef]
- Platt, J.R. Classification of Spectra of Cata-Condensed Hydrocarbons. J. Chem. Phys. 1949, 17, 484–495. [Google Scholar] [CrossRef]
- Negri, F.; Zgierski, M.Z. Vibronic structure of the emission spectra from single vibronic levels of the S1 manifold in naphthalene: Theoretical simulation. J. Chem. Phys. 1996, 104, 3486. [Google Scholar] [CrossRef]
- Knippenberg, S.; Starcke, J.H.; Wormit, M.; Dreuw, A. The low-lying excited states of neutral polyacenes and their radical cations: A quantum chemical study employing the algebraic diagrammatic construction scheme of second order. Mol. Phys. 2010, 108, 2801–2813. [Google Scholar] [CrossRef]
- Liang, J.; Feng, X.; Hait, D.; Head-Gordon, M. Revisiting the Performance of Time-Dependent Density Functional Theory for Electronic Excitations: Assessment of 43 Popular and Recently Developed Functionals from Rungs One to Four. J. Chem. Theory Comput. 2022, 18, 3460–3473. [Google Scholar] [CrossRef]
- Jacquemin, D.; Mennucci, B.; Adamo, C. Excited-state calculations with TD-DFT: From benchmarks to simulations in complex environments. Phys. Chem. Chem. Phys. 2011, 13, 16987. [Google Scholar] [CrossRef]
- Yamagata, H.; Pochas, C.M.; Spano, F.C. Designing J- and H-Aggregates through Wave Function Overlap Engineering: Applications to Poly(3-hexylthiophene). J. Phys. Chem. B 2012, 116, 14494–14503. [Google Scholar] [CrossRef]
- Plasser, F.; Lischka, H. Analysis of excitonic and charge transfer interactions from quantum chemical calculations. J. Chem. Theory Comput. 2012, 8, 2777–2789. [Google Scholar] [CrossRef]
- Casanova, D.; Krylov, A.I. Quantifying local exciton, charge resonance, and multiexciton character in correlated wave functions of multichromophoric systems. J. Chem. Phys. 2016, 144, 014102. [Google Scholar] [CrossRef]
- Bao, P.; Hettich, C.P.; Shi, Q.; Gao, J. Block-Localized Excitation for Excimer Complex and Diabatic Coupling. J. Chem. Theory Comput. 2021, 17, 240–254. [Google Scholar] [CrossRef]
- Shirai, S.; Kurashige, Y.; Yanai, T. Computational Evidence of Inversion of 1La and 1Lb-Derived Excited States in Naphthalene Excimer Formation from ab Initio Multireference Theory with Large Active Space: DMRG-CASPT2 Study. J. Chem. Theory Comput. 2016, 12, 2366–2372. [Google Scholar] [CrossRef]
- Chai, J.-D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615. [Google Scholar] [CrossRef] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Norton, J.E.; Brédas, J.-L. Theoretical characterization of titanyl phthalocyanine as a p-type organic semiconductor: Short intermolecular π-π interactions yield large electronic couplings and hole transport bandwidths. J. Chem. Phys. 2008, 128, 034701. [Google Scholar] [CrossRef] [Green Version]
- Löwdin, P. On the Non-Orthogonality Problem Connected with the Use of Atomic Wave Functions in the Theory of Molecules and Crystals. J. Chem. Phys. 1950, 18, 365–375. [Google Scholar] [CrossRef]
- Nottoli, M.; Jurinovich, S.; Cupellini, L.; Gardiner, A.T.; Cogdell, R.; Mennucci, B. The role of charge-transfer states in the spectral tuning of antenna complexes of purple bacteria. Photosynth. Res. 2018, 137, 215–226. [Google Scholar] [CrossRef]
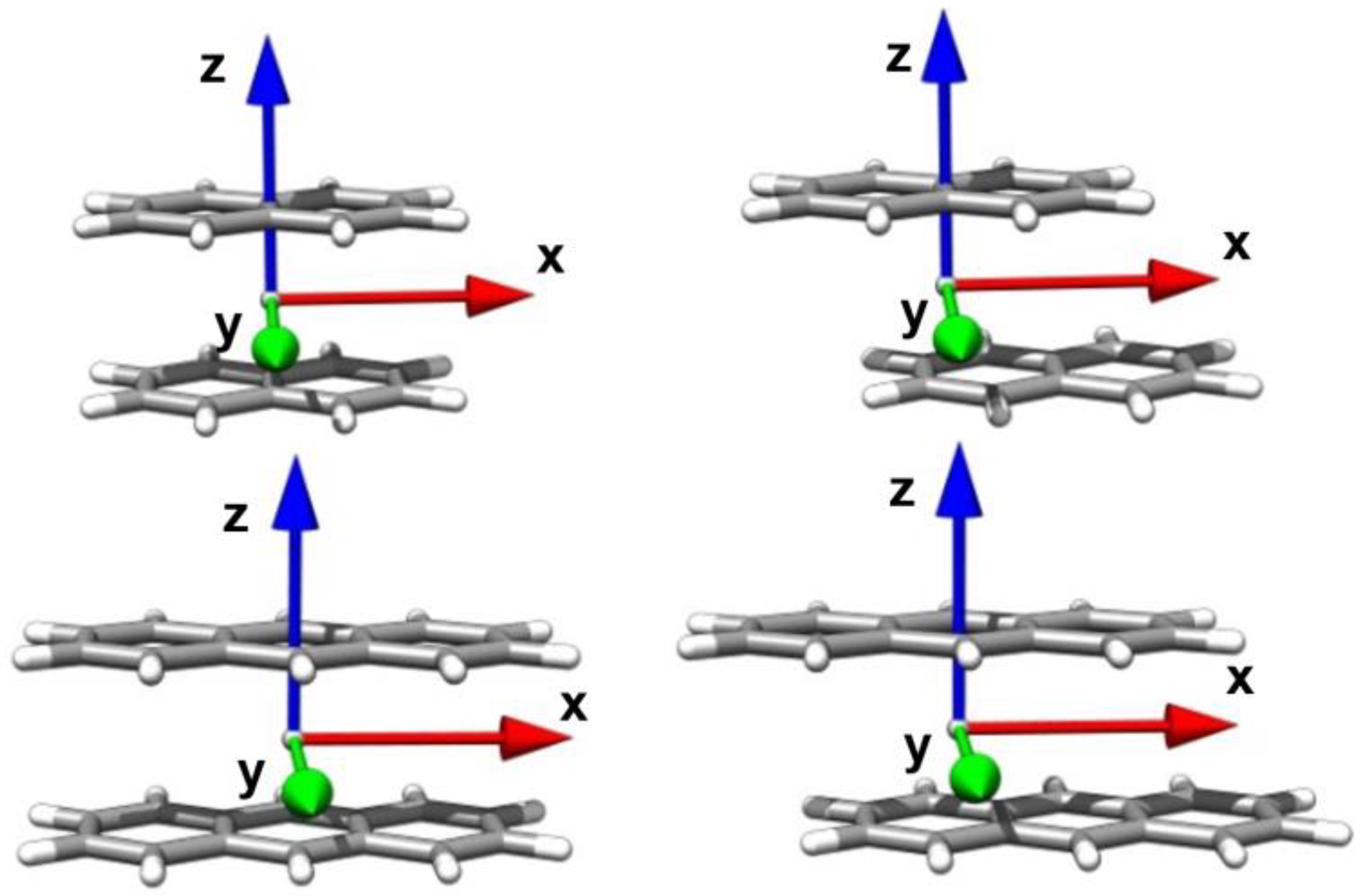



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dai, Y.; Calzolari, A.; Zubiria-Ulacia, M.; Casanova, D.; Negri, F. Intermolecular Interactions and Charge Resonance Contributions to Triplet and Singlet Exciton States of Oligoacene Aggregates. Molecules 2023, 28, 119. https://doi.org/10.3390/molecules28010119
Dai Y, Calzolari A, Zubiria-Ulacia M, Casanova D, Negri F. Intermolecular Interactions and Charge Resonance Contributions to Triplet and Singlet Exciton States of Oligoacene Aggregates. Molecules. 2023; 28(1):119. https://doi.org/10.3390/molecules28010119
Chicago/Turabian StyleDai, Yasi, Alessandro Calzolari, Maria Zubiria-Ulacia, David Casanova, and Fabrizia Negri. 2023. "Intermolecular Interactions and Charge Resonance Contributions to Triplet and Singlet Exciton States of Oligoacene Aggregates" Molecules 28, no. 1: 119. https://doi.org/10.3390/molecules28010119