
Synthesis, Cytotoxic Evaluation, and Structure-Activity Relationship of Substituted Quinazolinones as Cyclin-Dependent Kinase 9 Inhibitors
 , , , , , , , ,
, , , , , , , , 
Abstract
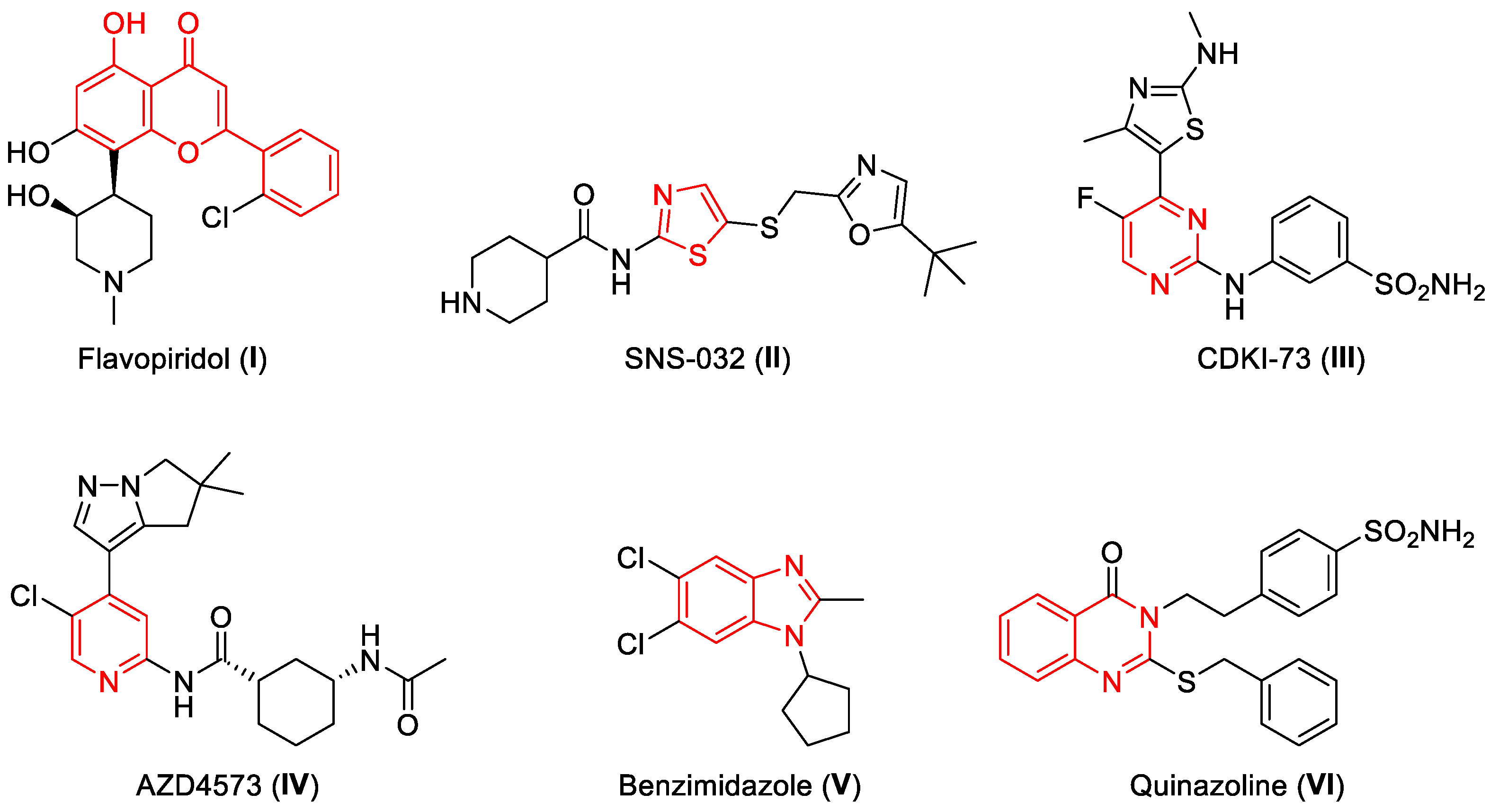
1. Introduction
2. Results and Discussion
2.1. Chemistry
2.2. Structure-Activity Relationship (SAR) Analysis
2.3. Molecular Docking
2.4. Analyses of the Physicochemical Properties of the Compounds
2.4.1. Lipinski’s Rule of Five
2.4.2. Ligand Efficiency (LE)
2.4.3. Ligand Lipophilic Efficiency (LLE)
3. Materials and Methods
3.1. Chemistry
3.2. Metabolic Assay
3.3. CDK9 Kinase Assay
3.4. Molecular Docking
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sánchez-Martínez, C.; Lallena, M.J.; Sanfeliciano, S.G.; de Dios, A. Cyclin dependent kinase (CDK) inhibitors as anticancer drugs: Recent advances (2015–2019). Bioorg. Med. Chem. Lett. 2019, 29, 126637. [Google Scholar] [CrossRef]
- Diab, S.; Yu, M.; Wang, S. CDK7 Inhibitors in Cancer Therapy: The Sweet Smell of Success? J. Med. Chem. 2020, 63, 7458–7474. [Google Scholar] [CrossRef] [PubMed]
- Cheng, W.; Yang, Z.; Wang, S.; Li, Y.; Wei, H.; Tian, X.; Kan, Q. Recent development of CDK inhibitors: An overview of CDK/inhibitor co-crystal structures. Eur. J. Med. Chem. 2019, 164, 615–639. [Google Scholar] [CrossRef]
- Tutone, M.; Almerico, A.M. Recent advances on CDK inhibitors: An insight by means of in silico methods. Eur. J. Med. Chem. 2017, 142, 300–315. [Google Scholar] [CrossRef] [PubMed]
- Asghar, U.; Witkiewicz, A.K.; Turner, N.C.; Knudsen, E.S. The history and future of targeting cyclin-dependent kinases in cancer therapy. Nat. Rev. Drug Discov. 2015, 14, 130–146. [Google Scholar] [CrossRef] [PubMed]
- Malumbres, M. Cyclin-dependent kinases. Genome Biol. 2014, 15, 122. [Google Scholar] [CrossRef]
- Sánchez-Martínez, C.; Gelbert, L.M.; Lallena, M.J.; de Dios, A. Cyclin dependent kinase (CDK) inhibitors as anticancer drugs. Bioorg. Med. Chem. Lett. 2015, 25, 3420–3435. [Google Scholar] [CrossRef]
- Krystof, V.; Baumli, S.; Furst, R. Perspective of Cyclin-dependent kinase 9 (CDK9) as a Drug Target. Curr. Pharm. Des. 2012, 18, 2883–2890. [Google Scholar] [CrossRef]
- Wang, S.; Fischer, P.M. Cyclin-dependent kinase 9: A key transcriptional regulator and potential drug target in oncology, virology and cardiology. Trends Pharmacol. Sci. 2008, 29, 302–313. [Google Scholar] [CrossRef]
- Lukasik, P.M.; Elabar, S.; Lam, F.; Shao, H.; Liu, X.R.; Abbas, A.Y.; Wang, S.D. Synthesis and biological evaluation of imidazo[4,5-b]pyridine and 4-heteroaryl-pyrimidine derivatives as anti-cancer agents. Eur. J. Med. Chem. 2012, 57, 311–322. [Google Scholar] [CrossRef]
- Alkahtani, H.M.; Abbas, A.Y.; Wang, S. Synthesis and biological evaluation of benzo[d]imidazole derivatives as potential anti-cancer agents. Bioorg. Med. Chem. Lett. 2012, 22, 1317–1321. [Google Scholar] [CrossRef]
- Shao, H.; Shi, S.H.; Foley, D.W.; Lam, F.; Abbas, A.Y.; Liu, X.R.; Huang, S.L.; Jiang, X.R.; Baharin, N.; Fischer, P.M.; et al. Synthesis, structure–activity relationship and biological evaluation of 2,4,5-trisubstituted pyrimidine CDK inhibitors as potential anti-tumour agents. Eur. J. Med. Chem. 2013, 70, 447–455. [Google Scholar] [CrossRef] [PubMed]
- Shao, H.; Shi, S.; Huang, S.; Hole, A.J.; Abbas, A.Y.; Baumli, S.; Liu, X.; Lam, F.; Foley, D.W.; Fischer, P.M.; et al. Substituted 4-(Thiazol-5-yl)-2-(phenylamino)pyrimidines Are Highly Active CDK9 Inhibitors: Synthesis, X-ray Crystal Structures, Structure–Activity Relationship, and Anticancer Activities. J. Med. Chem. 2013, 56, 640–659. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Wierda, W.G.; Chubb, S.; Hawtin, R.E.; Fox, J.A.; Keating, M.J.; Gandhi, V.; Plunkett, W. Mechanism of action of SNS-032, a novel cyclin-dependent kinase inhibitor, in chronic lymphocytic leukemia. Blood 2009, 113, 4637–4645. [Google Scholar] [CrossRef] [PubMed]
- Walsby, E.; Pratt, G.; Shao, H.; Abbas, A.Y.; Fischer, P.M.; Bradshaw, T.D.; Brennan, P.; Fegan, C.; Wang, S.; Pepper, C. A novel Cdk9 inhibitor preferentially targets tumor cells and synergizes with fludarabine. Oncotarget 2014, 5, 375–385. [Google Scholar] [CrossRef]
- Wu, T.; Qin, Z.; Tian, Y.; Wang, J.; Xu, C.; Li, Z.; Bian, J. Recent Developments in the Biology and Medicinal Chemistry of CDK9 Inhibitors: An Update. J. Med. Chem. 2020, 63, 13228–13257. [Google Scholar] [CrossRef]
- Kryštof, V.; Chamrád, I.; Jorda, R.; Kohoutek, J. Pharmacological targeting of CDK9 in cardiac hypertrophy. Med. Res. Rev. 2010, 30, 646–666. [Google Scholar] [CrossRef]
- Richter, A.; Schoenwaelder, N.; Sender, S.; Junghanss, C.; Maletzki, C. Cyclin-Dependent Kinase Inhibitors in Hematological Malignancies—Current Understanding, (Pre-)Clinical Application and Promising Approaches. Cancers 2021, 13, 2497. [Google Scholar] [CrossRef]
- Mandal, R.; Becker, S.; Strebhardt, K. Targeting CDK9 for Anti-Cancer Therapeutics. Cancers 2021, 13, 2181. [Google Scholar] [CrossRef]
- Mitra, P.; Yang, R.-M.; Sutton, J.; Ramsay, R.G.; Gonda, T.J. CDK9 inhibitors selectively target estrogen receptor-positive breast cancer cells through combined inhibition of MYB and MCL-1 expression. Oncotarget 2016, 7, 9069–9083. [Google Scholar] [CrossRef]
- Cheng, S.-S.; Qu, Y.-Q.; Wu, J.; Yang, G.-J.; Liu, H.; Wang, W.; Huang, Q.; Chen, F.; Li, G.; Wong, C.-Y.; et al. Inhibition of the CDK9–cyclin T1 protein–protein interaction as a new approach against triple-negative breast cancer. Acta Pharm. Sin. B 2022, 12, 1390–1405. [Google Scholar] [CrossRef] [PubMed]
- Deep, A.; Marwaha, R.K.; Marwaha, M.G.; Jyoti, J.; Nandal, R.; Sharma, A.K. Flavopiridol as cyclin dependent kinase (CDK) inhibitor: A review. New J. Chem. 2018, 42, 18500–18507. [Google Scholar] [CrossRef]
- Hameed, A.; Al-Rashida, M.; Uroos, M.; Ali, S.A.; Arshia; Ishtiaq, M.; Khan, K.M. Quinazoline and quinazolinone as important medicinal scaffolds: A comparative patent review (2011–2016). Expert Opin. Ther. Pat. 2018, 28, 281–297. [Google Scholar] [CrossRef] [PubMed]
- Alagarsamy, V.; Chitra, K.; Saravanan, G.; Solomon, V.R.; Sulthana, M.T.; Narendhar, B. An overview of quinazolines: Pharmacological significance and recent developments. Eur. J. Med. Chem. 2018, 151, 628–685. [Google Scholar] [CrossRef] [PubMed]
- Khan, I.; Zaib, S.; Batool, S.; Abbas, N.; Ashraf, Z.; Iqbal, J.; Saeed, A. Quinazolines and quinazolinones as ubiquitous structural fragments in medicinal chemistry: An update on the development of synthetic methods and pharmacological diversification. Bioorg. Med. Chem. 2016, 24, 2361–2381. [Google Scholar] [CrossRef]
- Alkahtani, H.M.; Abdalla, A.N.; Obaidullah, A.J.; Alanazi, M.M.; Almehizia, A.A.; Alanazi, M.G.; Ahmed, A.Y.; Alwassil, O.I.; Darwish, H.W.; Abdel-Aziz, A.A.-M.; et al. Synthesis, cytotoxic evaluation, and molecular docking studies of novel quinazoline derivatives with benzenesulfonamide and anilide tails: Dual inhibitors of EGFR/HER2. Bioorg. Chem. 2020, 95, 103461. [Google Scholar] [CrossRef]
- El-Azab, A.S.; Abdel-Aziz, A.A.-M.; Bua, S.; Nocentini, A.; El-Gendy, M.A.; Mohamed, M.A.; Shawer, T.Z.; AlSaif, N.A.; Supuran, C.T. Synthesis of benzensulfonamides linked to quinazoline scaffolds as novel carbonic anhydrase inhibitors. Bioorg. Chem. 2019, 87, 78–90. [Google Scholar] [CrossRef]
- Abdel-Aziz, A.A.-M.; Abou-Zeid, L.A.; ElTahir, K.E.H.; Mohamed, M.A.; Abu El-Enin, M.A.; El-Azab, A.S. Design, synthesis of 2,3-disubstitued 4(3H)-quinazolinone derivatives as anti-inflammatory and analgesic agents: COX-1/2 inhibitory activities and molecular docking studies. Bioorg. Med. Chem. 2016, 24, 3818–3828. [Google Scholar] [CrossRef]
- Abdel-Aziz, A.A.-M.; Abou-Zeid, L.A.; ElTahir, K.E.H.; Ayyad, R.R.; El-Sayed, M.A.-A.; El-Azab, A.S. Synthesis, anti-inflammatory, analgesic, COX-1/2 inhibitory activities and molecular docking studies of substituted 2-mercapto-4(3H)-quinazolinones. Eur. J. Med. Chem. 2016, 121, 410–421. [Google Scholar] [CrossRef]
- Alanazi, A.M.; Abdel-Aziz, A.A.-M.; Shawer, T.Z.; Ayyad, R.R.; Al-Obaid, A.M.; Al-Agamy, M.H.M.; Maarouf, A.R.; El-Azab, A.S. Synthesis, antitumor and antimicrobial activity of some new 6-methyl-3-phenyl-4(3H)-quinazolinone analogues: In silico studies. J. Enzym. Inhib. Med. Chem. 2016, 31, 721–735. [Google Scholar] [CrossRef]
- El-Azab, A.S.; Abdel-Hamide, S.G.; Sayed-Ahmed, M.M.; Hassan, G.S.; El-Hadiyah, T.M.; Al-Shabanah, O.A.; Al-Deeb, O.A.; El-Subbagh, H.I. Novel 4(3H)-quinazolinone analogs: Synthesis and anticonvulsant activity. Med. Chem. Res. 2013, 22, 2815–2827. [Google Scholar] [CrossRef]
- El-Azab, A.S.; ElTahir, K.E.H. Synthesis and anticonvulsant evaluation of some new 2,3,8-trisubstituted-4(3H)-quinazoline derivatives. Bioorg. Med. Chem. Lett. 2012, 22, 327–333. [Google Scholar] [CrossRef] [PubMed]
- El-Azab, A.S.; ElTahir, K.E. Design and synthesis of novel 7-aminoquinazoline derivatives: Antitumor and anticonvulsant activities. Bioorg. Med. Chem. Lett. 2012, 22, 1879–1885. [Google Scholar] [CrossRef]
- El-Azab, A.S.; ElTahir, K.E.H.; Attia, S.M. Synthesis and anticonvulsant evaluation of some novel 4(3H)-quinazolinones. Monatsh. Chem. 2011, 142, 837–848. [Google Scholar] [CrossRef]
- El-Azab, A.S.; Al-Omar, M.A.; Abdel-Aziz, A.A.-M.; Abdel-Aziz, N.I.; El-Sayed, M.A.-A.; Aleisa, A.M.; Sayed-Ahmed, M.M.; Abdel-Hamide, S.G. Design, synthesis and biological evaluation of novel quinazoline derivatives as potential antitumor agents: Molecular docking study. Eur. J. Med. Chem. 2010, 45, 4188–4198. [Google Scholar] [CrossRef]
- El-Azab, A.S. Synthesis of Some New Substituted 2-Mercaptoquinazoline Analogs as Potential Antimicrobial Agents. Phosphorus Sulfur Silicon Relat. Elem. 2007, 182, 333–348. [Google Scholar] [CrossRef]
- Aziza, M.A.; Nassar, M.W.; Abdel Hamide, S.G.; El Hakim, A.E.; El-Azab, A.S. Synthesis and antimicrobial activities of some new 3-heteroaryl-quinazolin-4-ones. Indian J. Heterocy. Ch. 1996, 6, 25–30. [Google Scholar]
- Alanazi, A.M.; Al-Suwaidan, I.A.; Abdel-Aziz, A.A.-M.; Mohamed, M.A.; El Morsy, A.M.; El-Azab, A.S. Design, synthesis and biological evaluation of some novel substituted 2-mercapto-3-phenethylquinazolines as antitumor agents. Med. Chem. Res. 2013, 22, 5566–5577. [Google Scholar] [CrossRef]
- Al-Suwaidan, I.A.; Abdel-Aziz, A.A.-M.; Shawer, T.Z.; Ayyad, R.R.; Alanazi, A.M.; El-Morsy, A.M.; Mohamed, M.A.; Abdel-Aziz, N.I.; El-Sayed, M.A.-A.; El-Azab, A.S. Synthesis, antitumor activity and molecular docking study of some novel 3-benzyl-4(3H)quinazolinone analogues. J. Enzym. Inhib. Med. Chem. 2016, 31, 78–89. [Google Scholar] [CrossRef]
- El-Azab, A.S.; Al-Dhfyan, A.; Abdel-Aziz, A.A.-M.; Abou-Zeid, L.A.; Alkahtani, H.M.; Al-Obaid, A.M.; Al-Gendy, M.A. Synthesis, anticancer and apoptosis-inducing activities of quinazoline–isatin conjugates: Epidermal growth factor receptor-tyrosine kinase assay and molecular docking studies. J. Enzym. Inhib. Med. Chem. 2017, 32, 935–944. [Google Scholar] [CrossRef]
- El-Azab, A.S.; Abdel-Aziz, A.A.-M.; Ghabbour, H.A.; Al-Gendy, M.A. Synthesis, in vitro antitumour activity, and molecular docking study of novel 2-substituted mercapto-3-(3,4,5-trimethoxybenzyl)-4(3H)-quinazolinone analogues. J. Enzym. Inhib. Med. Chem. 2017, 32, 1229–1239. [Google Scholar] [CrossRef] [PubMed]
- El-Azab, A.S.; Abdel-Aziz, A.A.-M.; Ghabbour, H.A.; Bua, S.; Nocentini, A.; Alkahtani, H.M.; Alsaif, N.A.; Al-Agamy, M.H.M.; Supuran, C.T. Carbonic Anhydrase Inhibition Activities of Schiff’s Bases Based on Quinazoline-Linked Benzenesulfonamide. Molecules 2022, 27, 7703. [Google Scholar] [CrossRef] [PubMed]
- El-Azab, A.S.; Abdel-Aziz, A.A.-M.; AlSaif, N.A.; Alkahtani, H.M.; Alanazi, M.M.; Obaidullah, A.J.; Eskandrani, R.O.; Alharbi, A. Antitumor activity, multitarget mechanisms, and molecular docking studies of quinazoline derivatives based on a benzenesulfonamide scaffold: Cell cycle analysis. Bioorg. Chem. 2020, 104, 104345. [Google Scholar] [CrossRef] [PubMed]
- Sancineto, L.; Iraci, N.; Massari, S.; Attanasio, V.; Corazza, G.; Barreca, M.L.; Sabatini, S.; Manfroni, G.; Avanzi, N.R.; Cecchetti, V.; et al. Computer-Aided Design, Synthesis and Validation of 2-Phenylquinazolinone Fragments as CDK9 Inhibitors with Anti-HIV-1 Tat-Mediated Transcription Activity. ChemMedChem 2013, 8, 1941–1953. [Google Scholar] [CrossRef]
- Abdalla, A.N.; Abdallah, M.E.; Aslam, A.; Bader, A.; Vassallo, A.; Tommasi, N.; Malki, W.H.; Gouda, A.M.; Mukhtar, M.H.; El-Readi, M.Z.; et al. Synergistic Anti Leukemia Effect of a Novel Hsp90 and a Pan Cyclin Dependent Kinase Inhibitors. Molecules 2020, 9, 25. [Google Scholar] [CrossRef]
- Bozdag, M.; Alafeefy, A.M.; Carta, F.; Ceruso, M.; Al-Tamimi, A.-M.S.; Al-Kahtani, A.A.; Alasmary, F.A.S.; Supuran, C.T. Synthesis 4-[2-(2-mercapto-4-oxo-4H-quinazolin-3-yl)-ethyl]-benzenesulfonamides with subnanomolar carbonic anhydrase II and XII inhibitory properties. Bioorg. Med. Chem. 2016, 24, 4100–4107. [Google Scholar] [CrossRef]
- Kulkarni, S.S.; Singh, S.; Shah, J.R.; Low, W.-K.; Talele, T.T. Synthesis and SAR optimization of quinazolin-4(3H)-ones as poly(ADP-ribose)polymerase-1 inhibitors. Eur. J. Med. Chem. 2012, 50, 264–273. [Google Scholar] [CrossRef]
- Alanazi, A.M.; Abdel-Aziz, A.A.M.; Al-Suwaidan, I.A.; Abdel-Hamide, S.G.; Shawer, T.Z.; El-Azab, A.S. Design, synthesis and biological evaluation of some novel substituted quinazolines as antitumor agents. Eur. J. Med. Chem. 2014, 79, 446–454. [Google Scholar] [CrossRef]
- Cho, N.-C.; Cha, J.H.; Kim, H.; Kwak, J.; Kim, D.; Seo, S.-H.; Shin, J.-S.; Kim, T.; Park, K.D.; Lee, J.; et al. Discovery of 2-aryloxy-4-amino-quinazoline derivatives as novel protease-activated receptor 2 (PAR2) antagonists. Bioorg. Med. Chem. 2015, 23, 7717–7727. [Google Scholar] [CrossRef]
- Hopkins, A.L.; Groom, C.R.; Alex, A. Ligand efficiency: A useful metric for lead selection. Drug Discov. Today 2004, 9, 430–431. [Google Scholar] [CrossRef]
- Abad-Zapatero, C.; Metz, J.T. Ligand efficiency indices as guideposts for drug discovery. Drug Discov. Today 2005, 10, 464–469. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R., Jr. Properties of FDA-approved small molecule protein kinase inhibitors: A 2020 update. Pharmacol. Res. 2020, 152, 104609. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, A.L.; Keserü, G.M.; Leeson, P.D.; Rees, D.C.; Reynolds, C.H. The role of ligand efficiency metrics in drug discovery. Nat. Rev. Drug Discov. 2014, 13, 105–121. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||
| Compd | Structure | R | CDK9 IC50 (μM)a | Cytotoxicity IC50 (μM), MCF-71,a |
| 1 | - | - | 0.644 ± 0.007 | 0.65 ± 0.15 |
| 2 | A | H | 0.454 ± 0.006 | 2.31 ± 0.39 |
| 3 | A | Ph | 0.421 ± 0.006 | 0.82 ± 0.08 |
| 4 | A | 4-tolyl | 0.788 ± 0.012 | 0.16 ± 0.02 |
| 5 | A | 4-acetylphenyl | 0.829 ± 0.014 | 0.18 ± 0.09 |
| 6 | A | 4-methoxyphenyl | 0.463 ± 0.007 | 4.65 ± 1.38 |
| 7 | A | 4-ethoxyphenyl | 0.115 ± 0.002 | 1.57 ± 0.04 |
| 8 | A | 3,4,5-trimethoxyphenyl | 0.501 ± 0.009 | 2.48 ± 0.32 |
| 9 | A | 4-bromophenyl | 0.131 ± 0.002 | 1.72 ± 0.26 |
| 10 | A | 4-chlorophenyl | 0.193 ± 0.003 | 1.04 ± 0.07 |
| 11 | A | 4-fluorophenyl | 0.336 ± 0.005 | 3.86 ± 0.78 |
| 12 | A | 4-flourobenzyl | 0.218 ± 0.004 | 0.69 ± 0.23 |
| 13 | A | 4-methoxybenzyl | 0.334 ± 0.006 | 0.84 ± 0.15 |
| 14 | A | 3,4-dimethoxybenzyl | 0.229 ± 0.004 | 0.59 ± 0.10 |
| 15 | B | Ph | 0.444 ± 0.007 | 0.67 ± 0.18 |
| 16 | B | 4-chlorophenyl | 0.350 ± 0.006 | 0.73 ± 0.01 |
| Flavopiridol | - | - | 0.020 ± 0.008 | 0.04 ± 0.001 |
 | ||||||
| Compound | X | Y | R1 | R2 | CDK9 (μM) a | Cytotoxicity IC50 (μM), MCF-7 1,a |
| 17 | O | NH | H | I | 0.639 ± 0.004 | 3.88 ± 0.12 |
| 18 | O | NH | 2-thienyl | I | 0.296 ± 0.003 | 21.4 ± 0.68 |
| 19 | O | NH | 4-tolyl | Cl | 0.282 ± 0.003 | 12.3 ± 0.39 |
| 20 | O | NH | 4-chlorophenyl | NO2 | 0.434 ± 0.003 | 6.07 ± 0.19 |
| 23 | - | - | - | - | 0.210 ± 0.003 | 11.5 ± 0.37 |
| 24 | O | O | 3-bromophenyl | CH3 | 0.486 ± 0.004 | 10.4 ± 0.33 |
| 25 | O | NH | 3-bromophenyl | CH3 | 0.142 ± 0.001 | 16.8 ± 0.54 |
| 26 | S | NH | 3-bromophenyl | CH3 | 0.289 ± 0.002 | 5.21 ± 0.17 |
| 27 | O | N-NH2 | 3-bromophenyl | CH3 | 0.835 ± 0.006 | 28.7 ± 0.92 |
| 28 | O | N-OH | 3-bromophenyl | CH3 | 0.210 ± 0.002 | 6.27 ± 0.2 |
| 29 | - | - | - | - | 0.589 ± 0.002 | 17.4 ± 0.56 |
| Flavopiridol | - | - | - | - | 0.020 ± 0.008 | 0.04 ± 0.001 |
| Compound | MW (Da) | CLogP | HBA | HBD | No. of Violations |
|---|---|---|---|---|---|
| 1 | 361 | 1.83 | 6 | 1 | 0 |
| 2 | 418 | 0.87 | 8 | 2 | 0 |
| 3 | 495 | 2.93 | 8 | 2 | 0 |
| 4 | 509 | 3.28 | 8 | 2 | 1 |
| 5 | 537 | 2.80 | 9 | 2 | 1 |
| 6 | 525 | 2.86 | 9 | 2 | 1 |
| 7 | 539 | 3.27 | 9 | 2 | 1 |
| 8 | 585 | 2.72 | 11 | 2 | 2 |
| 9 | 573 | 3.66 | 8 | 2 | 1 |
| 10 | 529 | 3.54 | 8 | 2 | 1 |
| 11 | 513 | 3.03 | 8 | 2 | 1 |
| 12 | 527 | 2.74 | 8 | 2 | 1 |
| 13 | 539 | 2.57 | 9 | 2 | 1 |
| 14 | 569 | 2.50 | 10 | 2 | 2 |
| 15 | 509 | 3.34 | 8 | 2 | 1 |
| 16 | 543 | 3.95 | 8 | 2 | 1 |
| 17 | 272 | 1.00 | 3 | 1 | 0 |
| 18 | 354 | 2.51 | 3 | 1 | 0 |
| 19 | 271 | 3.16 | 3 | 1 | 0 |
| 20 | 302 | 1.89 | 6 | 1 | 0 |
| 23 | 334 | 3.36 | 4 | 2 | 0 |
| 24 | 316 | 3.42 | 3 | 0 | 0 |
| 25 | 315 | 3.28 | 3 | 1 | 0 |
| 26 | 331 | 3.61 | 2 | 1 | 0 |
| 27 | 330 | 1.97 | 4 | 1 | 0 |
| 28 | 331 | 2.62 | 4 | 1 | 0 |
| 29 | 162 | 0.51 | 4 | 2 | 0 |
| Compound | IC50 (μM) | pIC50 | N | LE | LLE |
|---|---|---|---|---|---|
| 1 | 0.644 | 6.19 | 24 | 0.354 | 4.36 |
| 2 | 0.454 | 6.34 | 28 | 0.311 | 5.47 |
| 3 | 0.421 | 6.38 | 34 | 0.257 | 3.44 |
| 4 | 0.788 | 6.10 | 35 | 0.239 | 2.83 |
| 5 | 0.829 | 6.08 | 37 | 0.225 | 3.28 |
| 6 | 0.463 | 6.33 | 36 | 0.241 | 3.47 |
| 7 | 0.115 | 6.94 | 37 | 0.257 | 3.67 |
| 8 | 0.501 | 6.30 | 40 | 0.216 | 3.58 |
| 9 | 0.131 | 6.88 | 35 | 0.270 | 3.22 |
| 10 | 0.193 | 6.71 | 35 | 0.263 | 3.18 |
| 11 | 0.336 | 6.47 | 35 | 0.254 | 3.44 |
| 12 | 0.218 | 6.66 | 36 | 0.254 | 3.92 |
| 13 | 0.334 | 6.48 | 37 | 0.240 | 3.90 |
| 14 | 0.229 | 6.64 | 39 | 0.234 | 4.14 |
| 15 | 0.444 | 6.35 | 35 | 0.249 | 3.01 |
| 16 | 0.35 | 6.46 | 36 | 0.246 | 2.51 |
| 17 | 0.639 | 6.19 | 12 | 0.708 | 5.20 |
| 18 | 0.296 | 6.53 | 17 | 0.527 | 4.02 |
| 19 | 0.282 | 6.55 | 19 | 0.473 | 3.39 |
| 20 | 0.434 | 6.36 | 21 | 0.416 | 4.47 |
| 23 | 0.21 | 6.68 | 20 | 0.458 | 3.32 |
| 24 | 0.486 | 6.31 | 19 | 0.456 | 2.89 |
| 25 | 0.142 | 6.85 | 19 | 0.494 | 3.57 |
| 26 | 0.289 | 6.54 | 19 | 0.472 | 2.93 |
| 27 | 0.835 | 6.08 | 20 | 0.417 | 4.11 |
| 28 | 0.21 | 6.68 | 20 | 0.458 | 4.06 |
| 29 | 0.589 | 6.23 | 12 | 0.712 | 5.72 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alkahtani, H.M.; Zen, A.A.; Obaidullah, A.J.; Alanazi, M.M.; Almehizia, A.A.; Ansari, S.A.; Aleanizy, F.S.; Alqahtani, F.Y.; Aldossari, R.M.; Algamdi, R.A.; et al. Synthesis, Cytotoxic Evaluation, and Structure-Activity Relationship of Substituted Quinazolinones as Cyclin-Dependent Kinase 9 Inhibitors. Molecules 2023, 28, 120. https://doi.org/10.3390/molecules28010120
Alkahtani HM, Zen AA, Obaidullah AJ, Alanazi MM, Almehizia AA, Ansari SA, Aleanizy FS, Alqahtani FY, Aldossari RM, Algamdi RA, et al. Synthesis, Cytotoxic Evaluation, and Structure-Activity Relationship of Substituted Quinazolinones as Cyclin-Dependent Kinase 9 Inhibitors. Molecules. 2023; 28(1):120. https://doi.org/10.3390/molecules28010120
Chicago/Turabian StyleAlkahtani, Hamad M., Amer Alhaj Zen, Ahmad J. Obaidullah, Mohammed M. Alanazi, Abdulrahman A. Almehizia, Siddique Akber Ansari, Fadilah Sfouq Aleanizy, Fulwah Yahya Alqahtani, Rana M. Aldossari, Raghad Abdullah Algamdi, and et al. 2023. "Synthesis, Cytotoxic Evaluation, and Structure-Activity Relationship of Substituted Quinazolinones as Cyclin-Dependent Kinase 9 Inhibitors" Molecules 28, no. 1: 120. https://doi.org/10.3390/molecules28010120
APA StyleAlkahtani, H. M., Zen, A. A., Obaidullah, A. J., Alanazi, M. M., Almehizia, A. A., Ansari, S. A., Aleanizy, F. S., Alqahtani, F. Y., Aldossari, R. M., Algamdi, R. A., Al-Rasheed, L. S., Abdel-Hamided, S. G., Abdel-Aziz, A. A.-M., & El-Azab, A. S. (2023). Synthesis, Cytotoxic Evaluation, and Structure-Activity Relationship of Substituted Quinazolinones as Cyclin-Dependent Kinase 9 Inhibitors. Molecules, 28(1), 120. https://doi.org/10.3390/molecules28010120