Abstract
Wild edible plants, once consumed in times of famine or for health purposes, today represent an interesting dietary supplement, aimed at enriching local dishes and/or formulating healthy nutraceutical products. In fact, the broad content of different, and diversely bioactive, specialized metabolites therein suggests new scenarios of use which, in order to be as functional as possible, must maximize the bioactivity of these compounds while preserving their chemistry. In this context, based on a recent investigation on the metabolic profile of the organs of Calendula arvensis that highlighted that florets are abundant in flavonol glycosides and triterpene saponins, the freeze-drying encapsulation of their alcoholic extract (FE) into maltodextrin (MD) was investigated. FE-MD chemical composition was evaluated using Fourier Transform InfraRed spectroscopy (FTIR), while ultra-high performance liquid chromatography coupled with high-resolution tandem mass spectrometry (UHPLC-HRMS/MS) techniques were employed to unravel FE compound preservation also during in vitro simulated digestion. The establishment of H-bonds between FE compounds and MD hydroxyl groups was in line with FE-MD biocompatibility in Caco-2 cells, while in vitro digestion mostly affected structural integrity and/or diversity. Flavonol compounds underwent deglycosylation and demethylation, while deacylation, beyond oxidation, involved triterpene saponins, which massively preserve their aglycone core.
1. Introduction
The culture of consuming alimurgic plants dates back to ancient times. Nowadays, although their usage has been frequently undervalued, they are experiencing a renaissance [1,2] as a new trend in nutrition in contemporary European cuisine [3,4] and ecosystem service [5]. Alimurgic plants, also known as wild edible plants (WEPs), are recognized as a source of micro- and macro-nutrients for dietary requirements, as well as of bioactive compounds for a functional nutrition [6]. The scientific recognition of their healthy properties has been accompanied by different strategies aimed at increasing the conservation of biodiversity and promoting the rural areas where they grow [7,8]. The latter is in line with the goal of contributing to people’s local identity [9], which is largely made up of traditional cuisine, including cooking methods and the events during which they are consumed [2].
Among the WEPs, Calendula arvensis (Vaill.) L. (also known as field marigold) is a medicinal species used in the health field, and with an important place in popular tradition due to its pleasant sensory properties. In several Mediterranean regions, boiled leaves, basal rosettes, and tender shoots are consumed, including the aerial parts sometimes mixed with meatballs [10,11]. The flowers or inflorescences of C. arvensis are eaten raw in salads or used to make jams and even candied. In southern Italy the dried petals of C. arvensis are reported to flavor wine, which after being left in the sun for ten days becomes excellent vinegar [12]. C. arvensis is also used in a traditional ointment made from crushed flowers mixed with olive oil to treat wounds and mouth ulcers [13,14]. Recently, C. arvensis has also been reported for the Campania Region (Southern Italy), where in particular its florets and leaves, employed in salads or soups of local cuisine, have become part of the historical-cultural heritage of Cilento, Vallo di Diano, and Alburni National Park (PNVCDA), one of the most extensive protected areas in Italy [15]. The chemical composition of C. arvensis’ six organs has been very recently investigated, providing a deep insight into their diversity in specialized metabolites, mainly polyphenols and triterpene saponins, which will hopefully open new scenarios for its rational nutraceutical use [16]. Indeed, these compounds are prone to lose their structural goodness based on environmental factors (e.g., light, heat, oxygen degradation) and processing conditions [17,18] Moreover, further instability characterizes their digestion process at the gastrointestinal level after ingestion, resulting in a decrease in the putative bioactivity, as the native amount in the plant matrix barely corresponds to the bioaccessible and bioavailable fraction [19,20].
In this context, strategies capable of safeguarding the specialized bioactive metabolites from harmful chemical alterations, also responsible for their reduced efficacy, must be pursued. Encapsulation, defined as a process involving the incorporation of a bioactive compound (called core) into an inert coating (called wall or carrier material), is one of the most convincing procedures aimed at protecting bioactive compounds or promoting their controlled release in those districts where they exert a beneficial effect [21,22]. Several encapsulation methods have been proposed, with freeze-drying tools representing a valuable method for encapsulating polar and thermolabile pure compounds and/or extracts [23,24]. In addition, various coatings have been applied; regardless of the wall material (e.g., polysaccharides, proteins, or lipids), certain characteristics must be met, such as being food-grade and biodegradable. Among the saccharides, maltodextrin (MD), a non-sweet nutrient mixture consisting of d-glucose-based oligomers and polymers, is an advantageous choice due to the high availability of raw material, high water solubility, and low viscosity [25,26].
In light of the above, herein the MD encapsulation process via freeze-drying was exploited as a valuable strategy to preserve the integrity and antioxidant activity of C. arvensis polar specialized compounds and to mitigate the degradation process in the gastro-intestinal tract during digestion. For this purpose, florets of field marigold harvested in PNVCDA preliminarily underwent ultrasound-assisted maceration in MeOH. Encapsulates were analyzed using Fourier Transform Infra-Red (FTIR) spectroscopy before and after an in vitro digestion test to investigate the nature of interactions established, as well as the effect of the digestion process on the maintenance of the structure capable of guaranteeing bioactivity. The deep chemical profiling was achieved by means of ultra-high performance liquid chromatography hyphenated to high-resolution tandem mass spectrometry (UHPLC-HRMS/MS) tools. Compound bioaccessibility at the end of the simulated digestion process was also assessed using UHPLC-HRMS/MS to obtain information about the occurrence of bioactive compounds that are potentially available for carrying out their activity.
2. Results and Discussion
The encapsulation procedure of C. arvensis floret extract (FE) was carried out using 10% (w:v) maltodextrin (MD) as the wall material. The literature data on the different MD:core ratios have shown that an increase in this ratio is directly related to a higher encapsulation efficiency due to a higher viscosity, which hinders the migration of the compounds towards the surface [27]. Based on these previous findings, an MD:core ratio equal to 1:2 (w:w) was chosen. The FE-MD mixture was freeze-dried, producing a powder with good rehydration properties [28].
2.1. Encapsulation Efficiency and FTIR Analysis
The FE encapsulation efficiency in maltodextrin wall material was estimated by means of colorimetric assays, aimed at determining the amount of entrapped phenols and saponins inside the polysaccharide matrix compared to the amount located on the wall surface. In fact, a previous work [16], focused on the in-depth compositional analysis of C. arvensis organ methanolic extracts, revealed that florets are mainly composed of glycosylated flavonols, besides triterpene saponins, with a different abundance detected for each metabolite. Although the plant organ investigated herein was collected in a different geographic location but with a similar land use habitat, it was reasonable to assume a similar chemical composition from a qualitative point of view.
The results of the Folin–Ciocalteau assay for determining total phenol content (TPC), expressed as mg of gallic acid equivalents (GAEs) per g of extract, underlined a seven-fold lower content of these metabolites on the surface with respect to the internal core (2.6 ± 1.0 mg GAE), leading to a phenol encapsulation efficiency equal to about 85%. Saponins embedded in the matrix accounted for 245.48 ± 14.7 mg of oleanolic acid equivalents (OAEs) per g of extract. This value was about 15-fold higher when compared to the surface amount (Figure 1a). The overall encapsulation efficiency (%) was estimated to be 93.2 ± 6.0. These results suggest a strong interaction between the bioactive compounds and the polysaccharide matrix, leading us to hypothesize their protection during in vitro simulated digestion.
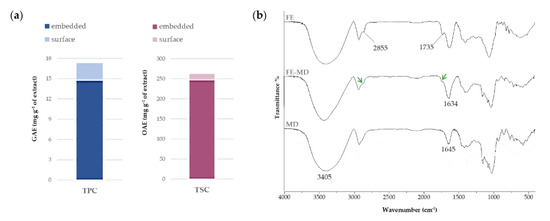
Figure 1.
(a) Total Phenol Content (TPC), expressed as mg GAE per g of extract, and Total Saponin Content (TSC), expressed as mg OAE per g of extract. The total content of each class of compounds takes into account relative surface and embedded contents. Values are reported as the mean ± SD of three independent measurements. (b) FTIR spectra of floret extract (FE), floret extract encapsulated in maltodextrin (FE-MD), and pure maltodextrin (MD). Green arrows focus on CH2 asymmetric and symmetric stretching, and estereal C=O symmetric stretching in FE-MD (please refer to the text).
The FTIR spectroscopy allowed us to unravel the nature of the bonds established in encapsulates between the MD wall material and the floret extract. In Figure 1b FTIR spectra recorded before and after the encapsulation procedure are reported. All spectra showed a broad band in the range 3600–3100 cm−1, attributable to the symmetric and asymmetric stretching of free hydroxyl groups, as well as of those employed in intra- and inter-molecular H-bonds, of carbohydrates, phenols, and polyphenols [29]. In particular, the OH vibrations shifted to lower (11–12 cm−1) wavelength values compared with the MD signal, indicating the formation of new H-bonds between extract constituents and the polysaccharidic carrier.
The deconvolution spectra (Figure 2) allowed us to better investigate the contribution of H-bonds with respect to free OH groups. Four components were revealed in the Gaussian deconvolution analysis. The peak at higher cm−1 values (3582–3578 cm−1) was assigned to OH stretching vibration of free OH groups in carbohydrates, phenols, and alcohols.
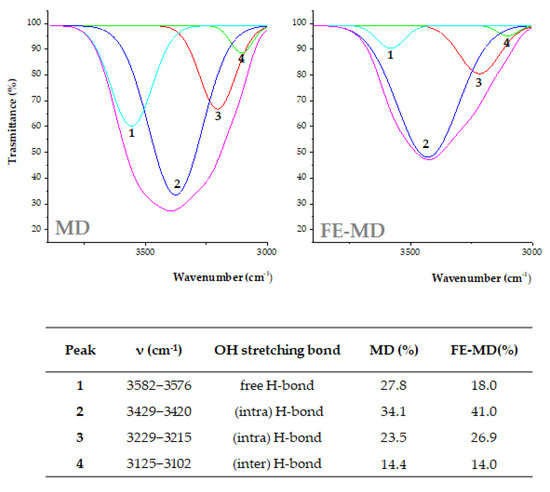
Figure 2.
OH stretching band deconvolution FTIR spectra from MD and FE-MD using the Gaussian model. Percentage area peaks from deconvoluted signals are also reported.
The peaks at 3429–3420 and 3229–3215 cm−1 were consistent with intra-molecular H-bonds, whereas the band at 3125–3102 cm−1 was likely due to the inter-molecular hydrogen bonds formed between the wall carrier and encapsulated extract. The percentage area of each OH stretching component was calculated (Figure 2). The free H-bond area was higher in MD and decreased when new H-bonds were established in FE-MD. This result also suggests the increase in H-bonds in the floret-based encapsulate [30].
Moreover, the bands at 2929 and 2845 cm−1 in Figure 1 were in accordance with CH2 asymmetric and symmetric stretching, whereas the peak at 1735 cm−1 was attributable to the stretching vibration of ester C=O groups. In the FE-MD spectrum the decrease and broadening of these peaks (labeled by green arrows) highlighted the encapsulation achievement in the MD wall with interaction that likely involved these functional groups. The bare maltodextrin spectrum shows its characteristic peaks in the fingerprint region. Indeed, the peaks at 1154 cm−1 and 1077 cm−1, and 1024 cm−1, were related to C-O stretching and the angular deformation of =CH and =CH2 bonds, respectively, typical of carbohydrate units, whereas the band at 1645 cm−1 could be assigned to deformation vibrations of –OH groups [31,32].
2.2. Evaluation of Cytotoxicity on the Caco-2 Cell Line
An MTT test was performed on the Caco-2 cell line after 3 h exposure time to investigate a potential cytotoxic effect exerted by the formulations in a time span that is slightly higher than that required by a typical simulated intestinal digestion phase.
A dose–response curve was prepared for FE in a wide concentration range, from 2.5 to 200 μg/mL (Figure 3a).
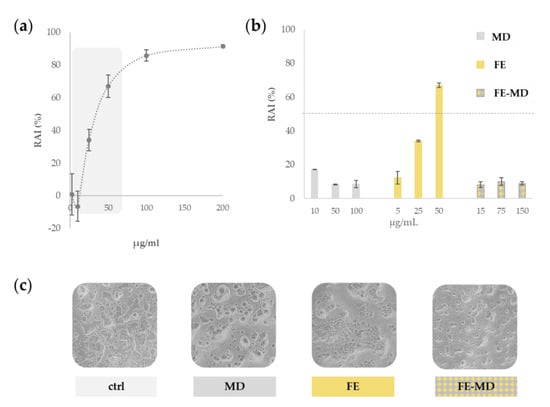
Figure 3.
(a) Preliminary assessment of redox activity inhibition (RAI %) of floret extract (FE) towards Caco-2 cells. (b) Comparison of RAI % due to FE, FE-MD formulation, and MD. (c) Representative images of untreated Caco-2 cell line, and cells treated with maltodextrin (MD), floret extract (FE), and FE-MD at the highest dose. Images were acquired with an Inverted Phase Contrast Brightfield Zeiss Primo Vert Microscope.
The results clearly indicated that floret extract had a dose-dependent cytotoxic effect, inhibiting significantly the cell mitochondrial redox activity (RAI) by more than 50%, when the concentration of 50 μg/mL was tested. Thus, this concentration was selected as the highest dose level of the core of MD encapsulates, and the role of the polysaccharide wall in the cell protection was investigated. Indeed, as depicted in Figure 3b, the field-marigold-MD formulation did not display cytotoxicity reduction, and its RAI value never exceeded 25%. This finding is in accordance with a high biocompatibility, as also revealed by morphologically observing treated cells (Figure 3c).
Maltodextrin, as an inert polysaccharide, is generally considered as safe by the US Food and Drug Administration [33]. In fact, it was previously reported that cell growth is usually not affected by the presence of this polymer, which acts only as a carrier [34], while exerting a protective role towards oxidation and increasing the stability of the encapsulated compounds [35]. A similar positive trend was found when microencapsulating stevia ethanolic extract in maltodextrin (ratio 1:2, w:w), which showed viability higher than 80%, strongly reducing the extract’s cytotoxicity even at the highest tested dose [36].
2.3. UHPLC-QqTOF-MS/MS Analysis before In Vitro Digestion
The chemical composition of C. arvensis alcoholic floret extract herein investigated, based on UHPLC-QqTOF-MS/MS data, is summarized in Table 1 and briefly discussed below.

Table 1.
Compounds tentatively identified in floret alcoholic extracts before in vitro digestion (base peaks in MS/MS experiments are reported in bold; RDB = Ring and Double Bonds, i.e., unsaturation degree; Rt = retention time). * Molecular ions detected as formate ([M + HCOO−]−) adducts. n.c. = not calculable.
Apart from compound 1, likely corresponding to a [M + Cl−]− adduct of a dihexose, the detected specialized metabolites were phenols, polyphenols, and triterpene saponins, as expected from the results of the TPC and TSC tests and previously published chemical profiles [16].
2.3.1. Phenols and Polyphenols
Compounds 2–4 and 13 were quinic acid esters, whose hydroxycinnamoyl moiety consisted of caffeoyl in 2 and 3 (at m/z 353.0880, C16H18O9) and feruloyl in 4 (at m/z 367.1043, C17H20O9). In their HR-MS/MS spectra, base peaks at m/z 191.056 derived from the neutral loss of dehydrated hydroxycinnamoyl moieties were observed, whereas the fragment ion at m/z 193.0505 in 4 was in agreement with ferulate ion. In particular, based on the fragmentation pattern and comparison with pure reference compounds, compounds 2 and 3 were identified as the geometrical isomers of 5-O-caffeoyl quinic acid [37]. Furthermore, the TOF-MS/MS spectrum of compound 13, showing a deprotonated molecular ion at m/z 515.1196, was in accordance with 3,5-di-O-caffeoylquinic acid [37].
Compounds 5–12 and 14–20 were tentatively identified as flavonol glycosides, with quercetin, kaempferol, and isorhamnetin as aglycone units. In particular, metabolites 5–9 and 12 were glycosylated quercetin derivatives whose elution order in reversed-phase chromatography was in accordance with a gradual decrease in polarity, mainly ascribable to the glyconic moiety. Thus, compounds 5–7 and 9 were quercetin diglycosides, bearing a dihexose, a hexosylpentose, and hexosyldeoxyhexoses, lost as dehydrated units (−324, −294, and −308 Da, respectively) [16]. The intensity ratio between the flavonol radical deprotonated ion (at m/z 300.027) and the related deprotonated aglycone (at m/z 301.035) allowed us to hypothesize the same glycosylation position (at C-3 hydroxyl group). The deprotonated molecular ion detected for metabolite 8 (m/z 463.0882, calc. mass) and the formula calculated therefrom (C21H20O12) suggested the occurrence of a quercetin hexoside, whereas compound 12 was putatively its malonyl derivative. In fact, the detection of fragment ions at m/z 505.0988 and 463.0885 in the TOF-MS/MS experiment was consistent with sequential losses of CO2 (44 Da) and C2H2O units (42 Da) from the precursor molecular ion to give quercetin hexoside (Figure S1). The latter compound was previously identified in C. officinalis flowers and petals and in C. morifolium [38], but, to the best of our knowledge, never in C. arvensis.
Based on our previous MS data, compounds 11 and 14 were identified as two isomers of kaempferol-3-O-hexoside, whereas compound 15 was in line with kaempferol-7-O-rutinoside [16].
Metabolites 10 and 16–20 were isorhamnetin glycosides. In particular, compounds 16 and 18, sharing the same molecular formula (C28H32O16), were tentatively identified as isorhamnetin-3-O-hexosyldeoxyhexoside and its 7-O isomer, respectively. The intensity ratio between the [aglycone–H]− (at m/z 315.051) and the [aglycone–H]•− (at m/z 314.043) ions, formed following the neutral loss of the disaccharide moiety (−308/309 Da), suggested a different glycosylation position. Compound 10 (at calc. m/z 639.1567, C28H32O16) was likely isorhamnetin-3-O-dihexoside, while metabolites 17 and 19 were two isomers of monohexosyl methoxyflavonol. Finally, the deprotonated compound 20 at m/z 563.1042, whose neutral losses resembled those observed for compound 12, was tentatively identified as the malonyl derivative of isorhamnetin-3-O-hexoside (Figure S1), and not reported before in field marigold florets.
2.3.2. Triterpene Saponins
Recently, deeply investigating the fragmentation pathways of C. arvensis triterpene saponins, we provided some useful information for their HR-MS and MS/MS straightforward chemical characterization in complex mixtures [16]. Herein, we applied those guidelines to identify all the detected compounds belonging to this class (21–38), among which seven saponins were not identified in our previous work. Thus, their identification is discussed herein in detail, providing hypothesized fragmentation pathways as supplementary material.
Saponins 21–26, 28, and 36 were echinocistic acid glycosides. Compound 21 with formula C47H74O19 and [M − H]− at m/z 941.4790 (4.1 ppm mass error) was tentatively identified as 3-O-(pentosyl)hexuronidyl 28-O-echinocystic acid hexosyl ester. Its fragmentation pattern resembles that described for calendasaponin B, differing in the presence of a pentose residue instead of a hexose, linked to the hexuronidyl moiety. Briefly, the [M − H]− ion, following the neutral loss of 162.05 Da, generated the fragment ion at m/z 779.4207. The latter provided an ion at m/z 717.4229 through the concerted loss of (44 + 18 = 62) Da on the hexuronidyl unit, or of the pentosyl unit (−150 Da) to provide the fragment ion at m/z 629.3699. Alternatively, from the ion at m/z 717.4229, following the neutral loss of 132 Da (pentose-H2O), the fragment at m/z 585.3797 was obtained. The deprotonated echinocystic acid at m/z 471.3480 could be finally revealed (Figure S2).
Compound 23 (C42H66O15, at m/z 809.4369) differed from the previous one in the absence of the pentosyl residue. Thus, it was tentatively identified as 3-O-hexuronidyl 28-O-echinocystic acid hexosyl ester, as reported by Fiorentino et al. [16]. Metabolite 25 seemed to be a derivative of the previous one bearing an acetylhexosyl instead of a hexosyl residue at the C-28 position. In fact, the first neutral loss of 42 Da from the deprotonated molecular ion gave rise to the ion at m/z 809.4329, and then the fragmentation pattern proceeded in a similar way (Figure S3). Neutral losses of 144 Da from compounds 26 (at m/z 953.4792, C48H74O45) and 36 (at m/z 791.4235, C42H64O14) were in accordance with a hydroxymethylglutaryl moiety on the molecular skeleton and differed in a hexose unit, as previously described [16].
Unlike the above-mentioned saponins, compounds 22, 24, and 28 were detected in the TOF-MS spectrum as formate adducts ([M + HCOO]−), giving evidence of the absence of a primarily acid site (neither free triterpene carboxylic group nor hexuronic acid). Thus, metabolite 24 was tentatively identified as 3-O-hexosyl 28-O-echinocystic acid hexosyl ester. In fact, the fragment ions at m/z 633.4030 and 587.3957 were generated following the neutral loss of the dehydrated hexosyl unit (−162 Da) at C-28 and the sequential loss of 46 Da. Then, the loss of the second hexosyl moiety (as 180 Da) gave rise to the ion at m/z 407.3348 (Figure S4). Moreover, following the same rationale, saponins 22 and 28 were tentatively identified as two isomers of 3-O-dihexosyl 28-O-echinocystic acid hexosyl ester (e.g., calendulastellaside B), whose fragmentation pathway is reported in Figure S5.
Saponins 27, 29–35, 37, and 38 were recognized as oleanolic acid glycosides, differing in the number and the nature of saccharidic units. Also within this subclass, for some compounds formate adducts were detected. In particular, TOF-MS and TOF-MS/MS spectra of compounds 27, 30, 33, and 38 were in accordance with the presence of 3-O-trihexosyl, dihexosyl (e.g., arvensoside A), and two monohexosyl derivatives of 28-O-oleanolic acid hexosyl ester, respectively, previously reported in C. arvensis organs [16].
All the other oleanolic acid glycosides showed deprotonated molecular ions. TOF-MS/MS fragmentation of saponins 29 and 31 (calc. mass at m/z 925.4802, C47H74O18) suggested the occurrence of two isomers of in which a pentose, a hexose, and a hexuronic acid were linked to oleanolic acid. Following the fragmentation route described for saponin 21, they were putatively assigned to 3-O-(pentosyl)hexuronidyl 28-O-oleanolic acid hexosyl esters. In both cases, the loss of the sugar units generated the deprotonated oleanolic acid at m/z 455.3531 (calc. mass) (Figure S6). Grati et al. [39] very recently reported oleanolic saponins in C. aegyptiaca fruits, providing a tentative structure in which both the pentose and the hexose were linked at the C-2 and C-3 positions of the uronic acid. Compound 32 was likely 3-O-hexuronidyl 28-O-oleanolic acid hexosyl ester (e.g., calenduloside F) or calenduloside G [39], whereas in metabolite 34 an acetylhexosyl moiety was supposed to be esterified at the C-28 position of 3-O-hexuronidyl oleanolic acid, in accordance with Fiorentino et al. [16]. The [M − H]− ion detected for compound 37 (at m/z 763.4303, C41H64O13) fragmented, losing a pentose unit (−150 Da) and generating the ion at m/z 613.3767, which in turn provided the oleanolate ion by the neutral loss of 158 Da, as described above (Figure S7). Thus, it was putatively identified as 3-O-(pentosyl)hexuronidyl oleanolic acid.
Finally, the fragmentation pattern of saponin 35 was consistent with the hydroxymethylglutaryl derivative of 34. In fact, after the loss of 144 Da from the deprotonated molecular ion (at m/z 937.4802, calc. mass) the two TOF-MS/MS spectra were almost superimposable.
2.4. Bioaccessibility Assessment of Encapsulated Bioactive Compounds by In Vitro Digestion
FE-MD underwent in vitro simulated digestion to obtain information about how the encapsulation in maltodextrin was able to influence the bioaccessibility in the gastro-intestinal tract of floret bioactive compounds.
FTIR spectra acquired after in vitro digestion gave preliminary insights about its capability to induce structural changes in encapsulates. In Figure 4 the zoomed region in the range 1800–500 cm−1 is reported for the digested formulation (FE-MDd), also in comparison with the native (undigested) sample. The most detectable change regards a decrease in the bands at 1734(5) cm−1 and 763 cm−1, attributable to the stretching vibration of carbonyl groups and the bending vibration of C-H in hydroxycinnamic acids and other phenolic acids [40], which could be in accordance with their lower occurrence in digestates. Moreover, the shoulder band at 993 cm−1, characteristic of C-O-C stretching vibration in polysaccharides, weakened, leading us to hypothesize the partial cleavage of α(1→4) glycosidic bonds within the maltodextrin wall [41].
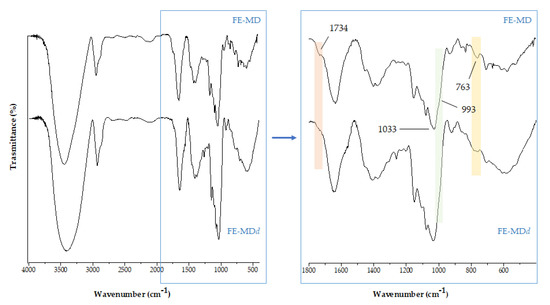
Figure 4.
The comparison of FTIR spectra of floret encapsulate before (FE-MD) and after in vitro digestion (FE-MDd).
UHPLC-HRMS tools allowed us to obtain more specific information about the fate of individual compounds at the end of the digestion process. However, a massive matrix effect was revealed in total ion current chromatograms of digesta, due to the presence of degradation products of maltodextrin, which eluted at the lowest retention time, resulting in metabolite ion suppression (data not shown). In order to avoid this problem, samples deriving from in vitro simulated digestion underwent a liquid/liquid extraction using n-butanol as the extracting solvent. The organic phases were collected and analyzed with hyphenated UHPLC and HRMS techniques compared to undigested samples.
A preliminary insight evidenced that (poly)phenols were massively affected by digestion degradation, whereas triterpenes were more preserved. Indeed, Bottcher et al. [42] reported that when saponins are dissolved in an aqueous system, such as digestion buffers, they are able to form a viscoelastic network through intermolecular hydrogen bonds with neighboring sugar residues, which is able to protect the phytocomplex embedded in the maltodextrin coating.
FE-MDd was the object of an in-depth UHPLC-HRMS study to analyze the bioaccessibility and putative biotransformations of its bioactive compounds during in vitro digestion. The relative amount of polyphenols and triterpene saponins before and after in vitro simulated digestion could be easily visualized through the heatmap analysis, reported in Figure 5.
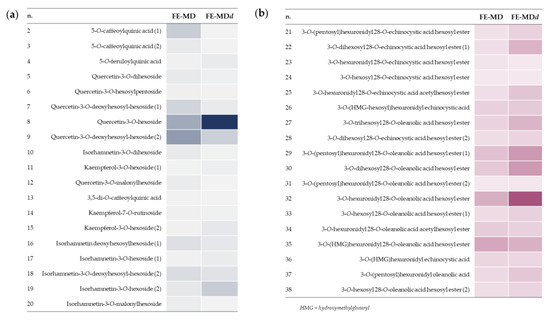
Figure 5.
Heatmaps of polyphenols (a) and triterpene saponins (b) in floret encapsulates before and after in vitro digestion (FE-MD and FE-MDd, respectively).
In particular, the hydroxycinnamoyl derivatives appeared particularly vulnerable, in accordance with hypotheses deriving from FTIR spectra. In fact, poor traces of 5-O-caffeoylquinic acid isomers, 5-O-feruloylquinic acid, as well as dicaffeoylquinic acid, were detected in the FE-MDd sample, in line with the ester bond cleavage that has been reported to occur in the simulated gastric acidic environment [43,44]. Flavonoid glycosides were partially degraded, also as a consequence of biotransformation reactions. In particular, a decrease in quercetin malonylhexosyl and diglycosyl derivatives, such as quercetin-3-O-dihexoside, quercetin-3-O-hexosylpentoside, and rutin, were observed in the digestate following deacylation and glycosidic bond hydrolysis to provide the quercetin 3-O-hexoside. Indeed, the latter was 2.5-fold more abundant in digested floret encapsulate compared with the undigested one. Similarly, signals assigned to diglycosyl derivates, as well as to the malonyl-hexosyl conjugate of isorhamnetin, strongly decreased, putatively giving rise to isorhamnetin-3-O-hexoside isomers (Figure S8). The occurrence of deacylation and deglycosylation reactions has been recently described as metabolic steps of flavonoid O-glycosides both at the gastric and at the intestinal level [45]. Moreover, it is worthy of note that quercetin 3-O-hexoside could also be generated following isorhamnetin 3-O-hexoside demethylation reactions at C-3′ position, or, alternatively, by hydroxylation of kaempferol 3-O-hexoside [46]. In line with the previous hypothesis, deacetylation and deglycosylation reactions, together with the loss of the hydroxymethylglutaryl moiety and hexose oxidation, were also assumed to explain structure modifications in triterpene saponins, leading to the formation of 3-O-hexuronidyl 28-O-oleanolic acid hexosyl ester (Figure 6).
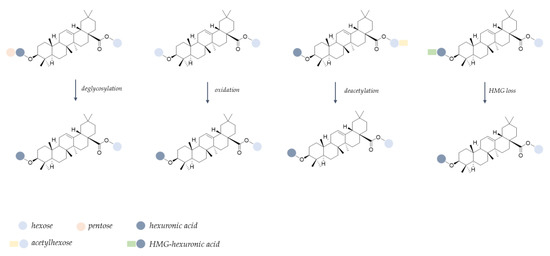
Figure 6.
Proposed biotransformation pattern for triterpene saponins during in vitro simulated digestion (HMG = hydroxymethylglutaryl).
2.5. Evaluation of Radical-Scavenging Capacity (RSC, %)
It is well known that the bioactivity of a compound closely depends on its chemical structure. The scenario is further complicated when a phytocomplex is considered and not pure molecules, as synergic or inhibiting effects could contribute to the obtained results. Thus, the bioactivity could be strongly affected by structural changes occurring during digestion steps.
With this awareness, FE samples were tested to assess their radical-scavenging capacity against DPPH• and ABTS•+ and the results were compared to FE-MD encapsulates, to evaluate the activity maintenance, and also to samples derived from the end of the simulated digestion procedure (FE-MDd) (Figure 7).
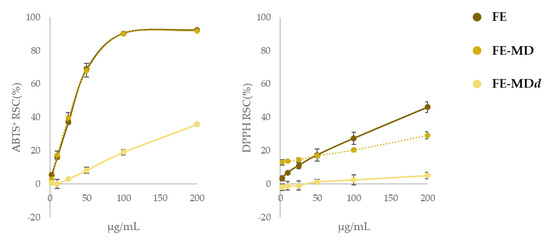
Figure 7.
Radical-scavenging activity (RSC%) of floret extract (FE) in comparison with its encapsulate (FE-MD) and digested formula (FE-MDd) using DPPH and ABTS tests. Values reported are the mean ± SD of three independent measurements.
The radical-scavenging efficiency of encapsulates against ABTS•+ was superimposable to that recorded for the naked extract, proving that the encapsulation process did not affect the capacity of bioactive compounds entrapped in the maltodextrin matrix to neutralize the presence of the radical probe. Considering that both ABTS and DPPH tests have been explained based on a mix of SET (Single Electron Transfer) and HAT (Hydrogen Atom Transfer) mechanisms, with ABTS radicals more prone to the former, and DPPH to the latter, it was not surprising that responses to the DPPH test were much lower than the previous ones. In fact, the formation of H-bonds between the core and the MD wall in encapsulates could be responsible for the compounds’ reduced attitude as H donors.
After the in vitro digestion a considerable reduction in the radical-scavenging activity was observed, again more pronounced vs. DPPH•. In this case, in line with data reported by Tarko et al. [47], the scenario is also worsened by the loss of a large portion of the polyphenols in the intestinal phase (only 10% of the total amount survived to the simulated digestion).
3. Materials and Methods
3.1. Plant Collection and Extraction
Calendula arvensis plants were collected in December 2021 at Ascea (Salerno, Italy), located in Cilento, Vallo di Diano, and Alburni National Park (PNCVDA) (40°09′45.2″ N 15°12′26.8″ E). A voucher specimen has been deposited in the Herbarium Austroitalicum (IT) of the Department of Environmental, Biological, and Pharmaceutical Sciences and Technologies of the University of Campania ‘Luigi Vanvitelli’ (Caserta, Italy). Fresh plants organs were dissected, immediately stored in liquid nitrogen, and transferred to the laboratory. Among them, after lyophilization, florets underwent a defatting extraction step in n-hexane and then were sonicated (Branson UltrasonicsTM BransonicTM M3800-E; Danbury, CT, USA) in methanol to collect polar bioactive compounds. The alcoholic extract thus obtained was dried using a rotary evaporator.
3.2. Floret Extract Encapsulation
Maltodextrin (MD, DE 16.5–19.5) powder (1.2 g) was dissolved in 12 mL of distilled water. Floret extract (FE; 600 mg) was solubilized in distilled water and then added drop by drop to the maltodextrin solution, reaching a FE:MD ratio of 1:2, under continuous stirring at 25 °C. The mixture was stored at −80 °C for 12 h and then was cryo-dried for 48 h using the FTS-System Flex-dryTM instrument 75 (SP Scientific, Stone Ridge, NY, USA). Finally, the obtained powdered sample was stored at 4 °C, sheltered from light, until use (Figure 8).
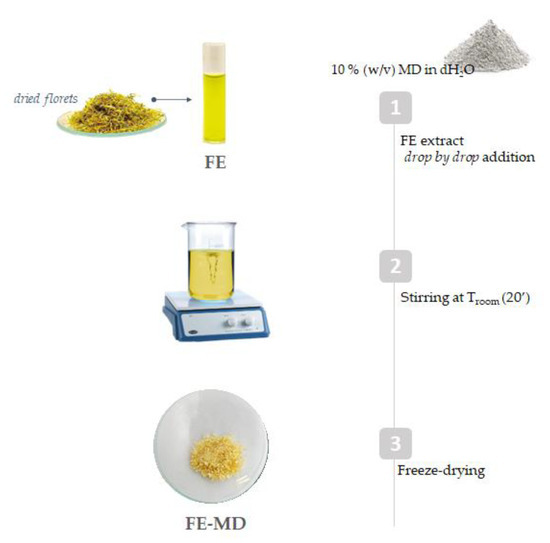
Figure 8.
Encapsulation scheme applied to field marigold floret extract (MD = maltodextrin; FE = floret extract).
3.2.1. Surface and Total Phenol Content in Encapsulates
Total phenol content (TPC) was determined according to the Folin–Ciocalteau method, as well as the surface phenol content. Briefly, for TPC, 2.5 and 5.0 mg of FE-MD were mixed with 2.25 mL of sodium carbonate (7.5% w/v) and 0.25 mL of Folin–Ciocalteau reagent. The mixtures were shaken for 3 h in the dark at room temperature. The absorbance was read at 765 nm using a Synergy spectrophotometer (Biotek, Winooski, VT, USA). Three replicates of three aliquots (n = 3) of each sample (in total, 3 × 3 measurements) were analyzed. A calibration curve of gallic acid was built up (0.78–50 µg/mL) and data were expressed as mg of gallic acid equivalents (GAEs) per g of sample (mean ± SD). For surface phenol content, FE-MD powder was mixed with ethyl acetate (1.0 mL), vortexed for 60 s, and centrifuged at 3000× g for 10 min. The surface phenol content was measured following the same Folin–Ciocalteau method. The embedded phenol content was calculated by subtracting the surface phenol content from the TPC value [48].
3.2.2. Surface and Total Saponin Content in Encapsulates
For the determination of total saponin content (TSC), 2.5 and 5.0 mg of FE-MD were mixed with 150 µL of isovanillin/glacial acetic acid (5% w/v) and 0.5 mL of perchloric acid. The mixture was incubated at 60 °C for 45 min and cooled down in an ice bath. The absorbance was spectrophotometrically read at 550 nm. The total saponins were quantified using a standard curve of calibration of oleanolic acid and expressed as mg of oleanolic acid equivalents (OAEs) per g of sample [49]. For surface saponin content, FE-MD and LE-MD encapsulates were mixed with butanol (1.0 mL), vortexed for 60 s, and centrifuged for 10 min at 3000× g. The surface saponin content was then measured following the method applicated for TSC, while embedded saponin content was calculated as the difference between TSC and the surface saponin content.
3.2.3. Evaluation of Encapsulation Efficiency
The overall encapsulation efficiency (EE %) in maltodextrin was calculated by measuring the amounts of phenols and saponins entrapped in the formulation over the total phenols and saponins revealed [48,50], following the equation:
where EPC and ESC are encapsulated phenol and saponin content, respectively, and correspond to (TPC—surface phenol content) and (TSC—surface saponin content) amounts.
EE % = [(EPC + ESC)/(TPC + TSC)] × 100
3.3. FTIR Analysis of the Floret Encapsulate
Fourier transform infrared (FTIR) spectra of FE-MD encapsulates and floret extract, used as a reference together with bare MD, were recorded using a Prestige 21 system (Shimadzu, Milan, Italy) with a deuterated triglycine sulfate detector with potassium bromide windows (DTGS KBr). The wavenumber range was 4000–400 cm−1 with a resolution of 2 cm−1 (60 scans). A 200 mg KBr pellet disk of each sample (1% in KBr) was prepared using a Specac manual hydraulic press (Orpington, UK) equipped with a cylindrical holder. FTIR spectra were processed with IR solution software (v.1.60, Shimadzu, Milan, Italy). The multiple peak fit tools of OriginPro 2015 software (OriginLab Corp., Northampton, MA, USA) was employed for the deconvolution analysis of the OH stretching band (3600–3100 cm−1): number of peaks = 4; fitting function: Gaussian; R-square ≥ 0.999; no parameters were fixed for analysis. Chi-sqr tolerance reached 1 × 10−9 for all deconvoluted spectra.
3.4. Cytotoxicity Evaluation
The cytotoxic effect on the gastrointestinal tract were evaluated on the human colorectal adenocarcinoma epithelial cell line Caco-2 (ATCC® HTB:37™, American Type Culture, Manassas, VA, USA), cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 50.0 U/mL penicillin, 100.0 µg/mL streptomycin, and 10% fetal bovine serum, at 37 °C in a 5% CO2 humidified atmosphere [51,52]. A total of 2.5 × 104 cells was seeded in each well of a 96-multiwell plate. After 24 h, cells were treated with different doses of the FE (2.5, 10, 25, 50, 100, and 200 µg/mL, final concentrations) for 3 h. Then, the inhibition of mitochondrial redox activity (RAI %) was determined with the MTT cell test. Based on the dose–response curves obtained for each sample, three different concentrations of the encapsulated extract were selected (15, 75, 150 µg/mL, containing 5, 25, 50 µg/mL of FE). Maltodextrin was used as the negative control sample, tested at the three different doses used in formulas to keep constant the 2:1 (MD:extract) ratio (10, 50, and 100 µg/mL). Two independent experiments were carried out with six replicate measurements in each of the three samples of the extract (in total, 6 × 3 measurements). Data were expressed as mean ± standard deviation (SD).
3.5. Bioaccessibility Study
The bioaccessibility of polyphenols and triterpene saponins in FE-MD was studied following the in vitro digestion procedure proposed by Minekus et al. [53]. Simulated salivary (SSF), gastric (SGF), and intestinal fluids (SIF) were employed for oral, gastric, and intestinal phases, respectively, prepared following the recommendations therein. One gram of FE-MD was suspended in SSF buffer, distilled water, and CaCl2 (0.3 M concentration in the mixture). A final ratio of encapsulates to SSF of 1:1 (w/v) was reached. After adding the porcine α-amylase enzyme (final concentration of 75 U/mL), the oral mixture was incubated for 2 min at 37 °C. The oral bolus was mixed with SGF with a final ratio of 1:1 v/v. The porcine pepsin enzyme and CaCl2 were added to achieve 2000 U/mL and 0.3 M in the final mixture, respectively. HCl (6 M) was added to obtain pH 3.0. The gastric phase step ended in 2 h under continuous stirring in the dark at 37 °C. Finally, the gastric chyme was mixed with SIF solution to obtain a 1:1 ratio v/v after the addition of pancreatin enzyme solution (100 U/mL based on trypsin activity, final concentration), 0.3 M CaCl2, bile salts (10 mM in the final mixture), and distilled water. NaOH 1 M solution was required to neutralize the mixture. The latter was incubated for 2 h under continuous stirring in the dark at 37 °C; after that the enzyme activity was blocked by immersion in liquid nitrogen.
Digested FE-MD was dissolved in water:butanol (1:1, v/v) and subjected to discontinuous liquid–liquid extraction (three cycles), in order to recover bioactive compounds from the maltodextrin wall material. Then, the obtained organic fractions were profiled using UHPLC-HRMS with the same experimental parameters used for native extracts.
3.6. UHPLC-ESI-MS and MS/MS Analyses
The chromatographic separation was performed with a Shimadzu NEXERA UHPLC system (Shimadzu, Tokyo, Japan). A Luna® Omega C18 (1.6 µm particle size, 50 × 2.1 mm i.d.) was employed, injecting 2.0 µL of each sample. The following linear gradient was built up, with mixtures of H2O (A) and CH3CN (B), both with 0.1% formic acid, as mobile phase: 0–1 min, 5% B; 1–7 min, 5→17.5% B; 7–9 min, 17.5→25% B; 9–18 min, 25→55% B; 18–20 min, 55→95% B; 20–21 min, 95% B. Then, the starting conditions were restored and maintained for 2 min for re-equilibration purposes. The flow rate was 500 µL/min. The MS analysis was carried out using a hybrid QqTOF MS instrument, the AB SCIEX Triple TOF® 4600 (AB Sciex, Concord, ON, Canada), equipped with a DuoSprayTM ion source, which operated in the negative ElectroSpray (ESI) mode. The HR-MS method consisted of a full-scan TOF survey (accumulation time 250 ms, 100–1500 Da) and eight IDA (information-dependent acquisition) scans (accumulation time 100 ms, 80–1300 Da). The MS parameters were as follows: curtain gas 35 psi, nebulizer gas 60 psi, heated gas 60 psi, ion spray voltage −4.5 kV, interface heater temperature 500 °C. The Analyst® TF 1.7 software was used for controlling the instrument, while data processing used PeakView® software version 2.2. The TOF-MS/MS parameters used for phenols and polyphenols (e.g., flavonoids, hydroxycinnamoyl compounds) were: −100 V declustering potential (DP), −40 V collision energy (CE), and −15 V collision energy spread (CES). DP of −120 V, CE of −100 V, and CES of −25 V were set for obtaining insight into triterpene saponins.
3.7. Radical-Scavenging Capacity Assessment
The radical-scavenging capacity of FE, and of its encapsulate and the digestate, was evaluated against the DPPH (2,2-diphenyl-1-picrylhydrazyl) radical and ABTS [2,2′-azinobis-(3-ethylbenzothiazolin-6-sulfonic acid)] radical cation, as previously reported [54]. Briefly, after aqueous solubilization, the samples were added to the radical solution to reach the tested concentrations ranging from 2.5 to 200 μg/mL. Three replicate measurements for each sample (three for each concentration) were tested in reference to a blank, in which samples were replaced with distilled water. Trolox® in the 2–32 μM final concentration range was employed as a positive standard. For FE-MD the tested doses referred to the floret concentration therein, taking into consideration the ratio FE (or LE):MD = 1:2. Data were expressed as mean ± standard deviation (SD) from three replicate measurements of three samples of the extract (in total, 3 × 3 measurements).
4. Conclusions
Calendula arvensis is locally used for health purposes, or for preparing traditional dishes. In order to promote the species’ valorization, allowing full usage of all its organs, the study of its polar bioactive compounds was previously investigated by means of extraction and fractionation strategies exploiting accelerated maceration with ultrasound.
The metabolic peculiarity of the floret alcoholic extract (FE) consisted of glycoside flavonols and triterpene saponins, mainly based on echinocystic and oleanolic acid, in line with its marked anti-radical capacity. In order to formulate an FE-based antioxidant preparation readily available for consumption, FE was screened for its cytotoxicity towards intestinal epithelial cells, and then encapsulated, at safe dose levels, into maltodextrin.
Encapsulation has proven to be an effective strategy to preserve the compounds in the extract, and the formation of complex-stabilizing hydrogen bonds was ascertained through FTIR spectroscopy, while the anti-radical efficacy was comparable to that exerted by FE. When in vitro simulated digestion was performed, a massive loss of the scavenging capacity was observed. This is largely due to the degradation of the flavonoid compounds, which are more responsive in DPPH and ABTS tube tests than triterpene saponins. The application of UHPLC HR MS/MS techniques downstream of the in vitro simulated digestion process shows that deacylation and oxidation reactions, with deglycosylation, impact the triterpenic saponins which, however, keep intact the aglycone core. This is not strictly true for flavonoids although deglycosylation and demethylation are key processes in the biotransformation of these compounds.
The constitution of a maltodextrin:FE formulation opens up to new investigations for its dedicated use in the nutraceutical, cosmeceutical, and phytoceutical sectors. The possibility of benefiting from non-invasive processing of the phytochemistry of a species realizes the optimization of short local supply chains that join together tradition and innovation.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/molecules28010199/s1. Figure S1. TOF-MS/MS spectra of compounds (a) 12 and (b) 20, and (c) their fragmentation pathways; Figure S2. (a) TOF-MS/MS spectrum of compound 21; (b) its hypothesized fragmentation pathway; Figure S3. (a) TOF-MS/MS spectrum of compound 25; (b) its hypothesized fragmentation pathway; Figure S4. (a) TOF-MS/MS spectrum of compound 24; (b) its hypothesized fragmentation pathway; Figure S5. TOF-MS/MS spectra of compounds (a) 22 and (b) 28, and (c) the putative fragmentation pathway; Figure S6. TOF-MS/MS spectra of compounds (a) 29 and (b) 31, and (c) the putative fragmentation pathway; Figure S7. (a) TOF-MS/MS spectrum of compound 37; (b) its hypothesized fragmentation pathway; Figure S8. Proposed biotransformation pattern for glycosylated flavonols during in vitro simulated digestion.
Author Contributions
Conceptualization, S.P. (Severina Pacifico); methodology, S.P. (Severina Pacifico), M.F. and S.P. (Simona Piccolella); formal analysis, M.F. and C.G.; investigation, M.F., C.G. and A.S.; data curation, M.F., S.P. (Simona Piccolella) and A.E.; writing—original draft preparation, M.F. and S.P. (Simona Piccolella); writing—review and editing, S.P. (Severina Pacifico); visualization, M.C., A.E. and S.P. (Simona Piccolella); supervision, A.E. and S.P. (Severina Pacifico). All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
The authors thank the PON Project “Valorizzazione delle specie officinali della flora autoctona del Parco Nazionale del Cilento, Vallo di Diano e Alburni (PNCVDA)” DOT13499551.
Conflicts of Interest
The authors declare no conflict of interest.
Sample Availability
Samples of the investigated extracts are available from the authors.
References
- Baldi, A.; Bruschi, P.; Campeggi, S.; Egea, T.; Rivera, D.; Obón, C.; Lenzi, A. The Renaissance of Wild Food Plants: Insights from Tuscany (Italy). Foods 2022, 11, 300. [Google Scholar] [CrossRef] [PubMed]
- Łuczaj, Ł.; Pieroni, A.; Tardío, J.; Pardo-de-Santayana, M.; Sõukand, R.; Svanberg, I.; Kalle, R. Wild food plant use in 21st century Europe: The disappearance of old traditions and the search for new cuisines involving wild edibles. Acta Soc. Bot. Pol. 2012, 81, 359–370. [Google Scholar] [CrossRef]
- Łuczaj, L.; Pieroni, A. Nutritional Ethnobotany in Europe: From Emergency Foods to Healthy Folk Cuisines and Contemporary Foraging Trends. In Mediterranean Wild Edible Plants; Sánchez-Mata, M., Tardío, J., Eds.; Springer: New York, NY, USA, 2016; Chapter 3; pp. 33–56. [Google Scholar] [CrossRef]
- Ceccanti, C.; Landi, M.; Benvenuti, S.; Pardossi, A.; Guidi, L. Mediterranean wild edible plants: Weeds or “new functional crops”? Molecules 2018, 23, 2299. [Google Scholar] [CrossRef] [PubMed]
- Schulp, C.J.; Thuiller, W.; Verburg, P.H. Wild food in Europe: A synthesis of knowledge and data of terrestrial wild food as an ecosystem service. Ecol. Econ. 2014, 105, 292–305. [Google Scholar] [CrossRef]
- Marrelli, M.; Statti, G.; Conforti, F. A Review of Biologically Active Natural Products from Mediterranean Wild Edible Plants: Benefits in the Treatment of Obesity and Its Related Disorders. Molecules 2020, 25, 649. [Google Scholar] [CrossRef] [PubMed]
- Pinela, J.; Carvalho, A.M.; Ferreira, I.C.F.R. Wild edible plants: Nutritional and toxicological characteristics, retrieval strategies and importance for today’s society. Food Chem. Toxicol. 2017, 110, 165–188. [Google Scholar] [CrossRef]
- Pasta, S.; La Rosa, A.; Garfì, G.; Marcenò, C.; Gristina, A.S.; Carimi, F.; Guarino, R. An updated checklist of the sicilian native edible plants: Preserving the traditional ecological knowledge of century-old agro-pastoral landscapes. Front. Plant Sci. 2020, 11, 388. [Google Scholar] [CrossRef]
- Bacchetta, L.; Visioli, F.; Cappelli, G.; Caruso, E.; Martin, G.; Nemeth, E.; Bacchetta, G.; Bedini, G.; Wezel, A.; van Asseldonk, T.; et al. A manifesto for the valorization of wild edible plants. J. Ethnopharmacol. 2016, 191, 180–187. [Google Scholar] [CrossRef]
- Tardio, J.; Pardo De Santayana, M.; Morales, R. Ethnobotanical review of wild edible plants in Spain. Bot. J. Linn. Soc. 2006, 152, 27–71. [Google Scholar] [CrossRef]
- Geraci, A.; Amato, F.; Di Noto, G.; Bazan, G.; Schicchi, R. The wild taxa utilized as vegetables in Sicily (Italy): A traditional component of the Mediterranean diet. J. Ethnobiol. Ethnomed. 2018, 14, 14. [Google Scholar] [CrossRef]
- Ranfa, A.; Bodesmo, M. An Ethnobotanical investigation of traditional knowledge and uses of edible wild plants in the Umbria Region, Central Italy. J. Appl. Bot. Food Qual. 2017, 90, 246–258. [Google Scholar] [CrossRef]
- Licata, M.; Tuttolomondo, T.; Leto, C.; Virga, G.; Bonsangue, G.; Cammaleri, I.; Gennaro, M.C.; La Bella, S. A survey of wild plant species for food use in Sicily (Italy)—Results of a 3-year study in four Regional Parks. J. Ethnobiol. Ethnomed. 2016, 12, 12. [Google Scholar] [CrossRef] [PubMed]
- Hadjichambis, A.C.; Paraskeva-Hadjichambi, D.; Della, A.; Giusti, M.E.; De Pasquale, C.; Lenzarini, C.; Censorii, E.; Gonzales-Tejero, M.R.; Sanchez-Rojas, C.P.; Ramiro-Gutierrez, J.M.; et al. Wild and semi-domesticated food plant consumption in seven circum-Mediterranean areas. Int. J. Food Sci. Nutr. 2008, 59, 383–414. [Google Scholar] [CrossRef] [PubMed]
- Motti, R.; Bonanomi, G.; Lanzotti, V.; Sacchi, R. The contribution of wild edible plants to the mediterranean diet: An ethnobotanical case study along the coast of Campania (Southern Italy). Econ. Bot. 2020, 74, 249–272. [Google Scholar] [CrossRef]
- Fiorentino, M.; Gravina, C.; Piccolella, S.; Pecoraro, M.T.; Formato, M.; Stinca, A.; Pacifico, S.; Esposito, A. Calendula arvensis (Vaill.) L.: A systematic plant analysis of the polar extracts from its organs by UHPLC-HRMS. Foods 2022, 11, 247. [Google Scholar] [CrossRef]
- McSweeney, M.; Seetharaman, K. State of polyphenols in the drying process of fruits and vegetables. Crit. Rev. Food Sci. Nutr. 2015, 55, 660–669. [Google Scholar] [CrossRef] [PubMed]
- Diaconeasa, Z. Time-dependent degradation of polyphenols from thermally-processed berries and their in vitro antiproliferative effects against melanoma. Molecules 2018, 23, 2534. [Google Scholar] [CrossRef]
- Duque-Soto, C.; Quirantes-Piné, R.; Borrás-Linares, I.; Segura-Carretero, A.; Lozano-Sánchez, J. Characterization and influence of static in vitro digestion on bioaccessibility of bioactive polyphenols from an olive leaf extract. Foods 2022, 11, 743. [Google Scholar] [CrossRef]
- Neto, J.; de Almeida, T.; de Medeiros, J.; Vieira, L.; Moreira, T.; Maia, A.; Ribeiro, P.; de Brito, E.; Farias, D.; Carvalho, A. Impact of bioaccessibility and bioavailability of phenolic compounds in biological systems upon the antioxidant activity of the ethanolic extract of Triplaris gardneriana seeds. Biomed. Pharmacother. 2017, 88, 999–1007. [Google Scholar] [CrossRef]
- Ferreira-Santos, P.; Ibarz, R.; Fernandes, J.-M.; Pinheiro, A.C.; Botelho, C.; Rocha, C.M.R.; Teixeira, J.A.; Martín-Belloso, O. Encapsulated pine bark polyphenolic extract during gastrointestinal digestion: Bioaccessibility, bioactivity and oxidative stress prevention. Foods 2021, 10, 328. [Google Scholar] [CrossRef]
- Mazumder, M.A.R.; Ranganathan, T.V. Encapsulation of isoflavone with milk, maltodextrin and gum acacia improves its stability. Curr. Res. Food Sci. 2020, 2, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Buljeta, I.; Pichler, A.; Šimunović, J.; Kopjar, M. Polysaccharides as carriers of polyphenols: Comparison of freeze-drying and spray-drying as encapsulation techniques. Molecules 2022, 27, 5069. [Google Scholar] [CrossRef] [PubMed]
- Yadav, K.; Bajaj, R.K.; Mandal, S.; Mann, B. Encapsulation of grape seed extract phenolics using whey protein concentrate, maltodextrin and gum arabica blends. J. Food Sci. Technol. 2020, 57, 426–434. [Google Scholar] [CrossRef] [PubMed]
- Pashazadeh, H.; Zannou, O.; Ghellam, M.; Koca, I.; Galanakis, C.M.; Aldawoud, T.M.S. Optimization and encapsulation of phenolic compounds extracted from maize waste by freeze-drying, spray-drying, and microwave-drying using maltodextrin. Foods 2021, 10, 1396. [Google Scholar] [CrossRef] [PubMed]
- Ravichandran, K.; Palaniraj, R.; Saw, N.M.; Gabr, A.M.; Ahmed, A.R.; Knorr, D.; Smetanska, I. Effects of different encapsulation agents and drying process on stability of betalains extract. J. Food Sci. Technol. 2014, 51, 2216–2221. [Google Scholar] [CrossRef] [PubMed]
- Ghandehari Yazdi, A.P.; Barzegar, M.; Sahari, M.A.; Ahmadi Gavlighi, H. Encapsulation of pistachio green hull phenolic compounds by spray drying. J. Agr. Sci. Technol. 2021, 23, 51–64. [Google Scholar]
- Grgić, J.; Šelo, G.; Planinić, M.; Tišma, M.; Bucić-Kojić, A. Role of the encapsulation in bioavailability of phenolic compounds. Antioxidants 2020, 9, 923. [Google Scholar] [CrossRef]
- Wongsa, P.; Phatikulrungsun, P.; Prathumthong, S. FT-IR characteristics, phenolic profiles and inhibitory potential against digestive enzymes of 25 herbal infusions. Sci. Rep. 2022, 12, 6631. [Google Scholar] [CrossRef]
- Djikanović, D.; Devečerski, A.; Steinbach, G.; Simonović, J.; Matović, B.; Garab, G.; Kalauzi, A.; Radotić, K. Comparison of macromolecular interactions in the cell walls of hardwood, softwood and maize by fluorescence and FTIR spectroscopy, differential polarization laser scanning microscopy and X-ray diffraction. Wood Sci. Technol. 2016, 50, 547–566. [Google Scholar] [CrossRef]
- Todorović, A.; Šturm, L.; Salević-Jelić, A.; Lević, S.; Osojnik Črnivec, I.G.; Prislan, I.; Skrt, M.; Bjeković, A.; Poklar Ulrih, N.; Nedović, V. Encapsulation of bilberry extract with maltodextrin and gum arabic by freeze-drying: Formulation, characterisation, and storage stability. Processes 2022, 10, 1991. [Google Scholar] [CrossRef]
- Matwijczuk, A.; Budziak-Wieczorek, I.; Czernel, G.; Karcz, D.; Barańska, A.; Jedlińska, A.; Samborska, K. Classification of honey powder composition by FTIR spectroscopy coupled with chemometric analysis. Molecules 2022, 27, 3800. [Google Scholar] [CrossRef] [PubMed]
- Arnold, A.R.; Chassaing, B. Maltodextrin, Modern Stressor of the Intestinal Environment. Cell. Mol. Gastroenterol. Hepatol. 2019, 7, 475–476. [Google Scholar] [CrossRef] [PubMed]
- Jovanović Galović, A.; Jovanović Lješković, N.; Vidović, S.; Vladić, J.; Jojić, N.; Ilić, M.; Srdić Rajić, T.; Kojić, V.; Jakimov, D. The Effects of Resveratrol-Rich Extracts of Vitis vinifera Pruning Waste on HeLa, MCF-7 and MRC-5 Cells: Apoptosis, Autophagia and Necrosis Interplay. Pharmaceutics 2022, 14, 2017. [Google Scholar] [CrossRef]
- Desai, N.M.; Haware, D.J.; Basavaraj, K.; Murthy, P.S. Microencapsulation of antioxidant phenolic compounds from green coffee. Prep. Biochem. Biotechnol. 2019, 49, 400–406. [Google Scholar] [CrossRef] [PubMed]
- Zorzenon, M.R.T.; Formigoni, M.; da Silva, S.B.; Hodas, F.; Piovan, S.; Ciotta, S.R.; Jansen, C.A.; Dacome, A.S.; Pilau, E.J.; Mareze-Costa, C.E.; et al. Spray drying encapsulation of stevia extract with maltodextrin and evaluation of the physicochemical and functional properties of produced powders. J. Food Sci. 2020, 85, 3590–3600. [Google Scholar] [CrossRef] [PubMed]
- Pacifico, S.; Piccolella, S.; Nocera, P.; Tranquillo, E.; Dal Poggetto, F.; Catauro, M. New insights into phenol and polyphenol composition of Stevia rebaudiana leaves. J. Pharm. Biomed. Anal. 2019, 163, 45–57. [Google Scholar] [CrossRef]
- Tsagkaris, A.S.; Louckova, A.; Jaegerova, T.; Tokarova, V.; Hajslova, J. The in vitro inhibitory effect of selected Asteraceae plants on pancreatic lipase followed by phenolic content identification through Liquid Chromatography High Resolution Mass Spectrometry (LC-HRMS). Int. J. Mol. Sci. 2022, 23, 11204. [Google Scholar] [CrossRef] [PubMed]
- Grati, W.; Samet, S.; Bouzayani, B.; Ayachi, A.; Treilhou, M.; Téné, N.; Mezghani-Jarraya, R. HESI-MS/MS Analysis of Phenolic Compounds from Calendula aegyptiaca fruits extracts and evaluation of their antioxidant activities. Molecules 2022, 27, 2314. [Google Scholar] [CrossRef]
- Kannan, R.; Arumugam, R.; Anantharaman, P. Fourier Transform Infrared Spectroscopy analysis of seagrass polyphenols. Curr. Bioact. Compd. 2011, 7, 118–125. [Google Scholar] [CrossRef]
- Sinyayev, V.; Gulparshyn, A.; Toxeitova, G.; Aigul, A.; Batyrbayeva, A.; Sassykova, L.; Azhigulova, R.; Sakhipov, Y. A comparative investigation of the IR spectra of a carbohydrate series. J. Chem. Technol. Metall. 2020, 55, 724–729. [Google Scholar]
- Böttcher, S.; Drusch, S. Saponins-Self-assembly and behavior at aqueous interfaces. Adv. Colloid Interface Sci. 2017, 243, 105–113. [Google Scholar] [CrossRef]
- Zeng, L.; Xiang, R.; Fu, C.; Qu, Z.; Liu, C. The Regulatory effect of chlorogenic acid on gut-brain function and its mechanism: A systematic review. Biomed. Pharmacother. 2022, 149, 112831. [Google Scholar] [CrossRef] [PubMed]
- Majdoub, Y.O.E.; Ginestra, G.; Mandalari, G.; Dugo, P.; Mondello, L.; Cacciola, F. The digestibility of Hibiscus sabdariffa L. polyphenols using an in vitro human digestion model and evaluation of their antimicrobial activity. Nutrients 2021, 13, 2360. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Deng, Z.; Zhang, J.; Dong, H.; Wang, W.; Xing, B.; Liu, X. Comparison of flavonoid O-glycoside, C-glycoside and their aglycones on antioxidant capacity and metabolism during in vitro digestion and in vivo. Foods 2022, 11, 882. [Google Scholar] [CrossRef] [PubMed]
- Spencer, J.P. Metabolism of tea flavonoids in the gastrointestinal tract. J. Nutr. 2003, 133, 3255S–3261S. [Google Scholar] [CrossRef]
- Tarko, T.; Duda-Chodak, A.; Zajac, N. Digestion and absorption of phenolic compounds assessed by in vitro simulation methods. A review. Rocz Panstw Zakl Hig. 2013, 64, 79–84. [Google Scholar]
- Vergara, C.; Pino, M.T.; Zamora, O.; Parada, J.; Pérez, R.; Uribe, M.; Kalazich, J. Microencapsulation of anthocyanin extracted from purple flesh cultivated potatoes by spray drying and its effects on in vitro gastrointestinal digestion. Molecules 2020, 25, 722. [Google Scholar] [CrossRef]
- Formato, M.; Piccolella, S.; Zidorn, C.; Vastolo, A.; Calabrò, S.; Cutrignelli, M.I.; Pacifico, S. UHPLC-ESI-QqTOF analysis and in vitro rumen fermentation for exploiting Fagus sylvatica leaf in ruminant diet. Molecules 2022, 27, 2217. [Google Scholar] [CrossRef]
- Abdel-Aty, A.M.; Barakat, A.Z.; Mohamed, S.A. Garden cress gum and maltodextrin as microencapsulation coats for entrapment of garden cress phenolic-rich extract: Improved thermal stability, storage stability, antioxidant and antibacterial activities. Food Sci. Biotechnol. 2022. [Google Scholar] [CrossRef]
- Borrelli, F.; Pagano, E.; Formisano, C.; Piccolella, S.; Fiorentino, A.; Tenore, G.C.; Izzo, A.A.; Rigano, D.; Pacifico, S. Hyssopus officinalis subsp. aristatus: An unexploited wild-growing crop for new disclosed bioactives. Ind. Crops Prod. 2019, 140, 111594. [Google Scholar] [CrossRef]
- Formato, M.; Piccolella, S.; Zidorn, C.; Pacifico, S. UHPLC-HRMS Analysis of Fagus sylvatica (Fagaceae) Leaves: A Renewable Source of Antioxidant Polyphenols. Antioxidants 2021, 10, 1140. [Google Scholar] [CrossRef] [PubMed]
- Minekus, M.; Alminger, M.; Alvito, P.; Balance, S.; Bohn, T.; Bourlieu, C.; Carrière, F.; Boutrou, R.; Corredig, M.; Dupont, D.; et al. A standardised static in vitro digestion method suitable for food—An international consensus. Food Funct. 2014, 5, 1113–1124. [Google Scholar] [CrossRef] [PubMed]
- Piccolella, S.; Bianco, A.; Crescente, G.; Santillo, A.; Chieffi Baccari, G.; Pacifico, S. Recovering Cucurbita pepo cv. ‘Lungo Fiorentino’ wastes: UHPLC-HRMS/MS metabolic profile, the basis for establishing their nutra- and cosmeceutical valorisation. Molecules 2019, 24, 1479. [Google Scholar] [CrossRef] [PubMed]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).