
Epimeric Mixture Analysis and Absolute Configuration Determination Using an Integrated Spectroscopic and Computational Approach—A Case Study of Two Epimers of 6-Hydroxyhippeastidine
 , , , and
, , , and
Abstract
:1. Introduction
2. Results
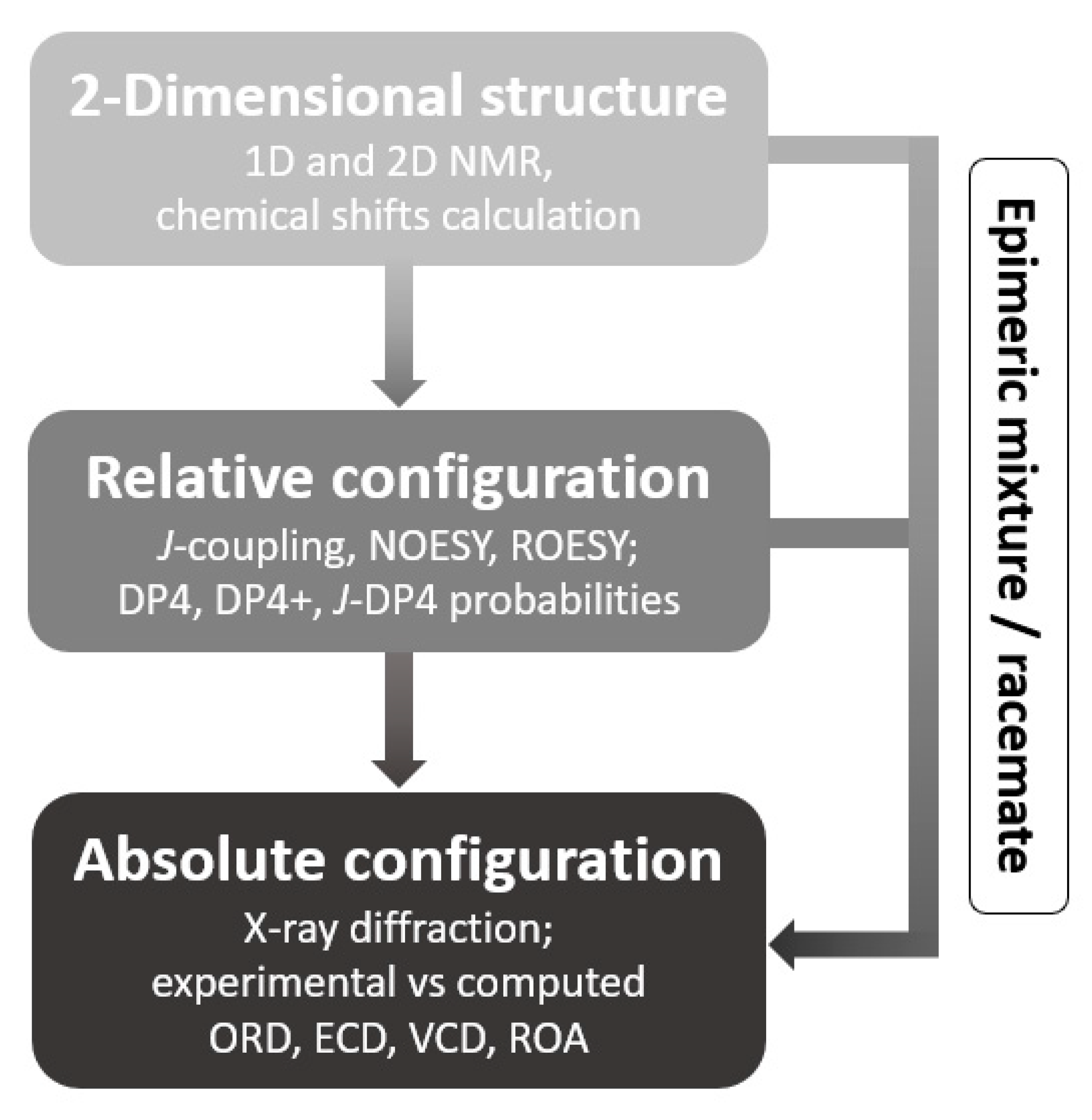
2.1. 2-Dimensional Structure Elucidation
2.2. Relative Configuration Determination
2.3. Absolute Configuration Determination
- -
- the averaged OR of the crinine-type = = 47.9;
- -
- the averaged OR of the haemanthamine-type = = −47.9.
- -
- the averaged OR of the crinine-type = = 47.8;
- -
- the averaged OR of the haemanthamine-type = = −47.9.
3. Discussion
4. Materials and Methods
4.1. General Experimental Procedure
4.2. Isolation and Purification
4.3. Computational Details
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Newman, D.J.; Cragg, G.M. Natural Products as Sources of New Drugs over the Nearly Four Decades from 01/1981 to 09/2019. J. Nat. Prod. 2020, 83, 770–803. [Google Scholar] [CrossRef] [Green Version]
- Lodewyk, M.W.; Siebert, M.R.; Tantillo, D.J. Computational Prediction of 1H and 13C Chemical Shifts: A Useful Tool for Natural Product, Mechanistic, and Synthetic Organic Chemistry. Chem. Rev. 2012, 112, 1839–1862. [Google Scholar] [CrossRef]
- Merrill, A.T.; Tantillo, D.J. Solvent Optimization and Conformational Flexibility Effects on 1H and 13C NMR Scaling Factors. Magn. Reson. Chem. 2020, 58, 576–583. [Google Scholar] [CrossRef]
- Smith, S.G.; Goodman, J.M. Assigning Stereochemistry to Single Diastereoisomers by GIAO NMR Calculation: The DP4 Probability. J. Am. Chem. Soc. 2010, 132, 12946–12959. [Google Scholar] [CrossRef]
- Ermanis, K.; Parkes, K.E.B.; Agback, T.; Goodman, J.M. Doubling the Power of DP4 for Computational Structure Elucidation. Org. Biomol. Chem. 2017, 15, 8998–9007. [Google Scholar] [CrossRef] [Green Version]
- Cooper, J.K.; Li, K.; Aubé, J.; Coppage, D.A.; Konopelski, J.P. Application of the DP4 Probability Method to Flexible Cyclic Peptides with Multiple Independent Stereocenters: The True Structure of Cyclocinamide A. Org. Lett. 2018, 20, 4314–4317. [Google Scholar] [CrossRef]
- Sarotti, A.M. In Silico Reassignment of (+)-Diplopyrone by NMR Calculations: Use of a DP4/J-DP4/DP4+/DIP Tandem to Revise Both Relative and Absolute Configuration. J. Org. Chem. 2020, 85, 11566–11570. [Google Scholar] [CrossRef]
- De Gussem, E.; Tehrani, K.A.; Herrebout, W.A.; Bultinck, P.; Johannessen, C. Comparative Study of the Vibrational Optical Activity Techniques in Structure Elucidation: The Case of Galantamine. ACS Omega 2019, 4, 14133–14139. [Google Scholar] [CrossRef] [Green Version]
- Grimblat, N.; Zanardi, M.M.; Sarotti, A.M. Beyond DP4: An Improved Probability for the Stereochemical Assignment of Isomeric Compounds Using Quantum Chemical Calculations of NMR Shifts. J. Org. Chem. 2015, 80, 12526–12534. [Google Scholar] [CrossRef]
- Zanardi, M.M.; Sarotti, A.M. Sensitivity Analysis of DP4+ with the Probability Distribution Terms: Development of a Universal and Customizable Method. J. Org. Chem. 2021, 86, 8544–8548. [Google Scholar] [CrossRef]
- Marcarino, M.O.; Cicetti, S.; Zanardi, M.M.; Sarotti, A.M. A Critical Review on the Use of DP4+ in the Structural Elucidation of Natural Products: The Good, the Bad and the Ugly. A Practical Guide. Nat. Prod. Rep. 2021, 39, 58–76. [Google Scholar] [CrossRef] [PubMed]
- Alexander, H.; Goodman, J.M. The DP5 probability, quantification and visualisation of structural uncertainty in single molecules. Chem. Sci. 2022, 13, 3507–3518. [Google Scholar]
- Howarth, A.; Ermanis, K.; Goodman, J.M. DP4-AI Automated NMR Data Analysis: Straight from Spectrometer to Structure. Chem. Sci. 2020, 11, 4351–4359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willoughby, P.H.; Jansma, M.J.; Hoye, T.R. A Guide to Small-Molecule Structure Assignment through Computation of (1 H and 13 C) NMR Chemical Shifts. Nat. Protoc. 2014, 9, 643–660. [Google Scholar] [CrossRef]
- Navarro-Vazquez, A.; Gil, R.R.; Blinov, K. Computer-Assisted 3D Structure Elucidation (CASE-3D) of Natural Products Combining Isotropic and Anisotropic NMR Parameters. J. Nat. Prod. 2018, 81, 203–210. [Google Scholar] [CrossRef]
- Troche-Pesqueira, E.; Anklin, C.; Gil, R.R.; Navarro-Vázquez, A. Computer-Assisted 3D Structure Elucidation of Natural Products Using Residual Dipolar Couplings. Angew. Chem. Int. Ed. 2017, 56, 3660–3664. [Google Scholar] [CrossRef]
- Soong, R.; Pautler, B.G.; Moser, A.; Jenne, A.; Lysak, D.H.; Adamo, A.; Simpson, A.J. CASE (Computer-Assisted Structure Elucidation) Study for an Undergraduate Organic Chemistry Class; ACS Publications: Washington, DC, USA, 2020. [Google Scholar]
- Nuzillard, J.; Plainchont, B. Tutorial for the Structure Elucidation of Small Molecules by Means of the LSD Software. Magn. Reson. Chem. 2018, 56, 458–468. [Google Scholar] [CrossRef]
- Lodewyk, M.W.; Tantillo, D.J. Prediction of the Structure of Nobilisitine A Using Computed NMR Chemical Shifts. J. Nat. Prod. 2011, 74, 1339–1343. [Google Scholar] [CrossRef]
- Burns, D.C.; Mazzola, E.P.; Reynolds, W.F. The Role of Computer-Assisted Structure Elucidation (CASE) Programs in the Structure Elucidation of Complex Natural Products. Nat. Prod. Rep. 2019, 36, 919–933. [Google Scholar] [CrossRef]
- Berova, N.; Polavarapu, P.L.; Nakanishi, K.; Woody, R.W. Comprehensive Chiroptical Spectroscopy: Applications in Stereochemical Analysis of Synthetic Compounds, Natural Products, and Biomolecules; John Wiley & Sons: Hoboken, NJ, USA, 2012; Volume 2. [Google Scholar]
- Superchi, S.; Rosini, C.; Mazzeo, G.; Giorgio, E. Determination of Molecular Absolute Configuration: Guidelines for Selecting a Suitable Chiroptical Approach. Compr. Chiroptical Spectrosc. 2012, 2, 421–448. [Google Scholar]
- Bohle, F.; Seibert, J.; Grimme, S. Automated Quantum Chemistry-Based Calculation of Optical Rotation for Large Flexible Molecules. J. Org. Chem. 2021, 86, 15522–15531. [Google Scholar] [CrossRef] [PubMed]
- Khatri Chhetri, B.; Lavoie, S.; Sweeney-Jones, A.M.; Mojib, N.; Raghavan, V.; Gagaring, K.; Dale, B.; McNamara, C.W.; Soapi, K.; Quave, C.L.; et al. Peyssonnosides A-B, Unusual Diterpene Glycosides with a Sterically Encumbered Cyclopropane Motif: Structure Elucidation Using an Integrated Spectroscopic and Computational Workflow. J. Org. Chem. 2019, 84, 8531–8541. [Google Scholar] [CrossRef] [PubMed]
- Batista, J.M.; Blanch, E.W.; Da Silva Bolzani, V. Recent Advances in the Use of Vibrational Chiroptical Spectroscopic Methods for Stereochemical Characterization of Natural Products. Nat. Prod. Rep. 2015, 32, 1280–1302. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Zhao, D.; Wu, S.-J.; Wang, J.; Jia, Y.-J.; Li, W.-X.; Zhu, H.-J.; Cao, F.; Li, W.; Pittman, C.U. Reassigning the Stereochemistry of Bioactive Cepharanthine Using Calculated versus Experimental Chiroptical Spectroscopies. Tetrahedron 2019, 75, 1194–1202. [Google Scholar] [CrossRef]
- Pettit, G.R.; Eastham, S.A.; Melody, N.; Orr, B.; Herald, D.L.; McGregor, J.; Knight, J.C.; Doubek, D.L.; Pettit, G.R.; Garner, L.C.; et al. Isolation and Structural Modification of 7-Deoxynarciclasine and 7-Deoxy-Trans-Dihydronarciclasine,1. J. Nat. Prod. 2006, 69, 7–13. [Google Scholar] [CrossRef]
- Chhetri, B.K.; Lavoie, S.; Sweeney-Jones, A.M.; Kubanek, J. Recent Trends in the Structural Revision of Natural Products. Nat. Prod. Rep. 2018, 35, 514–531. [Google Scholar] [CrossRef]
- Yoo, H.-D.; Nam, S.-J.; Chin, Y.-W.; Kim, M.-S. Misassigned Natural Products and Their Revised Structures. Arch. Pharm. Res. 2016, 39, 143–153. [Google Scholar] [CrossRef]
- Paul, D.; Kundu, A.; Saha, S.; Goswami, R.K. Total Synthesis: The Structural Confirmation of Natural Products. Chem. Commun. 2021, 57, 3307–3322. [Google Scholar] [CrossRef]
- Le, H.T.N.; Van Roy, E.; Dendooven, E.; Peeters, L.; Theunis, M.; Foubert, K.; Pieters, L.; Tuenter, E. Alkaloids from Lepidium Meyenii (Maca), Structural Revision of Macaridine and UPLC-MS/MS Feature-Based Molecular Networking. Phytochemistry 2021, 190, 112863. [Google Scholar] [CrossRef]
- Batista, A.N.L.; Angrisani, B.R.P.; Lima, M.E.D.; Da Silva, S.M.P.; Schettini, V.H.; Chagas, H.A.; dos Santos, F.M., Jr.; Batista, J.M., Jr.; Valverde, A.L. Absolute Configuration Reassignment of Natural Products: An Overview of the Last Decade. J. Braz. Chem. Soc. 2021, 32, 1499–1518. [Google Scholar] [CrossRef]
- Baldwin, S.W.; Debenham, J.S. Total Syntheses of (−)-Haemanthidine, (+)-Pretazettine, and (+)-Tazettine. Org. Lett. 2000, 2, 99–102. [Google Scholar] [CrossRef] [PubMed]
- Shawky, E.; Hammoda, H.; Abou Donia, A.; Toaima, S.; Kinoshita, E.; Takayama, H. Phytochemical and Biological Investigation of Narcissus Pseudonarcissus Cultivated in Egypt. Rec. Pharm. Biomed. Sci. 2018, 2, 26–34. [Google Scholar] [CrossRef]
- Shitara, N.; Hirasawa, Y.; Hasumi, S.; Sasaki, T.; Matsumoto, M.; Wong, C.P.; Kaneda, T.; Asakawa, Y.; Morita, H. Four New Amaryllidaceae Alkaloids from Zephyranthes Candida. J. Nat. Med. 2014, 68, 610–614. [Google Scholar] [CrossRef] [PubMed]
- Wagner, J.; Pham, H.L.; Döpke, W. Alkaloids from Hippeastrum Equestre Herb.—5. Circular Dichroism Studies. Tetrahedron 1996, 52, 6591–6600. [Google Scholar] [CrossRef]
- Bessa, C.D.P.B.; de Andrade, J.P.; de Oliveira, R.S.; Domingos, E.; Santos, H.; Romão, W.; Bastida, J.; Borges, W.S. Identification of Alkaloids from Hippeastrum Aulicum (Ker Gawl.) Herb.(Amaryllidaceae) Using CGC-MS and Ambient Ionization Mass Spectrometry (PS-MS and LS-MS). J. Braz. Chem. Soc. 2017, 28, 819–830. [Google Scholar] [CrossRef]
- Kepplinger, B.; Mardiana, L.; Cowell, J.; Morton-Laing, S.; Dashti, Y.; Wills, C.; Marrs, E.C.L.; Perry, J.D.; Gray, J.; Goodfellow, M.; et al. Discovery, Isolation, Heterologous Expression and Mode-of Action Studies of the Antibiotic Polyketide Tatiomicin from Amycolatopsis sp. DEM30355. Sci. Rep. 2022, 12, 15579. [Google Scholar] [CrossRef]
- Polavarapu, P.; Donahue, E.; Shanmugam, G.; Scalmani, G.; Hawkins, E.; Rizzo, C.; Ibnusaud, I.; Thomas, G.; Habel, D.; Sebastian, D. A single chiroptical spectroscopic method may not be able to establish the absolute configurations of diastereomers: Dimethylesters of hibiscus and garcinia acids. J. Phys. Chem. A 2011, 115, 5665–5673. [Google Scholar] [CrossRef] [Green Version]
- Prasad, P.L.; Covington, C.L. Comparison of experimental and calculated chiroptical spectra for chiral molecular structure determination. Chirality 2014, 26, 539–552. [Google Scholar]
- Polavarapu Prasad, L. Molecular structure determination using chiroptical spectroscopy: Where we may go wrong? Chirality 2012, 24, 909–920. [Google Scholar] [CrossRef]
- Polavarapu Prasad, L.; Santoro, E. Vibrational optical activity for structural characterization of natural products. Nat. Prod. Rep. 2020, 37, 1661–1699. [Google Scholar] [CrossRef]
- Pescitelli, G.; Bruhn, T. Good Computational Practice in the Assignment of Absolute Configurations by TDDFT Calculations of ECD Spectra. Chirality 2016, 28, 466–474. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Cao, Y.; Song, H.; Wang, X.; Yao, S. Calculations of Optical Rotation: Influence of Molecular Structure. J. Serbian Chem. Soc. 2012, 77, 887–898. [Google Scholar] [CrossRef]
- Stephens, P.J.; Devlin, F.J.; Cheeseman, J.R.; Frisch, M.J. Calculation of Optical Rotation Using Density Functional Theory. J. Phys. Chem. A 2001, 105, 5356–5371. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Experimental | Calculated | |||||
|---|---|---|---|---|---|---|
| Structure Proposal 1a | Structure Proposal 1b | |||||
| Position | δC, Type | δH (J in Hz) | δC | δH | δC | δH |
| 1α | 27.1, CH2 | 1.55 | 27.7 | 1.60 | 27.9 | 1.63 |
| 1β | 27.1, CH2 | 3.12 * | 27.7 | 2.99 | 27.9 | 2.94 |
| 2α | 27.9, CH2 | 1.90 m | 27.9 | 1.90 | 28.2 | 1.88 |
| 2β | 27.9, CH2 | 1.30 m | 27.9 | 1.32 | 28.2 | 1.29 |
| 3 | 77.6, CH | 3.10 * | 76.7 | 3.01 | 77.1 | 3.03 |
| 4α | 33.7, CH2 | 1.82 m | 33.2 | 1.76 | 33.5 | 1.76 |
| 4β | 33.7, CH2 | 1.05 q (11.8) | 33.2 | 1.10 | 33.5 | 1.08 |
| 4a | 61.3, CH | 3.07 * | 62.4 | 2.99 | 62.5 | 3.01 |
| 6 | 88.8, CH | 4.72 s | 88.9 | 4.68 | 89.0 | 4.75 |
| 6a | 132.0, C | - | 127.4 | - | 132.8 | - |
| 7 | 103.8, CH | 6.32 s | 106.2 | 6.45 | 103.5 | 6.36 |
| 8 | 150.9, C | - | 146.3 | - | 151.4 | - |
| 9 | 135.8, C | - | 144.6 | - | 134.8 | - |
| 10 | 147.7, C | - | 140.9 | - | 147.9 | - |
| 10a | 127.3, C | - | 137.2 | - | 128.4 | - |
| 10b | 43.3, C | - | 46.2 | - | 46.1 | - |
| 11 | 33.3, CH2 | 2.06 m | 34.8 | 2.11 | 34.5 | 2.08 |
| 11 | 33.3, CH2 | 1.51 * | 34.8 | 1.61 | 34.5 | 1.58 |
| 12 | 47.3, CH2 | 3.12 * | 47.5 | 3.11 | 47.8 | 3.13 |
| 12 | 47.3, CH2 | 2.52 | 47.5 | 2.55 | 47.8 | 2.56 |
| 3-OMe | 55.3, CH3 | 3.24 s | 53.2 | 3.20 | 53.5 | 3.22 |
| 8-OMe | 55.9, CH3 | 3.72 s | 52.9 | 3.68 | 53.2 | 3.65 |
| 9-OMe | 60.6, CH3 | 3.63 s | 55.7 | 3.70 | 57.8 | 3.74 |
| CMAE | 3.0 | 0.06 | 1.04 | 0.06 | ||
| Max. outlier | 9.9 | 0.13 | 2.83 | 0.18 | ||
| Experimental | Calculated | |||||
|---|---|---|---|---|---|---|
| Structure Proposal 2a | Structure Proposal 2b | |||||
| Position | δC, Type | δH (J in Hz) | δC | δH | δC | δH |
| 1α | 27.1 | 1.55 | 27.6 | 1.63 | 28.0 | 1.66 |
| 1β | 27.1 | 3.12 * | 27.6 | 2.98 | 28.0 | 2.95 |
| 2α | 27.9 | 1.90 m | 27.8 | 1.89 | 28.0 | 1.88 |
| 2β | 27.9 | 1.30 m | 27.8 | 1.32 | 28.0 | 1.29 |
| 3 | 77.3 | 3.10 * | 76.6 | 3.01 | 76.8 | 3.03 |
| 4α | 33.9 | 1.88 m | 33.4 | 1.85 | 33.7 | 1.84 |
| 4β | 33.9 | 1.13 q (11.8) | 33.4 | 1.16 | 33.7 | 1.16 |
| 4a | 66.3 | 2.95 | 67.0 | 2.85 | 67.1 | 2.88 |
| 6 | 86.8 | 5.43 s | 87.8 | 5.37 | 87.9 | 5.42 |
| 6a | 133.1 | - | 128.5 | - | 134.0 | - |
| 7 | 102.3 | 6.44 s | 104.3 | 6.63 | 101.7 | 6.50 |
| 8 | 150.8 | - | 146.3 | - | 151.3 | - |
| 9 | 135.6 | - | 144.4 | - | 134.6 | - |
| 10 | 147.4 | - | 140.7 | - | 147.7 | - |
| 10a | 126.5 | - | 136.0 | - | 127.0 | - |
| 10b | 44.3 | - | 47.2 | - | 47.2 | - |
| 11 | 35.0 | 2.13 m | 37.1 | 2.20 | 36.6 | 2.17 |
| 11 | 35.0 | 1.48 * | 37.1 | 1.54 | 36.6 | 1.52 |
| 12 | 41.9 | 3.17 * | 41.6 | 3.20 | 42.2 | 3.21 |
| 12 | 41.9 | 2.76 | 41.6 | 2.69 | 42.2 | 2.72 |
| 3-OMe | 55.3 | 3.24 s | 53.1 | 3.21 | 53.4 | 3.23 |
| 8-OMe | 55.9 | 3.72 s | 52.8 | 3.69 | 53.1 | 3.65 |
| 9-OMe | 60.6 | 3.63 s | 55.6 | 3.70 | 57.7 | 3.74 |
| CMAE | 3.1 | 0.06 | 1.09 | 0.06 | ||
| Max. outlier | 9.5 | 0.16 | 2.94 | 0.17 | ||
| Compound 1 | Compound 2 | ||
|---|---|---|---|
| Diastereomer | Probability (%) | Diastereomer | Probability (%) |
| RRRRR | 100 | RRRRR | 0 |
| RRRSR | 0 | RRRSR | 100 |
| RSRRR | 0 | RSRRR | 0 |
| RSRSR | 0 | RSRSR | 0 |
| SRRRR | 0 | SRRRR | 0 |
| SRRSR | 0 | SRRSR | 0 |
| SSRRR | 0 | SSRRR | 0 |
| SSRSR | 0 | SSRSR | 0 |
| Diastereomer | OR | Theory Level | |
|---|---|---|---|
| Experimental | 43.7 | ||
| Computed | RRRRR | 80.3 | B3LYP/6-31++G(d,p)// 6-311++G(3df,2dp) |
| RRRSR | −54.2 | ||
| SSSSS | −80.3 | ||
| SSSRS | 54.1 | ||
| RRRRR | 80.1 | B3LYP/6-31++G(d,p)// aug-cc-pVTZ | |
| RRRSR | −53.7 | ||
| SSSSS | −80.1 | ||
| SSSRS | 53.5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Le, N.-T.-H.; Vermeyen, T.; Aerts, R.; Herrebout, W.A.; Pieters, L.; Tuenter, E. Epimeric Mixture Analysis and Absolute Configuration Determination Using an Integrated Spectroscopic and Computational Approach—A Case Study of Two Epimers of 6-Hydroxyhippeastidine. Molecules 2023, 28, 214. https://doi.org/10.3390/molecules28010214
Le N-T-H, Vermeyen T, Aerts R, Herrebout WA, Pieters L, Tuenter E. Epimeric Mixture Analysis and Absolute Configuration Determination Using an Integrated Spectroscopic and Computational Approach—A Case Study of Two Epimers of 6-Hydroxyhippeastidine. Molecules. 2023; 28(1):214. https://doi.org/10.3390/molecules28010214
Chicago/Turabian StyleLe, Ngoc-Thao-Hien, Tom Vermeyen, Roy Aerts, Wouter A. Herrebout, Luc Pieters, and Emmy Tuenter. 2023. "Epimeric Mixture Analysis and Absolute Configuration Determination Using an Integrated Spectroscopic and Computational Approach—A Case Study of Two Epimers of 6-Hydroxyhippeastidine" Molecules 28, no. 1: 214. https://doi.org/10.3390/molecules28010214