
Traditional Machine and Deep Learning for Predicting Toxicity Endpoints
Abstract
:1. Introduction
2. Materials and Methods
2.1. CATMoS Datasets
2.2. Feature Generation
2.2.1. Structure Standardization
2.2.2. RDKit Descriptors
2.2.3. CDDD Descriptors
2.3. Traditional Machine Learning
2.4. Deep Learning
2.4.1. MolBERT
2.4.2. Molecular-Graph-BERT
2.5. Conformal Prediction
3. Results and Discussion

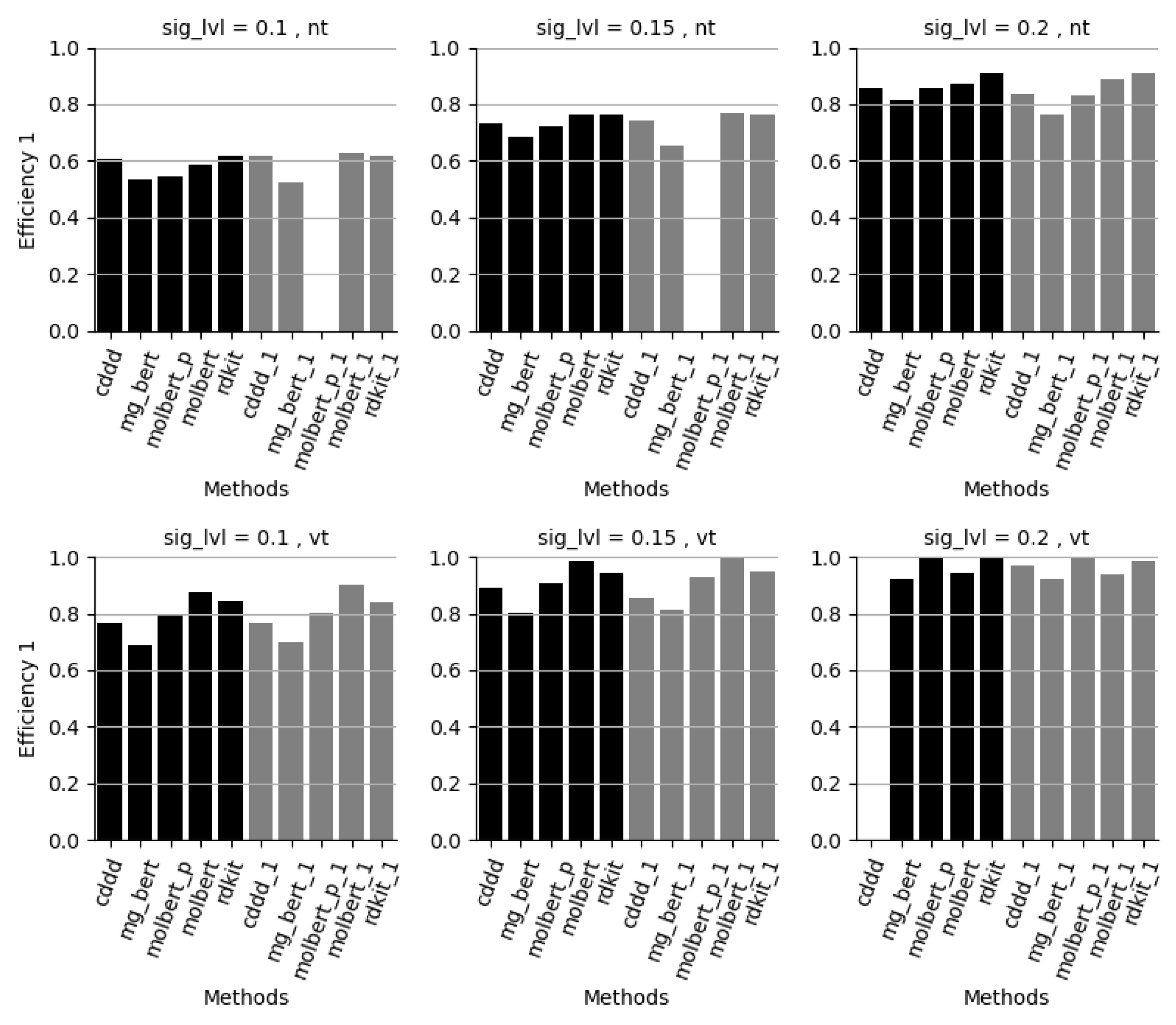
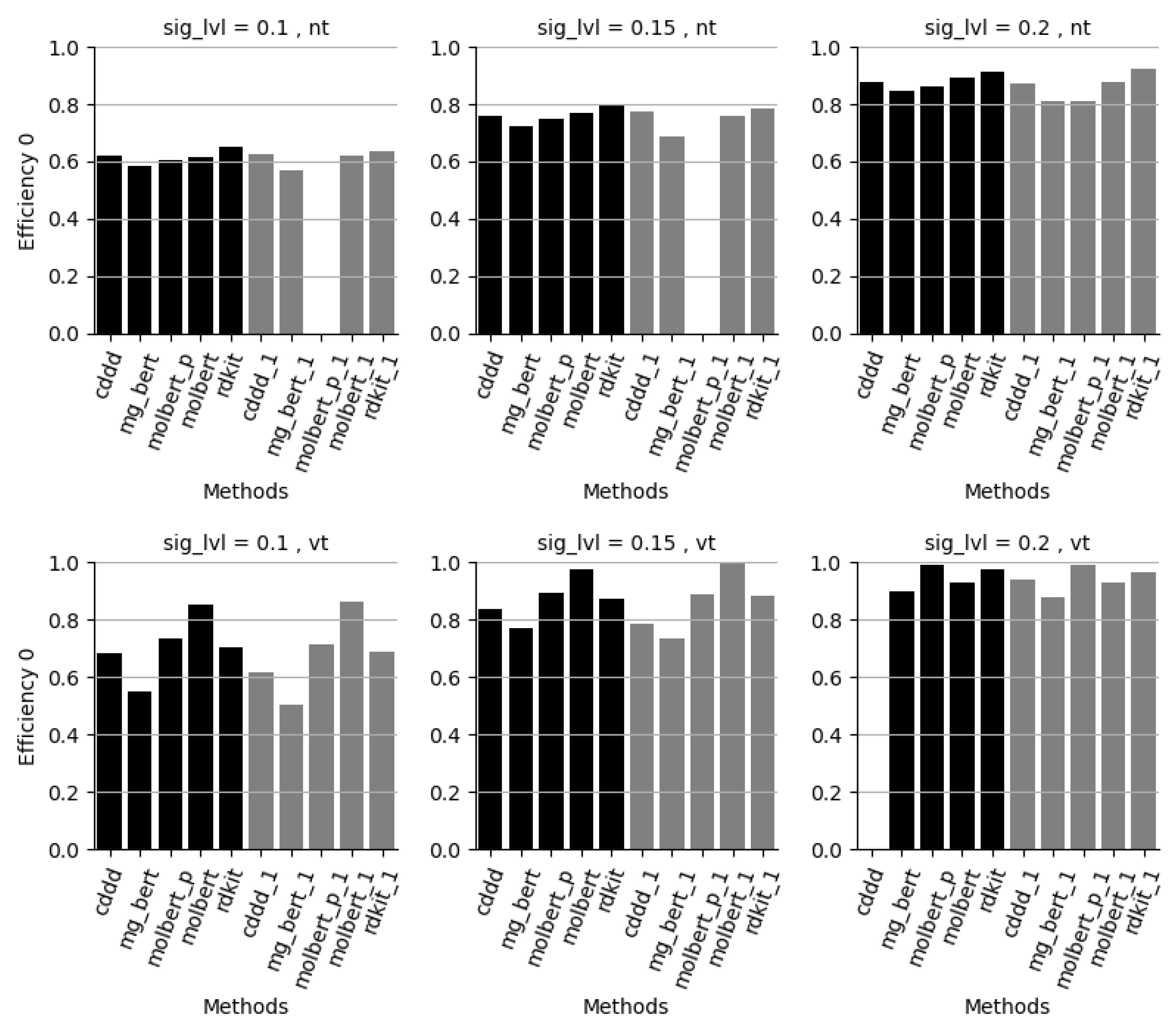
4. Conclusions
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- DiMasi, J.A.; Grabowski, H.G.; Hansen, R.W. Innovation in the pharmaceutical industry: New estimates of R&D costs. J. Health Econ. 2016, 47, 20–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, T.J.; Carpenter, D.; Lauffenburger, J.; Wang, B.; Franklin, J.M.; Kesselheim, A. Failure of Investigational Drugs in Late-Stage Clinical Development and Publication of Trial Results. JAMA Intern. Med. 2016, 176, 1826–1833. [Google Scholar] [CrossRef] [PubMed]
- Schaduangrat, N.; Lampa, S.; Simeon, S.; Gleeson, M.P.; Spjuth, O.; Nantasenamat, C. Towards reproducible computational drug discovery. J. Cheminform. 2020, 12, 9. [Google Scholar] [CrossRef] [Green Version]
- Sabe, V.T.; Ntombela, T.; Jhamba, L.A.; Maguire, G.E.; Govender, T.; Naicker, T.; Kruger, H.G. Current trends in computer aided drug design and a highlight of drugs discovered via computational techniques: A review. Eur. J. Med. Chem. 2021, 224, 113705. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Li, X.; Lin, X. A Review on Applications of Computational Methods in Drug Screening and Design. Molecules 2020, 25, 1375. [Google Scholar] [CrossRef] [Green Version]
- Brogi, S.; Ramalho, T.C.; Kuca, K.; Medina-Franco, J.L.; Valko, M. Editorial: In silico Methods for Drug Design and Discovery. Front. Chem. 2020, 8, 612. [Google Scholar] [CrossRef] [PubMed]
- Muratov, E.N.; Bajorath, J.; Sheridan, R.P.; Tetko, I.V.; Filimonov, D.; Poroikov, V.; Oprea, T.I.; Baskin, I.I.; Varnek, A.; Roitberg, A.; et al. QSAR without borders. Chem. Soc. Rev. 2020, 49, 3525–3564. [Google Scholar] [CrossRef]
- Cox, P.B.; Gupta, R. Contemporary Computational Applications and Tools in Drug Discovery. ACS Med. Chem. Lett. 2022, 13, 1016–1029. [Google Scholar] [CrossRef]
- Idakwo, G.; Luttrell, J.; Chen, M.; Hong, H.; Zhou, Z.; Gong, P.; Zhang, C. A review on machine learning methods for in silico toxicity prediction. J. Environ. Sci. Health C Environ. Carcinog. Ecotoxicol. Rev. 2018, 36, 169–191. [Google Scholar] [CrossRef]
- Pérez, S.E.; Rodríguez, S.R.; González, G.M.; Del Mar García Suárez, M.; Díaz, G.D.B.; Cabal, M.D.C.; Rojas, J.M.M.; López Sánchez, J.I. Toxicity prediction based on artificial intelligence: A multidisciplinary overview. WIREs Comput. Mol. Sci. 2021, 11, e1516. [Google Scholar]
- Dara, S.; Dhamercherla, S.; Jadav, S.S.; Babu, C.M.; Ahsan, M.J. Machine Learning in Drug Discovery: A Review. Artif. Intell. Rev. 2021, 55, 1947–1999. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaka, Y.; Yashiro, R. Applications of Deep Learning for Drug Discovery Systems with BigData. Biomedinformatics 2022, 2, 603–624. [Google Scholar] [CrossRef]
- Young, T.; Hazarika, D.; Poria, S.; Cambria, E. Recent Trends in Deep Learning Based Natural Language Processing. IEEE Comput. Intell. Mag. 2018, 13, 55–75. [Google Scholar] [CrossRef]
- Yang, B.; Xu, S.; Chen, H.; Zheng, W.; Liu, C. Reconstruct Dynamic Soft-Tissue With Stereo Endoscope Based on a Single-Layer Network. IEEE Trans. Image Process. 2022, 31, 5828–5840. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Engkvist, O.; Wang, Y.; Olivecrona, M.; Blaschke, T. The rise of deep learning in drug discovery. Drug Discov. Today 2018, 23, 1241–1250. [Google Scholar] [CrossRef] [PubMed]
- Weininger, D. SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules. J. Chem. Inf. Model. 1988, 28, 31–36. [Google Scholar] [CrossRef]
- Jastrzebski, S.; Lesniak, D.; Czarnecki, W.M. Learning to SMILE(S). arXiv 2016, arXiv:1602.06289. [Google Scholar]
- Winter, R.; Montanari, F.; Noé, F.; Clevert, D.A. Learning continuous and data-driven molecular descriptors by translating equivalent chemical representations. Chem. Sci. 2018, 10, 1692–1701. [Google Scholar] [CrossRef] [Green Version]
- Yang, K.; Swanson, K.; Jin, W.; Coley, C.; Eiden, P.; Gao, H.; Guzman-Perez, A.; Hopper, T.; Kelley, B.; Mathea, M.; et al. Analyzing Learned Molecular Representations for Property Prediction. J. Chem. Inf. Model. 2019, 59, 3370–3388. [Google Scholar] [CrossRef] [Green Version]
- Goh, G.B.; Siegel, C.M.; Vishnu, A.; Hodas, N.O. Using Rule-Based Models for Weak Supervised Learning: A ChemNet for Transferable Chemical Property Prediction. In Proceedings of the 24th ACM SIGKDD International Conference on Knowledge Discovery & Data Mining, London, UK, 19 August 2018; pp. 302–310. [Google Scholar]
- Goh, G.B.; Siegel, C.M.; Vishnu, A.; Hodas, N.O.; Baker, N. Chemception: A Deep Neural Network with Minimal Chemistry Knowledge Matches the Performance of Expert-developed QSAR/QSPR Models. arXiv 2017, arXiv:1706.06689. [Google Scholar]
- Li, X.; Fourches, D. Inductive transfer learning for molecular activity prediction: Next-Gen QSAR Models with MolPMoFiT. J. Cheminform. 2020, 12, 27. [Google Scholar] [CrossRef] [PubMed]
- Maziarka, Ł.; Danel, T.; Mucha, S.; Rataj, K.; Tabor, J.; Jastrzebski, S. Molecule Attention Transformer. arXiv 2020, arXiv:2002.08264. [Google Scholar]
- Wang, S.; Guo, Y.; Wang, Y.; Sun, H.; Huang, J. SMILES-BERT: Large Scale Unsupervised Pre-Training for Molecular Property Prediction. In Proceedings of the 10th ACM International Conference on Bioinformatics, Computational Biology and Health Informatics, New York, NY, USA, 7 September 2019; pp. 429–436. [Google Scholar]
- Maziarka, Ł.; Majchrowski, D.; Danel, T.; Gaiński, P.; Tabor, J.; Podolak, I.; Morkisz, P.; Jastrzębski, S. Relative Molecule Self-Attention Transformer. arXiv 2021, arXiv:2110.05841. [Google Scholar]
- Zhang, X.-C.; Wu, C.-K.; Yang, Z.-J.; Wu, Z.-X.; Yi, J.-C.; Hsieh, C.-Y.; Hou, T.-J.; Cao, D.-S. MG-BERT: Leveraging unsupervised atomic representation learning for molecular property prediction. Brief. Bioinform. 2021, 22, bbab152. [Google Scholar] [CrossRef] [PubMed]
- Fabian, B.; Edlich, T.; Gaspar, H.; Segler, M.H.; Meyers, J.; Fiscato, M.; Ahmed, M. Molecular representation learning with language models and domain-relevant auxiliary tasks. arXiv 2020, arXiv:2011.13230. [Google Scholar]
- Mistra SafeChem. Available online: https://www.ivl.se/projektwebbar/mistra-safechem.html (accessed on 26 October 2022).
- Mansouri, K.; Karmaus, A.L.; Fitzpatrick, J.; Patlewicz, G.; Pradeep, P.; Alberga, D.; Alepee, N.; Allen, T.E.H.; Allen, D.; Alves, V.M.; et al. CATMoS: Collaborative Acute Toxicity Modeling Suite. Environ. Health Perspect. 2021, 129, 47013. [Google Scholar] [CrossRef]
- Continuous and Data-Driven Descriptors (CDDD). Available online: https://github.com/jrwnter/cddd (accessed on 11 August 2019).
- RDKit: Open-Source Cheminformatics. version 2020.09.1.0. Available online: https://www.rdkit.org (accessed on 28 January 2021).
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-learn: Machine Learning in Python. J. Mach. Learn Res. 2011, 12, 2825–2830. Available online: https://scikit-learn.org (accessed on 28 January 2021).
- MolBERT. Available online: https://github.com/BenevolentAI/MolBERT (accessed on 21 August 2022).
- MolBERT Pre-Trained Model. Available online: https://ndownloader.figshare.com/files/25611290 (accessed on 21 August 2022).
- Molecular-Graph-BERT. Available online: https://github.com/zhang-xuan1314/Molecular-graph-BERT (accessed on 21 August 2022).
- Vovk, V.; Gammerman, A.; Shafer, G. Algorithmic Learning in a Random World; Springer: New York, NY, USA, 2005; pp. 1–324. [Google Scholar]
- Cortés-Ciriano, I.; Bender, A. Concepts and applications of conformal prediction in computational drug discovery. In Artificial Intelligence in Drug Discovery; Nathan, B., Ed.; The Royal Society of Chemistry: Cambridge, UK, 2021; pp. 63–101. [Google Scholar]
- Carlsson, L.; Eklund, M.; Norinder, U. Aggregated Conformal Prediction. In Artificial Intelligence Applications and Innovations. AIAI 2014. IFIP Advances in Information and Communication Technology; Iliadis, L., Maglogiannis, I., Papadopoulos, H., Sioutas, S., Makris, C., Eds.; Springer: Berlin/Heidelberg, Germany, 2014; Volume 437, pp. 231–240. [Google Scholar]
- Nonconformist. Available online: https://github.com/donlnz/nonconformist (accessed on 28 January 2021).
- Norinder, U.; Myatt, G.; Ahlberg, E. Predicting Aromatic Amine Mutagenicity with Confidence: A Case Study Using Conformal Prediction. Biomolecules 2018, 8, 85. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Wong, A.K.C.; Kamel, M.S. Classification of imbalanced data: A review. Int. J. Pattern Recognit. Artif. Intell. 2009, 23, 687–719. [Google Scholar] [CrossRef]
- Johnson, J.M.; Khoshgoftaar, T.M. Survey on deep learning with class imbalance. J. Big Data 2019, 6, 27. [Google Scholar] [CrossRef] [Green Version]
- Korkmaz, S. Deep Learning-Based Imbalanced Data Classification for Drug Discovery. J. Chem. Inf. Model. 2020, 60, 4180–4190. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dataset | Training Set | DL Validation Set | Evaluation Set | CP Calibration Set a |
|---|---|---|---|---|
| catmos_nt | 6004 | 662 | 2776 | 1670 |
| catmos_vt | 6449 | 717 | 2985 | 1789 |
| Dataset | Method a | Significance Level b | Validity Minority Class 1 | Validity Majority Class 0 | Efficiency Minority Class 1 | Efficiency Majority Class 0 | Sensitivity (SE) | Specificity (SP) | Balanced Accuracy (BA) |
|---|---|---|---|---|---|---|---|---|---|
| catmos_nt | cddd | 0.2 | 0.802 | 0.824 | 0.855 | 0.879 | 0.769 | 0.800 | 0.785 |
| catmos_nt | mg_bert | 0.2 | 0.798 | 0.848 | 0.814 | 0.845 | 0.751 | 0.821 | 0.786 |
| catmos_nt | molbert_p | 0.2 | 0.791 | 0.830 | 0.856 | 0.864 | 0.756 | 0.803 | 0.779 |
| catmos_nt | molbert | 0.2 | 0.797 | 0.830 | 0.873 | 0.892 | 0.767 | 0.810 | 0.788 |
| catmos_nt | rdkit | 0.2 | 0.800 | 0.805 | 0.909 | 0.912 | 0.780 | 0.786 | 0.783 |
| catmos_vt | cddd | 0.2 | 0.770 | 0.817 | |||||
| catmos_vt | mg_bert | 0.2 | 0.843 | 0.829 | 0.923 | 0.900 | 0.830 | 0.810 | 0.820 |
| catmos_vt | molbert_p | 0.2 | 0.798 | 0.819 | 0.996 | 0.991 | 0.798 | 0.820 | 0.809 |
| catmos_vt | molbert | 0.2 | 0.815 | 0.818 | 0.944 | 0.931 | 0.863 | 0.878 | 0.871 |
| catmos_vt | rdkit | 0.2 | 0.819 | 0.821 | 0.996 | 0.979 | 0.822 | 0.839 | 0.830 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Norinder, U. Traditional Machine and Deep Learning for Predicting Toxicity Endpoints. Molecules 2023, 28, 217. https://doi.org/10.3390/molecules28010217
Norinder U. Traditional Machine and Deep Learning for Predicting Toxicity Endpoints. Molecules. 2023; 28(1):217. https://doi.org/10.3390/molecules28010217
Chicago/Turabian StyleNorinder, Ulf. 2023. "Traditional Machine and Deep Learning for Predicting Toxicity Endpoints" Molecules 28, no. 1: 217. https://doi.org/10.3390/molecules28010217
APA StyleNorinder, U. (2023). Traditional Machine and Deep Learning for Predicting Toxicity Endpoints. Molecules, 28(1), 217. https://doi.org/10.3390/molecules28010217