Abstract
Amides, anhydrides, esters, and thioesters of 2H-azirine-2-carboxylic acids were prepared by a rapid procedure at room temperature involving FeCl2-catalyzed isomerization of 5-chloroisoxazoles to 2H-azirine-2-carbonyl chlorides, followed by reaction with N-, O-, or S-nucleophiles mediated by an ortho-substituted pyridine. With readily available chloroisoxazoles and a nucleophile, 2-picoline can be used as an inexpensive base. When a high yield of the acylation product is important, the reagent 2-(trimethylsilyl)pyridine/ethyl chloroformate is more suitable for the acylation with 2H-azirine-2-carbonyl chlorides.
1. Introduction
2H-Azirine-2-carboxylic acid derivatives are not only useful synthetic blocks but also exhibit various biological activities [1,2,3]. Accordingly, 2H-azirine-2-carboxylic acid esters show antibacterial properties [4,5,6,7,8]. Recently, 2H-azirine-2-carboxylates, azirine-containing depsipeptides, 2H-azirine-2-carboxamides, and azirine-containing dipeptides, which showed activity against ESKAPE pathogens, were prepared using the Passerini and Ugi reactions. [9]. In addition to the Ugi reaction (Scheme 1, Equation (1)), 2H-azirine-2-carboxamides were prepared by thermal [10,11], metal-catalyzed [12,13,14,15], and photo-isomerization [11,16,17] of 5-amino-substituted isoxazoles (Scheme 1, Equation (2)). A limitation of the isomerization approach is that the required 5-amino-substituted isoxazoles can only be prepared by the reaction of 5-halo-substituted isoxazoles with highly nucleophilic amines, while non-nucleophilic amines, in particular anilines, do not react [18]. In addition, UV irradiation may be incompatible with UV-sensitive substituents, while the use of visible light requires an expensive Hoveyda-Grubbs II (HG-II) catalyst as a photocatalyst [17].
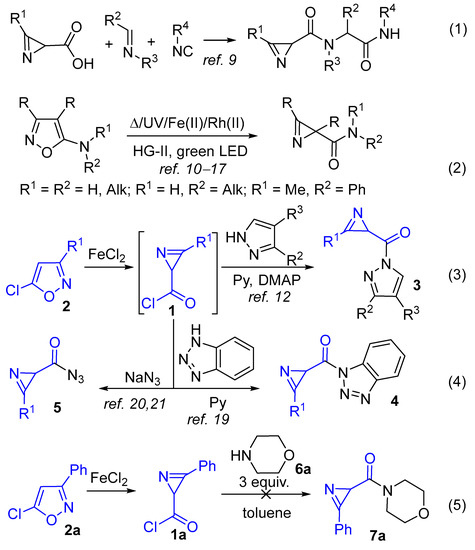
Scheme 1.
Approaches to the synthesis of 2H-azirine-2-carboxamides.
2H-Azirine-2-carbonyl chloride 1, prepared by Fe(II)-catalyzed isomerization of 5-chloroisoxazoles 2 (for the mechanism of isomerization, see [12]), react with some N-nucleophiles [12,19,20,21] to give the products of the nucleophilic acyl substitution reaction. Thus, 2H-azirine-2-carbonyl chlorides react with pyrazoles to give 2-(1H-pyrazol-1-ylcarbonyl)-2H-azirines 3 [12] (Scheme 1, Equation (3)), benzotriazole to give 1-(2H-azirine-2-carbonyl)benzotriazoles 4 [19], and sodium azide to give 2H-azirine-2-carbonyl azides 5 [20,21] (Scheme 1, Equation (4)). Surprisingly, the treatment of 2H-azirine-2-carbonyl chloride 1a with 3 equiv. of morpholine 6a gives no amide 7a (Scheme 1, Equation (5)). Taking into account the limited availability of 2H-azirine-2-carboxamides noted above, we set ourselves the task of finding optimal conditions for the acylation of primary and secondary amines with 2H-azirine-2-carbonyl chlorides. Here we report the preparation of 2H-azirine-2-carboxamides from various amines and 2H-azirine-2-carbonyl chlorides, as well as the conversion of the latter into anhydride, esters, and thioesters of 2H-azirine-2-carboxylic/thiocarboxylic acids by reactions with O- and S-nucleophiles.
2. Results and Discussion
Since the reaction of 2H-azirine-2-carbonyl chloride 1a with 3 equiv. of morpholine, 6a, gives within 3 min a complex mixture of products, without any amide 7a among them, the amount of used morpholine was varied. It resulted in the presence of 2 equiv. of morpholine, amide 7a was formed at 70% yield. Moreover, it was found that when 1 equiv. of morpholine was added to the amide 7a, its complete destruction occurred within 10 min. This means that the absence of amide 7a among products of the reaction of 2H-azirine-2-carbonyl chloride 1a with 3 equiv. of morpholine 6a is due to the destruction of the azirine core of amide 7a and possibly the azirine core of the starting chloride 1a. Then, the effect of adding other bases to trap HCl on the yield of amide 7a was investigated (Table 1). The best yield of amide 7a (89–90%) was obtained with 2,6-lutidine and 2-picoline (Table 1, entries 4, 5). The use of pyridines without an ortho-substituent gave a significantly lower yield of amide 7a (Table 1, entries 2, 6). Tertiary amines also turned out to be less effective in the reaction (Table 1, entries 7, 8).

Table 1.
Optimization of a base for the synthesis of morpholide 7.
We also tested some other approaches to amide 7a. Thus, heating 5-chloroisoxazole 2a with morpholine 6a to form 5-morpholinoisoxazole 8a, followed by its isomerization catalyzed by Fe(II), gave amide 7a only with an isolated yield of 60% (Scheme 2). Hydrolysis of carbonyl chloride 1a to azirine-2-carboxylic acid 9a, followed by amide coupling using HATU/DIPEA reagents, gave amide 7a in a 77% yield (Scheme 2). These results indicated that using 1 equiv. of morpholine in a reaction with chloride 1a in the presence of 1 equiv. of the ortho-substituted pyridine is the most efficient approach in converting chloroisoxazole 2a to amide 7a.
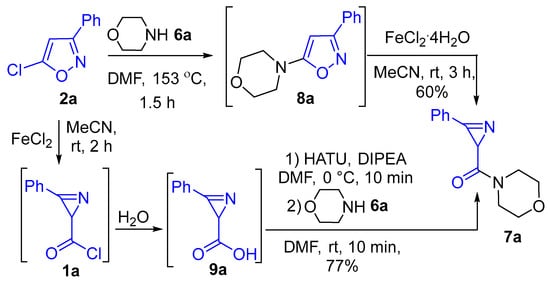
Scheme 2.
Other approaches to morpholide 7.
Therefore, using the cheapest 2-picoline, a number of cyclic (6b,c), secondary (6d,e), and primary (6f–h) amines were reacted with chloride 1a (Scheme 3) to give amides 7b–h.
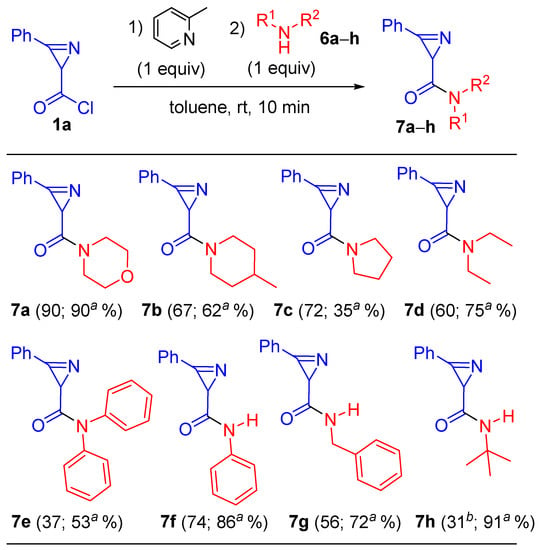
Scheme 3.
Synthesis of amides 7a–h. a Under the conditions of entry 3 of Table 2 (vide infra); b 35% with 2 equiv. of amine 6h.
It resulted that the yields of amides 7b–h obtained from amines 6b–h were significantly lower than the yield of morpholide 7a. This may be due to the complex effect of amine nucleophilicity on the competition of amine attack on the carbonyl of the acid chloride group and on the C=N azirine bond, which affects the ratio of the processes of amide formation and azirine ring destruction. In addition, the difference in the basicity of amines affected the formation and ratio of hydrochlorides of amine 6 and 2-picoline, which complicated the protonation of the azirine nitrogen of compounds 1 and 7. In turn, this catalyzed the reaction of the amine with the azirine core, and this catalysis accelerated the decomposition of azirines.
Surprisingly when chloride 1a was treated with 2-picoline without the subsequent addition of any amine, anhydride 10a was obtained in a 40% yield (Scheme 4). Compound 10a was obtained as a mixture of two diastereomers (~1:1.1). (RS,SR)-isomer crystallized from the mixture, and its amazing π-stacked structure was confirmed by X-ray analysis (Figure 1) (See the Supplementary Materials). It was found that chloride 1a does not react with acid 9a to give anhydride 10a in the absence of 2-picoline. A plausible mechanism of the formation of anhydride 10 includes the reaction of salt 11 with acid 9 promoted by 2-picoline. Acid 9 was formed by the hydrolysis of chloride 1 with water adsorbed on the wall of the glassware (the glassware was not specially dried) and traces of water entering the reaction mixture during the isolation of chloride 1 from the mixture obtained after the isomerization of chloroisoxazole 2 (extraction with ether, filtration through celite). Since the scale of the reaction of chloroisoxazole 2a was 1 mmol, only 3.6 mg of water was required to obtain a 40% yield of anhydride 10a. When chloroisoxazole 2b was used in the reaction, anhydride 10b was obtained at 66% (Scheme 4). The addition of 0.2 equiv. of water, together with 2-picoline, increased the yield of anhydride 10b to 78%. Both the excess and deficiency of water in the reaction mixture will reduce the yield of anhydride, but it is difficult to control the exact amount of water required for the reaction on a 1 mmol scale without technically complicating the reaction procedure.
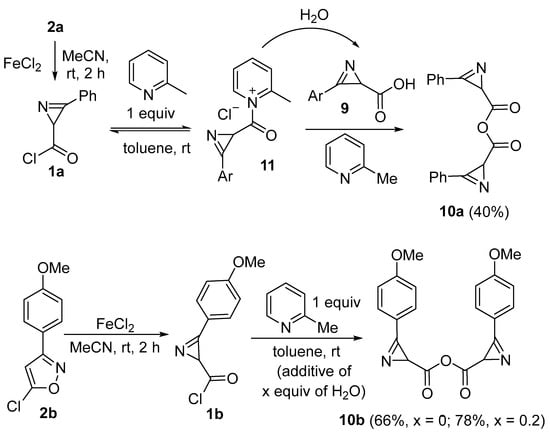
Scheme 4.
Synthesis of anhydrides 10.
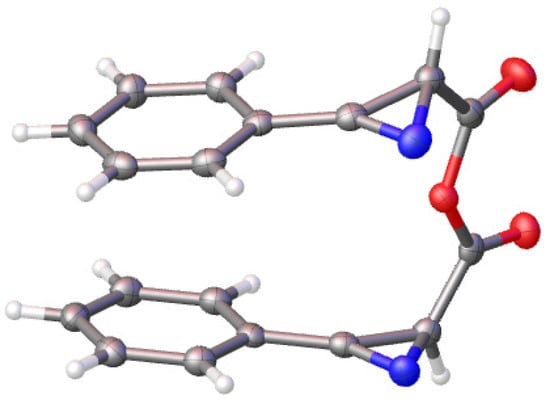
Figure 1.
Perspective view of (RS,SR)-isomer of anhydride 10a showing thermal ellipsoids at 50% probability level.
The formation of anhydride 10 due to the presence of traces water suggests an additional route for the formation of amides through the reaction of an amine with an anhydride. Thus, the formation of amides 7 can proceed via several mechanisms that can operate simultaneously: (1) direct interaction of chloride 1a with amines 6 (Scheme 5, route a); (2) pathways mediated by a pyridine via salt A (Scheme 5, route b); and (3) formation of anhydride 10, initiated by traces of water, followed by reaction of this anhydride with amines 6 (Scheme 5, route c).
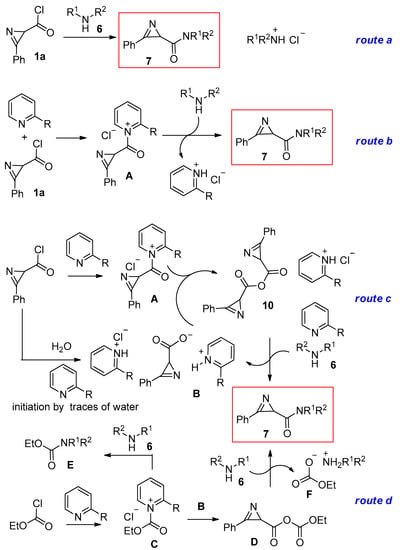
Scheme 5.
Plausible mechanisms for the formation of amides 7.
Amines 6 differ considerably in nucleophilicity [22] and basicity [23], which affects the rate of their acylation with azirinecarbonyl chlorides 1 and the relative rate of decomposition of the starting azirines 1 and products 7 (vide supra). For instance, the difference in the relative rate of decomposition of azirine 7h in the presence of different amines was demonstrated by 1H NMR analysis of equimolar mixtures of azirine 7h with tert-butylamine and azirine 7h with morpholine in C6D6. In the first mixture, primarily starting substances were detected after 1 h, and there was 85% decomposition of azirine 7h with the formation of a complex mixture of products after 10 h, while in the second mixture, complete decomposition of azirine 7h was recorded after 1 h.
In order to partially smooth out the differences in the nucleophilicity and basicity of the amines, we decided to increase the excess of amine to 3 equiv. to accelerate the rate of amide formation and reduce the effect of this excess on the decomposition of azirines and add ethyl chloroformate. We assumed that such an additive could, on the one hand, create an additional pathway for the formation of amide 7 and, on the other hand, neutralize the unfavorable effect of an excess of amine by converting it into amide E and a salt of weak acid F. The latter, in contrast to the corresponding hydrochloride salt, should not protonate azirines and thus initiate their decomposition by amines (Scheme 5, route d). To check this hypothesis, additional experiments were carried out to prepare amide 7a (Table 2). The best results in terms of the yield of amide 7a and the economical use of reagents correspond to experiment 3. It was also seen that the use of acetyl chloride (Table 2, entry 6) for the above purposes was much less efficient than ethyl chloroformate, which may be due to the fact that salt F, with AcO- instead of EtO2CO-, can better catalyze the decomposition of azirines, being a salt of a stronger acid.
Table 2.
Optimization of the reaction conditions for the synthesis of morpholide 7.
High yields of amides 7 using pyridines with bulky ortho-substituents (Table 2) may be because, on the one hand, the C(O)–N bond in salt A (Scheme 5) in such compounds is weaker, making the respective pyridinium a better leaving group, and on the other hand, the pyridinium chloride with such ortho-substituents has a lower protonation capacity for azirines.
Then, experiments were carried out with various amines, 6a–h, under optimal conditions (entry 3 of Table 2). The use of these reaction conditions allowed for the preparation of amides 7 in higher yields (excluding 7c): 7a (90%), 7b (62%), 7c (35%), 7d (75%), 7e (53%), 7f (86%), 7g (72%), and 7h (91%) (Scheme 3, footnote a). A dramatic increase in yield was significant in the case of tert-butylamine (6h). We noted that the yield of amide 7h was only 42% in the direct reaction of chloride 1a with 2 equiv. of 6h. Importantly, the reaction with aniline gave anilide 7f, which was not available by other approaches [10,11,12,13,14,15,16,17], in an 86% yield.
The scope of the reaction with various 2H-azirine-2-carbonyl chlorides 1 was evaluated using tert-butyl amine (6h) as the N-nucleophile. Amides 7h–q were obtained in 60–91% yields (Scheme 6), starting from 3-aryl/hetaryl/alkyl-2H-azirine-2-carbonyl chlorides 1a–i, as well from 2,2,3-trisubstituted derivative 1j. All new compounds were characterized by 1H, 13C NMR, and HRMS methods. Moreover, the structure of 7n was also confirmed by single-crystal X-ray diffraction analysis (Figure 2) (See the Supplementary Materials).
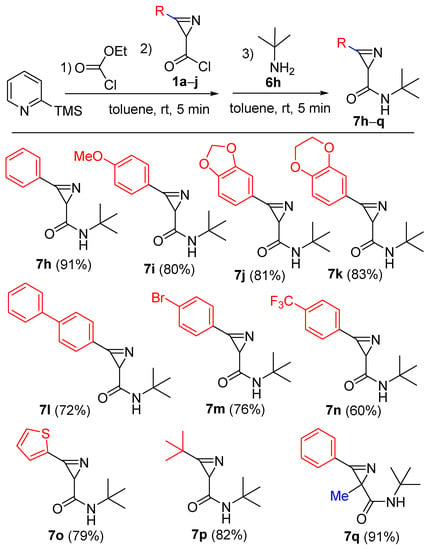
Scheme 6.
Synthesis of amides 7h–q.
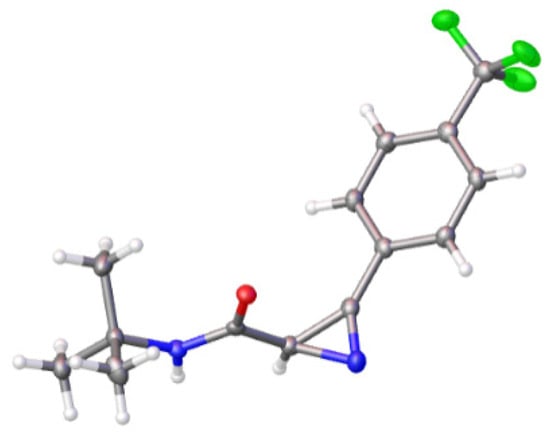
Figure 2.
Perspective view of amide 7n showing thermal ellipsoids at 50% probability level.
Additionally, 2H-azirine-2-carbonyl chloride 1a was reacted with alkyl- and aryl-substituted O- and S-nucleophiles, such as phenol 12a, methyl 2-(2-hydroxyphenyl)acetate 12b, propargyl 12c, and benzyl 12d alcohols, as well as thiophenol 13a, cyclopentanethiol 13b, and N-(sulfanylmethyl)benzamide 13c. The reactions were carried out under standard conditions but used 1.5 equiv. of O- or S-nucleophile and with the DMAP addition to trap HCl. Azirinecarboxylic esters 14a-d were obtained in 77-92% isolated yields (Scheme 7). S-Phenyl and S-cyclopentyl azirinecarbothioates 15a,b were obtained in 81% and 73% yields; the yield of S-(benzamidomethyl) azirinecarbothioate 15c was lower, only 21%, likely due to multifunctionality of the starting N-(sulfanylmethyl)benzamide (Scheme 7). The yields of 14a/15a were significantly lower when using 2 equiv. of 2-picoline and 1 equiv. of nucleophile 12a/13a (cf. Table 2).
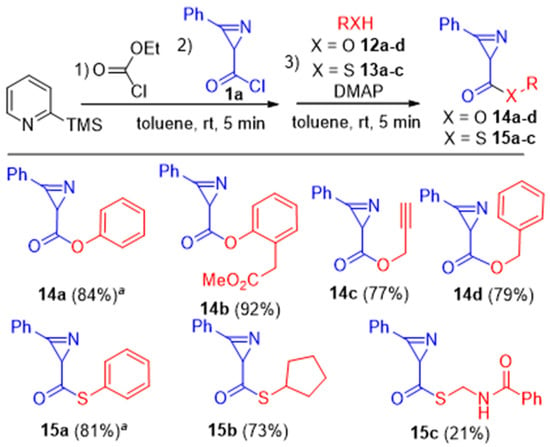
Scheme 7.
Synthesis of azirinecarboxylic esters 14a–d and azirinecarbothioates 15a–c. a The yields in the reaction with phenol and thiophenol were 43% and 53%, respectively, under the condition where 2 equiv. of 2-picoline and 1 equiv. of nucleophile 12a or 13a were used (toluene, rt, 10 min).
3. Materials and Methods
3.1. General Instrumentation
Melting points were determined on a melting point apparatus SMP30. 1H-NMR (400 MHz) and 13C-NMR (100 MHz) spectra were recorded on a Bruker AVANCE 400 spectrometer in CDCl3 or C6D6. Chemical shifts (δ) were reported in parts per million downfield from tetramethylsilane (TMS, δ = 0.00). 1H-NMR spectra were calibrated according to the residual peak of CDCl3 (7.28 ppm) and C6D6 (7.16 ppm). For all new compounds, 13C{1H} spectra were recorded and calibrated according to the peak of CDCl3 (77.00 ppm) and C6D6 (128.00 ppm). Electrospray ionization (ESI), positive mode, and mass spectra were measured on a Bruker MaXis mass spectrometer, HRMS-ESI-QTOF, using MeOH for the dilution of samples. Single-crystal X-ray data were collected by means of “XtaLAB Synergy” and “SuperNova” diffractometers. The crystals of 7n and 10a were measured at a temperature of 99.9(8) K. Crystallographic data for the structures 7n (CCDC 2214535) and 10a (CCDC 2215155) have been deposited with the Cambridge Crystallographic Data Centre. Thin-layer chromatography (TLC) was conducted on aluminum sheets precoated with SiO2 ALUGRAM SIL G/UV254. Column chromatography was performed on Macherey-Nagel silica gel 60M (0.04–0.063 mm). All solvents were distilled and dried prior to use. Toluene and diethyl ether were distilled and stored over sodium metal. MeCN was distilled from P2O5 and redistilled from K2CO3. 5-Chloroisoxazoles 2a [18], 2b [24], 2f,g,i [19], 2h [20], and 2j [25] are known compounds and were prepared by the reported procedures.
3.2. General Experimental Procedures
3.2.1. General Procedure A (GP-A) for the Synthesis of 5-Chloroisoxazoles (2)
Triethylamine (253 mg, 2.5 mmol, 0.35 mL) was added dropwise at 0 °C to a stirring suspension of isoxazol-5(4H)-one (3 mmol) in POCl3 (4 mL). The mixture was stirred at 75 °C for 12 h, poured into ice (300 g), and extracted with EtOAc (3 × 30 mL). The organic layer was washed with brine and dried over Na2SO4. The solvent was evaporated under reduced pressure, and the product was purified by column chromatography (petroleum ether–EtOAc, 10:1) to give chloroisoxazole 2.
3.2.2. General Procedure B (GP-B) for the Synthesis of Amides (7)
Anhydrous FeCl2 (51 mg, 0.4 mmol, 0.2 equiv.) was added to a solution of 5-chloroisoxazole 2 (2 mmol) in dry acetonitrile (4 mL) under an Ar atmosphere. The mixture was stirred at room temperature for 2 h until 5-chloroisoxazole 2 was consumed (monitored by TLC). The solvent was evaporated, the residue was diluted with dry diethyl ether (100 mL), and the precipitated iron chloride was filtered off through celite. The ether was evaporated under reduced pressure, and the formed 2H-azirine-2-carbonyl chloride 1 was dissolved in anhydrous toluene (4 mL). Then amine 6 (4 mmol, 2 equiv.) was added, and the reaction mixture was stirred at room temperature for 3 min. The solvent was evaporated, and the residue was purified by column chromatography (petroleum ether–EtOAc).
3.2.3. General Procedure C (GP-C) for the Synthesis of Amides (7)
Anhydrous FeCl2 (51 mg, 0.4 mmol, 0.2 equiv.) was added to a solution of 5-chloroisoxazole 2 (2 mmol) in dry acetonitrile (4 mL) under an Ar atmosphere. The mixture was stirred at room temperature for 2 h until 5-chloroisoxazole 2 was consumed (monitored by TLC). The solvent was evaporated, the residue was diluted with dry diethyl ether (100 mL), and the precipitated iron chloride was filtered off through celite. The ether was evaporated under reduced pressure, and the formed 2H-azirine-2-carbonyl chloride 1 was dissolved in anhydrous toluene (4 mL). Then 2-methylpyridine (186 mg, 2 mmol, 1 equiv.) and amine 6 (2 mmol, 1 equiv.) were added successively. The reaction mixture was stirred for 10 min at room temperature, the solvent was evaporated, and the residue was purified by column chromatography (petroleum ether–EtOAc).
3.2.4. General Procedure D (GP-D) for the Synthesis of Amides (7)
Anhydrous FeCl2 (51 mg, 0.4 mmol, 0.2 equiv.) was added to a solution of 5-chloroisoxazole 2 (2 mmol) in dry acetonitrile (4 mL) under an Ar atmosphere. The mixture was stirred at room temperature for 2 h until 5-chloroisoxazole 2 was consumed (monitored by TLC). The solvent was evaporated, the residue was diluted with dry diethyl ether (100 mL), and the precipitated iron chloride was filtered off through celite. The ether was evaporated under reduced pressure, and the formed 2H-azirine-2-carbonyl chloride 1 was dissolved in anhydrous toluene (2 mL). The resulting solution was added to a stirred mixture of 2-(trimethylsilyl)pyridine (151 mg, 1 mmol, 0.5 equiv.) and ethyl chloroformate (109 mg, 1 mmol, 0.5 equiv.) at room temperature. Then amine 6 (6 mmol, 3 equiv.) was added, and the reaction mixture was stirred for 5 min. The solvent was evaporated, and the residue was purified by column chromatography (petroleum ether–EtOAc).
3.2.5. General Procedure E (GP-E) for the Synthesis of Amides (7)
A mixture of 5-chloroisoxazole 2 (2 mmol), amine (4 mmol, 2 equiv.), and K2CO3 (828 mg, 6 mmol, 3 equiv.) in DMF (10 mL) was refluxed while being stirred for 1.5 h. The reaction mixture was diluted with water (30 mL), extracted with EtOAc (3 × 15 mL), washed with brine (20 mL), and dried over Na2SO4. The solvent was evaporated, the residue was dissolved in acetonitrile (10 mL), and FeCl2 × 4H2O (80 mg, 20 mol %) was added. The mixture was stirred at room temperature for 3 h, the solvent was evaporated, and the residue was purified by column chromatography (petroleum ether–EtOAc).
3.2.6. General Procedure F (GP-F) for the Synthesis of Amides (7)
Anhydrous FeCl2 (51 mg, 0.4 mmol, 0.2 equiv.) was added to a solution of 5-chloroisoxazole 2 (2 mmol) in dry acetonitrile (25 mL) under an Ar atmosphere. The mixture was stirred at room temperature for 2 h until 5-chloroisoxazole 2 was consumed (monitored by TLC). Water (25 mL) was added, and the mixture was stirred at room temperature for 15 min. The 2H-azirine-2-carboxylic acid was extracted with EtOAc (325 mL), washed with water (25 mL) and brine (10 mL), and dried over Na2SO4. The solvent was evaporated, and the residue was diluted with DMF (5 mL) and cooled to 0 °C. Then HATU (1.52 g, 4 mmol, 2 equiv.) and DIPEA (774 mg, 6 mmol, 3 equiv.) were added, and the reaction mixture was stirred at 0 °C for 10 min. After that, amine 6 (2.4 mmol, 1.2 equiv) was added, and the reaction mixture was stirred at room temperature for 10 min. The reaction mixture was then diluted with water (75 mL), extracted with EtOAc (3 × 25 mL), washed with brine (30 mL), and dried over Na2SO4. The solvent was evaporated, and the residue was purified by column chromatography (petroleum ether–EtOAc).
3.2.7. General Procedure G (GP-G) for the Synthesis of Anhydrides (10)
Anhydrous FeCl2 (51 mg, 0.4 mmol, 0.2 equiv.) was added to a solution of 5-chloroisoxazole 2 (2 mmol) in dry acetonitrile (4 mL) under an Ar atmosphere. The mixture was stirred at room temperature for 2 h until 5-chloroisoxazole 2 was consumed (monitored by TLC). The solvent was evaporated, the residue was diluted with dry diethyl ether (100 mL), and the precipitated iron chloride was filtered off through celite. The ether was evaporated, and the formed 2H-azirine-2-carbonyl chloride 1 was dissolved in anhydrous toluene (4 mL). Then 2-methylpyridine (186 mg, 2 mmol, 1 equiv.) was added. The reaction mixture was stirred for 10 min at room temperature, the solvent was evaporated, and the residue was purified by column chromatography (petroleum ether–EtOAc).
3.2.8. General Procedure H (GP-H) for the Synthesis of Esters (14) and Thioesters (15)
Anhydrous FeCl2 (51 mg, 0.4 mmol, 0.2 equiv.) was added to a solution of 5-chloroisoxazole 2a (359 mg, 2 mmol) in dry acetonitrile (4 mL) under an Ar atmosphere. The mixture was stirred at room temperature for 2 h until 5-chloroisoxazole 2a was consumed (monitored by TLC). The solvent was evaporated, the residue was diluted with dry diethyl ether (100 mL), and the precipitated iron chloride was filtered off through celite. The ether was evaporated, anhydrous toluene (2 mL) was added, and the resulting solution was added to a stirring mixture of 2-(trimethylsilyl)pyridine (151 mg, 1 mmol, 0.5 equiv.) and ethyl chloroformate (109 mg, 1 mmol, 0.5 equiv) at room temperature. Then the corresponding nucleophile (3 mmol, 1.5 equiv.) and N,N-dimethylpyridin-4-amine (733 mg, 6 mmol, 3 equiv.) base were added, and the reaction mixture was stirred for 10 min. The solvent was evaporated, and the residue was purified by column chromatography (petroleum ether–EtOAc).
3.2.9. Specific Procedures and Characterization
3-(Benzo[d][1,3]dioxol-5-yl)-5-chloroisoxazole (2c). Compound 2c was prepared following the general procedure of GP-A from 3-(benzo[d][1,3]dioxol-5-yl)isoxazol-5(4H)-one (616 mg, 3 mmol) as colorless solid (443 mg, yield 66%). Mp: 82–83 °C (Et2O–hexane). 1H NMR (400 MHz, CDCl3) δ 7.28 (s, 1H), 7.25–7.20 (m, 1H), 6.93–6.85 (m, 1H), 6.41 (s, 1H), 6.05 (s, 2H). 13C{1H} NMR (100 MHz, CDCl3) δ 163.7, 154.9, 149.6, 148.3, 122.1, 121.2, 108.7, 106.6, 101.6, 99.4. HRMS-ESI [M + H]+ calcd for C10H735ClNO3+, 224.0109; found, 224.0112.
5-Chloro-3-(2,3-dihydrobenzo[b][1,4]dioxin-6-yl)isoxazole (2d). Compound 2d was prepared following the general procedure of GP-A from 3-(2,3-dihydrobenzo[b][1,4]dioxin-6-yl)isoxazol-5(4H)-one (658 mg, 3 mmol) as colorless solid (506 mg, yield 71%). Mp: 78–79 °C (Et2O–hexane). 1H NMR (400 MHz, CDCl3) δ 7.29 (s, 1H), 7.26–7.21 (m, 1H), 6.96–6.89 (m, 1H), 6.39 (s, 1H), 4.36–4.25 (s, 4H). 13C{1H} NMR (100 MHz, CDCl3) δ 163.6, 154.7, 145.6, 143.9, 121.4, 120.0, 117.8, 115.6, 99.4, 64.5, 64.2. HRMS-ESI [M + Na]+ calcd for C11H835ClNNaO3+, 260.0085; found, 260.0079.
5-Chloro-3-(2,3-dihydrobenzo[b][1,4]dioxin-6-yl)isoxazole (2e). Compound 2e was prepared following the general procedure of GP-A from 3-([1,1’-biphenyl]-4-yl)isoxazol-5(4H)-one (712 mg, 3 mmol) as colorless solid (506 mg, yield 66%). Mp: 139–140 °C (Et2O–hexane). 1H NMR (400 MHz, CDCl3) δ 7.89–7.83 (m, 2H), 7.75–7.71 (m, 2H), 7.68–7.63 (m, 2H), 7.53–7.47 (m, 2H), 7.46–7.39 (m, 1H), 6.54 (s, 1H). 13C{1H} NMR (100 MHz, CDCl3) δ 163.9, 155.1, 143.4, 140.0, 128.9, 127.9, 127.7 (2C), 127.1, 127.0, 99.6. HRMS-ESI [M + H]+ calcd for C15H1135ClNO+, 256.0524; found, 256.0527.
Morpholino(3-phenyl-2H-azirin-2-yl)methanone (7a). Compound 7a was prepared following GP-B–F procedure from 5-chloro-3-phenylisoxazole 2a (359 mg, 2 mmol, 1 equiv.) with morpholine 6a (GP-B and GP-E: 348 mg, 4 mmol, 2 equiv.; GP-C: 174 mg, 2 mmol, 1 equiv.; GP-D: 522 mg, 6 mmol, 3 equiv.; GP-F: 209 mg, 2.4 mmol, 1.2 equiv.) as a nucleophile. A mixture of PE–EtOAc (from 5:1 to 1:1) was used as an eluent for chromatography. Orange solid (GP-B: 322 mg, yield 70%; GP-C and GP-D: 506 mg, yield 90%; GP-E: 276 mg, yield 60%; GP-F: 355 mg, yield 77%). Mp: 73–75 °C (Et2O–hexane). 1H NMR (400 MHz, CDCl3) δ 7.96–7.82 (m, 2H), 7.65–7.51 (m, 3H), 3.97–3.57 (m, 8H), 3.04 (s, 1H). 13C{1H} NMR (100 MHz, CDCl3) δ 169.0, 159.3, 133.5, 130.2, 129.1, 122.9, 66.7 (2C), 45.9, 42.6, 28.5. HRMS-ESI [M + Na]+ calcd for C13H14N2NaO2+, 253.0948; found, 253.0949.
(4-Methylpiperidin-1-yl)(3-phenyl-2H-azirin-2-yl)methanone (7b). Compound 7b was prepared following GP-C and GP-D procedures from 5-chloro-3-phenylisoxazole 2a (359 mg, 2 mmol, 1 equiv) with 4-methylpiperidine 6b (GP-C: 198 mg, 2 mmol, 1 equiv; GP-D: 595 mg, 6 mmol, 3 equiv) as a nucleophile. A mixture of PE–EtOAc (from 5:1 to 1:1) was used as an eluent for chromatography. Orange oil (GP-C: 325 mg, yield 67%; GP-D: 300 mg, yield 62%). 1H NMR (400 MHz, CDCl3) δ 7.99–7.86 (m, 2H), 7.66–7.52 (m, 3H), 4.64–4.39 (m, 2H), 3.38–3.20 (m, 1H), 3.10 (s, 1H), 2.78–2.60 (m, 1H), 1.95–1.79 (m, 1H), 1.77–1.65 (m, 2H), 1.41–1.13 (m, 2H), 1.02 (d, J = 6.2 Hz, 3H). 13C{1H} NMR (100 MHz, CDCl3) δ 168.5, 159.8 (d, J = 17.5 Hz), 133.3, 130.2, 129.1, 123.3, 45.9 (d, J = 11.6 Hz), 42.8 (d, J = 13.9 Hz), 34.9 (d, J = 13.3 Hz), 33.6, 31.1 (d, J = 13.1 Hz), 29.0, 21.7. HRMS-ESI [M + H]+ calcd for C15H19N2O+, 243.1492; found, 243.1494.
(3-Phenyl-2H-azirin-2-yl)(pyrrolidin-1-yl)methanone (7c). Compound 7c was prepared following GP-C and GP-D procedures from 5-chloro-3-phenylisoxazole 2a (359 mg, 2 mmol, 1 equiv.) with pyrrolidine 6c (GP-C: 142 mg, 2 mmol, 1 equiv.; GP-D: 427 mg, 6 mmol, 3 equiv.) as a nucleophile. A mixture of PE–EtOAc (from 5:1 to 1:1) was used as an eluent for chromatography. Orange solid (GP-C: 309 mg, yield 72%; GP-D: 150 mg, yield 35%). Mp: 58–59 °C (Et2O–hexane). 1H NMR (400 MHz, CDCl3) δ 7.94–7.84 (m, 2H), 7.64–7.50 (m, 3H), 3.93–3.74 (m, 2H), 3.61–3.49 (m, 2H), 2.97 (s, 1H), 2.13–1.83 (m, 4H). 13C{1H} NMR (100 MHz, CDCl3) δ 168.6, 159.4, 133.3, 130.2, 129.0, 123.1, 46.4, 46.3, 30.0, 26.2, 24.1. HRMS-ESI [M + H]+ calcd for C13H15N2O+, 215.1179; found, 215.1181.
N,N-Diethyl-3-phenyl-2H-azirine-2-carboxamide (7d). Compound 7d was prepared following GP-C and GP-D procedures from 5-chloro-3-phenylisoxazole 2a (359 mg, 2 mmol, 1 equiv.) with diethylamine 6d (GP-C: 146 mg, 2 mmol, 1 equiv.; GP-D: 439 mg, 6 mmol, 3 equiv.) as a nucleophile. A mixture of PE–EtOAc (from 5:1 to 1:1) was used as an eluent for chromatography. Orange solid (GP-C: 260 mg, yield 60%; GP-D: 324 mg, yield 75%). Mp: 53–54 °C (Et2O–hexane). 1H NMR (400 MHz, CDCl3) δ 7.92–7.86 (m, 2H), 7.62–7.51 (m, 3H), 3.80–3.70 (m, 1H), 3.70–3.59 (m, 1H), 3.58–3.48 (m, 1H), 3.44–3.34 (m, 1H), 3.04 (s, 1H), 1.40 (t, J = 7.1 Hz, 3H), 1.16 (t, J = 7.1 Hz, 3H). 13C{1H} NMR (100 MHz, CDCl3) δ 169.4, 159.4, 133.2, 130.1, 129.0, 123.3, 42.0, 41.1, 29.0, 15.1, 13.0. HRMS-ESI [M + H]+ calcd for C13H17N2O+, 217.1335; found, 217.1338.
N,N,3-Triphenyl-2H-azirine-2-carboxamide (7e). Compound 7e was prepared following GP-C and GP-D procedures from 5-chloro-3-phenylisoxazole 2a (359 mg, 2 mmol, 1 equiv.) with diphenylamine 6e (GP-C: 334 mg, 2 mmol, 1 equiv.; GP-D: 1.02 g, 6 mmol, 3 equiv.) as a nucleophile. A mixture of PE–EtOAc (from 5:1 to 1:1) was used as an eluent for chromatography. Orange solid (GP-C: 231 mg, yield 37%; GP-D: 331 mg, yield 53%). Mp: 189–190 °C (Et2O–hexane). 1H NMR (400 MHz, CDCl3) δ 7.99–7.87 (m, 2H), 7.67–7.56 (m, 3H), 7.53–7.24 (m, 10H), 2.82 (s, 1H). 13C{1H} NMR (100 MHz, CDCl3) δ 170.4, 158.6, 142.4, 133.4, 130.2, 129.4, 129.1, 127.1, 123.0, 31.3. HRMS-ESI [M + H]+ calcd for C21H17N2O+, 313.1335; found, 313.1340.
N,3-Diphenyl-2H-azirine-2-carboxamide (7f). Compound 7f was prepared following GP-C and GP-D procedures from 5-chloro-3-phenylisoxazole 2a (359 mg, 2 mmol, 1 equiv.) with aniline 6f (GP-C: 186 mg, 2 mmol, 1 equiv; GP-D: 559 mg, 6 mmol, 3 equiv.) as a nucleophile. A mixture of PE–EtOAc (from 5:1 to 1:1) was used as an eluent for chromatography. Orange solid (GP-C: 350 mg, yield 74%; GP-D: 406 mg, yield 86%). Mp: 179–180 °C (Et2O–hexane). 1H NMR (400 MHz, CDCl3) δ 8.01–7.93 (m, 2H), 7.71–7.65 (m, 1H), 7.63–7.57 (m, 2H), 7.55–7.50 (m, 2H), 7.43 (s, 1H), 7.34–7.26 (m, 2H), 7.13–7.07 (m, 1H), 2.94 (s, 1H). 13C{1H} NMR (100 MHz, CDCl3) δ 168.6, 162.2, 137.4, 134.3, 130.6, 129.4, 128.9, 124.4, 122.3, 119.7, 32.0. HRMS-ESI [M + Na]+ calcd for C15H12N2NaO+, 259.0842; found, 259.0842.
N-Benzyl-3-phenyl-2H-azirine-2-carboxamide (7g) [16,17]. Compound 7g was prepared following GP-C and GP-D procedures from 5-chloro-3-phenylisoxazole 2a (359 mg, 2 mmol, 1 equiv.) with phenylmethanamine 6g (GP-C: 214 mg, 2 mmol, 1 equiv.; GP-D: 643 mg, 6 mmol, 3 equiv.) as a nucleophile. A mixture of PE–EtOAc (from 5:1 to 1:1) was used as an eluent for chromatography. Light yellow solid (GP-C: 280 mg, yield 56%; GP-D: 360 mg, yield 72%). Mp: 145–146 °C (Et2O–hexane). 1H NMR (400 MHz, CDCl3) δ 7.96–7.89 (m, 2H), 7.70–7.65 (m, 1H), 7.63–7.57 (m, 2H), 7.33–7.20 (m, 5H), 5.90 (s, 1H), 4.54–4.35 (m, 2H), 2.85 (s, 1H). 13C{1H} NMR (100 MHz, CDCl3) δ 170.5, 162.1, 137.9, 134.1, 130.5, 129.4, 128.6, 127.6, 127.4, 122.4, 43.4, 31.4. HRMS-ESI [M + Na]+ calcd for C16H14N2NaO+, 273.0998; found, 273.1001.
N-(tert-Butyl)-3-phenyl-2H-azirine-2-carboxamide (7h). Compound 7h was prepared following methods GP-B, GP-C and GP-D from 5-chloro-3-phenylisoxazole 2a (359 mg, 2 mmol, 1 equiv.) with 2-methylpropan-2-amine 6h (GP-B: 293 mg, 4 mmol, 2 equiv.; GP-C: 146 mg, 2 mmol, 1 equiv.; GP-D: 439 mg, 6 mmol, 3 equiv.) as a nucleophile. A mixture of PE–EtOAc (from 5:1 to 1:1) was used as an eluent for chromatography. Colorless solid (GP-B: 182 mg, yield 42%; GP-C: 134 mg, yield 31%; GP-D: 394 mg, yield 91%). Mp: 171–172 °C (Et2O–hexane). 1H NMR (400 MHz, CDCl3) δ 7.95–7.89 (m, 2H), 7.69–7.63 (m, 1H), 7.63–7.56 (m, 2H), 5.32 (s, 1H), 2.69 (s, 1H), 1.32 (s, 9H). 13C{1H} NMR (100 MHz, CDCl3) δ 169.6, 162.6, 133.9, 130.3, 129.3, 122.7, 51.3, 32.1, 28.7. HRMS-ESI [M + H]+ calcd for C13H17N2O+, 217.1335; found, 217.1338.
N-(tert-Butyl)-3-(4-methoxyphenyl)-2H-azirine-2-carboxamide (7i). Compound 7i was prepared following GP-D procedure from 5-chloro-3-(4-methoxyphenyl)isoxazole 2b (419 mg, 2 mmol, 1 equiv.) with 2-methylpropan-2-amine 6h (439 mg, 6 mmol, 3 equiv.) as a nucleophile. A mixture of PE–EtOAc (from 5:1 to 1:1) was used as an eluent for chromatography. Colorless solid (394 mg, yield 80%). Mp: 157–158 °C (Et2O–hexane). 1H NMR (400 MHz, CDCl3) δ 7.88–7.82 (m, 2H), 7.10–7.04 (m, 2H), 5.26 (s, 1H), 3.91 (s, 3H), 2.63 (s, 1H), 1.30 (s, 9H). 13C{1H} NMR (100 MHz, CDCl3) δ 170.0, 164.1, 161.3, 132.5, 115.0, 114.9, 55.6, 51.1, 31.8, 28.6. HRMS-ESI [M + Na]+ calcd for C14H18N2NaO2+, 269.1260; found, 269.1262.
3-(Benzo[d][1,3]dioxol-5-yl)-N-(tert-butyl)-2H-azirine-2-carboxamide (7j). Compound 7j was prepared following GP-D procedure from 3-(benzo[d][1,3]dioxol-5-yl)-5-chloroisoxazole 2c (447 mg, 2 mmol, 1 equiv.) with 2-methylpropan-2-amine 6h (439 mg, 6 mmol, 3 equiv.) as a nucleophile. A mixture of PE–EtOAc (from 5:1 to 1:1) was used as an eluent for chromatography. Light yellow solid (422 mg, yield 81%). Mp: 150–151 °C (Et2O–hexane). 1H NMR (400 MHz, CDCl3) δ 7.45–7.41 (m, 1H), 7.38–7.35 (m, 1H), 7.01–6.96 (m, 1H), 6.11 (s, 2H), 5.27 (s, 1H), 2.63 (s, 1H), 1.31 (s, 9H). 13C{1H} NMR (100 MHz, CDCl3) δ 169.7, 161.6, 152.6, 148.6, 127.2, 116.5, 109.1, 109.0, 102.2, 51.2, 32.3, 28.6. HRMS-ESI [M + H]+ calcd for C14H17N2O3+, 261.1234; found, 261.1237.
N-(tert-Butyl)-3-(2,3-dihydrobenzo[b][1,4]dioxin-6-yl)-2H-azirine-2-carboxamide (7k). Compound 7k was prepared following GP-D procedure from 5-chloro-3-(2,3-dihydrobenzo[b][1,4]dioxin-6-yl)isoxazole 2d (475 mg, 2 mmol, 1 equiv.) with 2-methylpropan-2-amine 6h (439 mg, 6 mmol, 3 equiv.) as a nucleophile. A mixture of PE–EtOAc (from 5:1 to 1:1) was used as an eluent for chromatography. Colorless solid (455 mg, yield 83%). Mp: 123–124 °C (Et2O–hexane). 1H NMR (400 MHz, CDCl3) δ 7.46–7.39 (m, 2H), 7.08–7.00 (m, 1H), 5.24 (s, 1H), 4.38–4.29 (m, 4H), 2.61 (s, 1H), 1.31 (s, 9H). 13C{1H} NMR (100 MHz, CDCl3) δ 169.9, 161.5, 148.7, 144.1, 124.4, 119.3, 118.4, 115.7, 64.7, 64.0, 51.2, 32.0, 28.6. HRMS-ESI [M + Na]+ calcd for C15H18N2NaO3+, 297.1210; found, 297.1212.
3-([1,1’-Biphenyl]-4-yl)-N-(tert-butyl)-2H-azirine-2-carboxamide (7l). Compound 7l was prepared following GP-D procedure from 3-([1,1’-biphenyl]-4-yl)-5-chloroisoxazole 2e (511 mg, 2 mmol, 1 equiv.) with 2-methylpropan-2-amine 6h (439 mg, 6 mmol, 3 equiv.) as a nucleophile. A mixture of PE–EtOAc (from 5:1 to 1:1) was used as an eluent for chromatography. Light yellow solid (421 mg, yield 72%). Mp: 161–162 °C (Et2O–hexane). 1H NMR (400 MHz, CDCl3) δ 8.02–7.98 (m, 2H), 7.84–7.80 (m, 2H), 7.68–7.64 (m, 2H), 7.54–7.42 (m, 3H), 5.33 (s, 1H), 2.72 (s, 1H), 1.35 (s, 9H). 13C{1H} NMR (100 MHz, CDCl3) δ 169.7, 162.3, 146.8, 139.5, 130.8, 129.0, 128.6, 128.0, 127.3, 121.3, 51.3, 32.1, 28.7. HRMS-ESI [M + H]+ calcd for C19H21N2O+, 293.1648; found, 293.1652.
3-(4-Bromophenyl)-N-(tert-butyl)-2H-azirine-2-carboxamide (7m). Compound 7m was prepared following GP-D procedure from 3-(4-bromophenyl)-5-chloroisoxazole 2f (517 mg, 2 mmol, 1 equiv.) with 2-methylpropan-2-amine 6h (439 mg, 6 mmol, 3 equiv.) as a nucleophile. A mixture of PE–EtOAc (from 5:1 to 1:1) was used as an eluent for chromatography. Colorless solid (449 mg, yield 76%). Mp: 171–172 °C (Et2O–hexane). 1H NMR (400 MHz, CDCl3) δ 7.81–7.72 (m, 4H), 5.34 (s, 1H), 2.69 (s, 1H), 1.33 (s, 9H). 13C{1H} NMR (100 MHz, CDCl3) δ 169.2, 162.1, 132.8, 131.5, 129.1, 121.6, 51.4, 32.3, 28.7. HRMS-ESI [M + H]+ calcd for C13H1679BrN2O+, 295.0441; found, 295.0443.
N-(tert-butyl)-3-(4-(trifluoromethyl)phenyl)-2H-azirine-2-carboxamide (7n). Compound 7n was prepared following GP-D procedure from 5-chloro-3-(4-(trifluoromethyl)phenyl)isoxazole 2g (495 mg, 2 mmol, 1 equiv.) with 2-methylpropan-2-amine 6h (439 mg, 6 mmol, 3 equiv.) as a nucleophile. A mixture of PE–EtOAc (from 5:1 to 1:1) was used as an eluent for chromatography. Colorless solid (341 mg, yield 60%). Mp: 142–143 °C (Et2O–hexane). 1H NMR (400 MHz, CDCl3) δ 8.07–8.01 (m, 2H), 7.88–7.83 (m, 2H), 5.47 (s, 1H), 2.75 (s, 1H), 1.34 (s, 9H). 13C{1H} NMR (100 MHz, CDCl3) δ 168.9, 162.2, 135.3 (q, J = 33.0 Hz), 130.6, 126.4 (q, J = 3.7 Hz), 126.2, 123.3 (q, J = 272.9 Hz), 51.6, 32.6, 28.7. HRMS-ESI [M + H]+ calcd for C14H16F3N2O+, 285.1209; found, 285.1213.
N-(tert-butyl)-3-(thiophen-2-yl)-2H-azirine-2-carboxamide (7o). Compound 7o was prepared following GP-D procedure from 5-chloro-3-(thiophen-2-yl)isoxazole 2h (371 mg, 2 mmol, 1 equiv.) with 2-methylpropan-2-amine 6h (439 mg, 6 mmol, 3 equiv.) as a nucleophile. A mixture of PE–EtOAc (from 5:1 to 1:1) was used as an eluent for chromatography. Colorless solid (351 mg, yield 79%). Mp: 135–136 °C (Et2O–hexane). 1H NMR (400 MHz, CDCl3) δ 7.89 (d, J = 3.9 Hz, 1H), 7.74 (d, J = 2.7 Hz, 1H), 7.28 (dd, J = 5.0, 3.7 Hz, 1H), 5.35 (s, 1H), 2.70 (s, 1H), 1.32 (s, 9H). 13C{1H} NMR (100 MHz, CDCl3) δ 169.2, 155.8, 135.6, 135.5, 128.6, 125.1, 51.3, 32.8, 28.6. HRMS-ESI [M + H]+ calcd for C11H15N2Os+, 223.0900; found, 223.0903.
N,3-Di-tert-butyl-2H-azirine-2-carboxamide (7p). Compound 7p was prepared following GP-D procedure from 3-(tert-butyl)-5-chloroisoxazole 2i (319 mg, 2 mmol, 1 equiv.) with 2-methylpropan-2-amine 6h (439 mg, 6 mmol, 3 equiv.) as a nucleophile. A mixture of PE–EtOAc (from 5:1 to 1:1) was used as an eluent for chromatography. Colorless solid (322 mg, yield 82%). Mp: 129–130 °C (Et2O–hexane). 1H NMR (400 MHz, CDCl3) δ 5.15 (s, 1H), 2.34 (s, 1H), 1.31 (s, 9H), 1.30 (s, 9H). 13C{1H} NMR (100 MHz, CDCl3) δ 13C NMR (101 MHz, CDCl3) δ 171.9, 170.0, 51.1, 33.3, 32.3, 28.7, 25.7. HRMS-ESI [M + H]+ calcd for C11H21N2O+, 197.1648; found, 197.1651.
N-(tert-Butyl)-2-methyl-3-phenyl-2H-azirine-2-carboxamide (7q). Compound 7q was prepared following GP-D procedure from 5-chloro-4-methyl-3-phenylisoxazole 2j (387 mg, 2 mmol, 1 equiv.) with 2-methylpropan-2-amine 6h (439 mg, 6 mmol, 3 equiv.) as a nucleophile. A mixture of PE–EtOAc (from 10:1 to 5:1) was used as an eluent for chromatography. Colorless solid (419 mg, yield 91%). Mp: 84–85 °C (Et2O–hexane).1H NMR (400 MHz, CDCl3) δ 7.91–7.85 (m, 2H), 7.68–7.63 (m, 1H), 7.62–7.56 (m, 2H), 5.22 (s, 1H), 1.60 (s, 3H), 1.27 (s, 9H). 13C{1H} NMR (100 MHz, CDCl3) δ 171.5, 168.0, 133.8, 130.1, 129.4, 122.7, 51.0, 37.1, 28.5, 17.4. HRMS-ESI [M + H]+ calcd for C14H19N2O+, 231.1492; found, 231.1495.
3-Phenyl-2H-azirine-2-carboxylic anhydride (10a). Compound 10a was prepared following GP-G procedure from 5-chloro-3-phenylisoxazole 2a (359 mg, 2 mmol, 1 equiv.). A mixture of PE–EtOAc (from 10:1 to 5:1) was used as an eluent for chromatography. Colorless solid (122 mg, yield 40%). Mp: 92–93 °C (Et2O–hexane). 1H NMR (400 MHz, C6D6) δ 7.32–7.23 (m, 4H), 7.03–6.95 (m, 2H), 6.92–6.83 (m, 4H), 2.80–2.74 (m, 2H). 13C{1H} NMR (100 MHz, C6D6) δ 174.21, 174.18, 158.0 (2C), 134.2, 131.1, 130.7 (2C), 129.44, 129.41, 121.10, 121.07, 37.7 (2C). HRMS-ESI [M + Na]+ calcd for C18H12N2NaO3+, 327.0740; found, 327.0733.
3-(4-Methoxyphenyl)-2H-azirine-2-carboxylic anhydride (10b). Compound 10b was prepared following GP-G procedure from 2b (419 mg, 2 mmol, 1 equiv.). A mixture of PE–EtOAc (from 10:1 to 2:1) was used as an eluent for chromatography. Colorless solid (242 mg, yield 66%). Mp: 96–98 °C (Et2O–hexane). 1H NMR (400 MHz, C6D6) δ 7.49–7.37 (m, 4H), 6.53–6.43 (m, 4H), 3.12 (s, 6H), 2.78–2.60 (m, 2H). 13C{1H} NMR (100 MHz, CDCl3) δ 177.3 (2C), 168.1, 167.9, 164.4, 164.3, 155.8, 155.7, 132.82, 132.79, 115.0, 114.9, 113.4, 113.3, 55.62, 55.60, 29.7, 28.7. HRMS-ESI [M + Na]+ calcd for C20H16N2NaO5+, 387.0951; found, 387.0955.
Phenyl 3-phenyl-2H-azirine-2-carboxylate (14a). Compound 14a was prepared following GP-H procedure from 5-chloro-3-phenylisoxazole 2a (359 mg, 2 mmol, 1 equiv.) with phenol 12a (282 mg, 3 mmol, 1.5 equiv.) as a nucleophile and N,N-dimethylpyridin-4-amine (733 mg, 6 mmol, 3 equiv.) as a base. A mixture of PE–EtOAc (from 20:1 to 5:1) was used as an eluent for chromatography. Colorless solid (399 mg, yield 84%). Mp: 82–83 °C (Et2O–hexane). 1H NMR (400 MHz, CDCl3) δ 8.04–7.95 (m, 2H), 7.74–7.59 (m, 3H), 7.43–7.34 (m, 2H), 7.28–7.21 (m, 1H), 7.18–7.08 (m, 2H), 3.09 (s, 1H). 13C{1H} NMR (100 MHz, CDCl3) δ 170.1, 158.2, 150.6, 134.1, 130.5, 129.4, 129.3, 125.9, 122.0, 121.3, 29.6. HRMS-ESI [M + Na]+ calcd for C15H11NNaO2+, 260.0682; found, 260.0686.
2-(2-Methoxy-2-oxoethyl)phenyl 3-phenyl-2H-azirine-2-carboxylate (14b). Compound 14b was prepared following GP-H procedure from 5-chloro-3-phenylisoxazole 2a (359 mg, 2 mmol, 1 equiv.) with methyl 2-(2-hydroxyphenyl)acetate 12b (499 mg, 3 mmol, 1.5 equiv.) as a nucleophile and N,N-dimethylpyridin-4-amine (733 mg, 6 mmol, 3 equiv.) as a base. A mixture of PE–EtOAc (from 20:1 to 5:1) was used as an eluent for chromatography. Colorless solid (835 mg, yield 92%). Mp: 52–53 °C (Et2O–hexane). 1H NMR (400 MHz, CDCl3) δ 8.08–7.99 (m, 2H), 7.74–7.61 (m, 3H), 7.35–7.29 (m, 2H), 7.25–7.15 (m, 2H), 3.64 (s, 3H), 3.58 (AB-q, J = 15.6 Hz, 2H), 3.09 (s, 1H). 13C{1H} NMR (100 MHz, CDCl3) δ 170.9, 169.8, 158.1, 148.9, 134.1, 131.3, 130.6, 129.4, 128.5, 126.3, 126.2, 122.4, 122.0, 52.0, 36.1, 29.5. HRMS-ESI [M + H]+ calcd for C18H16NO4+, 310.1074; found, 310.1077.
Prop-2-yn-1-yl 3-phenyl-2H-azirine-2-carboxylate (14c). Compound 14c was prepared following GP-H procedure from 5-chloro-3-phenylisoxazole 2a (359 mg, 2 mmol, 1 equiv.) with prop-2-yn-1-ol 12c (168 mg, 3 mmol, 1.5 equiv.) as a nucleophile and N,N-dimethylpyridin-4-amine (733 mg, 6 mmol, 3 equiv.) as a base. A mixture of PE–EtOAc (from 20:1 to 5:1) was used as an eluent for chromatography. Colorless solid (307 mg, yield 77%). Mp: 42–43 °C (Et2O–hexane). 1H NMR (400 MHz, CDCl3) δ 7.93–7.87 (m, 2H), 7.71–7.64 (m, 1H), 7.63–7.56 (m, 2H), 4.77 (dd, J = 2.4, 1.0 Hz, 2H), 2.91 (s, 1H), 2.50 (t, J = 2.4 Hz, 1H). 13C{1H} NMR (100 MHz, CDCl3) δ 170.9, 158.0, 134.0, 130.5, 129.3, 121.9, 77.2, 75.2, 52.6, 29.3. HRMS-ESI [M + Na]+ calcd for C12H9NNaO2+, 222.0526; found, 222.0527.
Benzyl 3-phenyl-2H-azirine-2-carboxylate (14d) [26]. Compound 14d was prepared following GP-H procedure from 5-chloro-3-phenylisoxazole 2a (359 mg, 2 mmol, 1 equiv.) with phenylmethanol 12d (324 mg, 3 mmol, 1.5 equiv.) as a nucleophile and N,N-dimethylpyridin-4-amine (733 mg, 6 mmol, 3 equiv.) as a base. A mixture of PE–EtOAc (from 20:1 to 5:1) was used as an eluent for chromatography. Colorless solid (636 mg, yield 79%). Mp: 65–66 °C (Et2O–hexane). 1H NMR (400 MHz, CDCl3) δ 7.92–7.88 (m, 2H), 7.69–7.64 (m, 1H), 7.62–7.57 (m, 2H), 7.39–7.34 (m, 5H), 5.23 (AB-q, J = 12.4 Hz, 2H), 2.93 (s, 1H). 13C{1H} NMR (100 MHz, CDCl3) δ 171.5, 158.4, 135.6, 133.9, 130.4, 129.3, 128.5, 128.2, 128.1, 122.2, 66.9, 29.6. HRMS-ESI [M + Na]+ calcd for C16H13NNaO2+, 274.0839; found, 274.0841.
S-Phenyl 3-phenyl-2H-azirine-2-carbothioate (15a). Compound 15a was prepared following GP-H procedure from 5-chloro-3-phenylisoxazole 2a (359 mg, 2 mmol, 1 equiv.) with benzenethiol 13a (331 mg, 3 mmol, 1.5 equiv.) as a nucleophile and N,N-dimethylpyridin-4-amine (733 mg, 6 mmol, 3 equiv.) as a base. A mixture of PE–EtOAc (from 20:1 to 5:1) was used as an eluent for chromatography. Colorless solid (410 mg, yield 81%). Mp: 54–55 °C (Et2O–hexane). 1H NMR (400 MHz, CDCl3) δ 8.01–7.96 (m, 2H), 7.74–7.69 (m, 1H), 7.67–7.61 (m, 2H), 7.45–7.39 (m, 5H), 3.22 (s, 1H). 13C{1H} NMR (100 MHz, CDCl3) δ 196.8, 158.8, 134.6, 134.3, 130.7, 129.5, 129.4, 129.1, 126.9, 121.7, 38.0. HRMS-ESI [M + Na]+ calcd for C15H11NNaOS+, 276.0454; found, 276.0456.
S-Cyclopentyl 3-phenyl-2H-azirine-2-carbothioate (15b). Compound 15b was prepared following GP-H procedure from 5-chloro-3-phenylisoxazole 2a (359 mg, 2 mmol, 1 equiv.) with cyclopentanethiol 13b (306 mg, 3 mmol, 1.5 equiv.) as a nucleophile and N,N-dimethylpyridin-4-amine (733 mg, 6 mmol, 3 equiv.) as a base. A mixture of PE–EtOAc (from 20:1 to 5:1) was used as an eluent for chromatography. Yellow oil (358 mg, yield 73%). 1H NMR (400 MHz, CDCl3) δ 7.97–7.86 (m, 2H), 7.70–7.57 (m, 3H), 3.83–3.71 (m, 1H), 3.11 (s, 1H), 2.19–2.01 (m, 2H), 1.70–1.45 (m, 6H). 13C{1H} NMR (100 MHz, CDCl3) δ 199.3, 158.7, 134.0, 130.6, 129.4, 121.9, 42.4, 38.1, 33.3, 33.1, 24.7, 24.7. HRMS-ESI [M + H]+ calcd for C14H16NOS+, 246.0947; found, 246.0951.
S-(Benzamidomethyl) 3-phenyl-2H-azirine-2-carbothioate (15c). Compound 15c was prepared following GP-H procedure from 5-chloro-3-phenylisoxazole 2a (359 mg, 2 mmol, 1 equiv.) with N-(mercaptomethyl)benzamide 13c (502 mg, 3 mmol, 1.5 equiv.) as a nucleophile and N,N-dimethylpyridin-4-amine (733 mg, 6 mmol, 3 equiv.) as a base. A mixture of PE–EtOAc (from 20:1 to 5:1) was used as an eluent for chromatography. Light yellow solid (130 mg, yield 21%). Mp: 112–113 °C (Et2O–hexane). 1H NMR (400 MHz, CDCl3) δ 8.15 (s, 1H), 7.95–7.88 (m, 2H), 7.75–7.86 (m, 1H), 7.65–7.58 (m, 2H), 7.51–7.44 (m, 2H), 7.37–7.30 (m, 2H), 7.18–7.08 (m, 1H), 3.70 (s, 2H), 3.24 (s, 1H). 13C{1H} NMR (100 MHz, CDCl3) δ 199.8, 166.0, 158.2, 137.5, 134.6, 130.8, 129.6, 128.9, 124.5, 121.1, 119.9, 37.9, 33.6. HRMS-ESI [M + H]+ calcd for C17H14N2OS+, 311.0849; found, 311.0852.
4. Conclusions
Mild and rapid procedures for the preparation of amides, anhydrides, esters, and thioesters of 2H-azirine-2-carboxylic acids involving FeCl2-catalyzed isomerization of 5-chloroisoxazoles to 2H-azirine-2-carbonyl chlorides, followed by reaction with N-, O-, or S-nucleophiles mediated by an ortho-substituted pyridine have been developed. In the case of readily available chloroisoxazoles and nucleophiles, inexpensive 2-picoline is a suitable base. To achieve the maximum yield of the acylation product with 2H-azirine-2-carbonyl chlorides, it is recommended to use the reagent 2-(trimethylsilyl)pyridine/ethyl chloroformate, which often made it possible to increase the yield of acylation products by 1.5–3 times. The mechanism of acylation with 2H-azirine-2-carbonyl chlorides was discussed.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/molecules28010275/s1, X-ray diffraction experiments (Figures S1, S2, Tables S1–S14 [27,28,29]) and NMR spectra of compounds 2c–e; 7a–q; 10a, b; 14a–d; 15a–c.
Author Contributions
Conceptualization, A.F.K. and M.S.N.; Methodology, A.F.K. and A.V.A.; synthesis, A.V.A.; writing, A.F.K., A.V.A. and M.S.N.; funding acquisition, A.F.K. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by the Russian Science Foundation, grant number 22-13-00011.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data are contained within the article.
Acknowledgments
This research was carried out using resources of the Centre for Magnetic Resonance, the Research Centre for X-ray Diffraction Studies, and the Centre for Chemical Analysis and Materials of the Science Park of Saint Petersburg State University.
Conflicts of Interest
The authors declare no conflict of interest.
Sample Availability
Samples of the compounds 2, 7, 10, 14, and 15 are available from the authors.
References
- Khlebnikov, A.F.; Novikov, M.S.; Rostovskii, N.V. Advances in 2H-azirine chemistry: A seven-year update. Tetrahedron 2019, 75, 2555–2624. [Google Scholar] [CrossRef]
- Huang, C.-Y.; Doyle, A.G. The Chemistry of Transition Metals with Three-Membered Ring Heterocycles. Chem. Rev. 2014, 114, 8153–8198. [Google Scholar] [CrossRef] [PubMed]
- Sakharov, P.A.; Novikov, M.S.; Rostovskii, N.V. 2H-Azirines in medicinal chemistry. Chem. Heterocycl. Compd. 2021, 57, 512–521. [Google Scholar] [CrossRef]
- Molinski, T.F.; Ireland, C.M. Dysidazirine, a cytotoxic azacyclopropene from the marine sponge Dysidea fragilis. J. Org. Chem. 1988, 53, 2103–2105. [Google Scholar] [CrossRef]
- Salomon, C.E.; Williams, D.H.; Faulkner, D.J. New azacyclopropene derivatives from Dysidea fragilis collected in Pohnpei. J. Nat. Prod. 1995, 58, 1463–1466. [Google Scholar] [CrossRef]
- Keffer, J.L.; Plaza, A.; Bewley, C.A. Motualevic Acids A− F, Antimicrobial Acids from the Sponge Siliquariaspongia sp. Org. Lett. 2009, 11, 1087–1090. [Google Scholar] [CrossRef]
- Skepper, C.K.; Dalisay, D.S.; Molinski, T.F. Synthesis and antifungal activity of (−)-(Z)-dysidazirine. Org. Lett. 2008, 10, 5269–5271. [Google Scholar] [CrossRef] [PubMed]
- Skepper, C.K.; Dalisay, D.S.; Molinski, T.F. Synthesis and chain-dependent antifungal activity of long-chain 2H-azirine-carboxylate esters related to dysidazirine. Bioorg. Med. Chem. Lett. 2010, 20, 2029–2032. [Google Scholar] [CrossRef]
- Rostovskii, N.V.; Koronatov, A.N.; Sakharov, P.A.; Agafonova, A.V.; Novikov, M.S.; Khlebnikov, A.F.; Rogacheva, E.V.; Kraeva, L.A. Azirine-containing dipeptides and depsipeptides: Synthesis, transformations and antibacterial activity. Org. Biomol. Chem. 2020, 18, 9448–9460. [Google Scholar] [CrossRef]
- Lipshutz, B.H.; Reuter, D.C. Cyclopeptide alkaloid model studies. A two-step conversion of 5-aminoisoxazoles to amino acid bis-amides. Tetrahedron Lett. 1988, 29, 6067–6070. [Google Scholar] [CrossRef]
- Himbert, G.; Kuhn, H.; Barz, M. Cycloadditionen, 18. 5-Aminoisoxazole durch Cycloaddition von Nitriloxiden an Inamine. Liebigs Ann. Chem. 1990, 1990, 403–407. [Google Scholar] [CrossRef]
- Mikhailov, K.I.; Galenko, E.E.; Galenko, A.V.; Novikov, M.S.; Ivanov, A.Y.; Starova, G.L.; Khlebnikov, A.F. Fe(II)-Catalyzed isomerization of 5-chloroisoxazoles to 2H-azirine-2-carbonylchlorides as a key stage in the synthesis of pyrazole-nitrogen heterocycle dyads. J. Org. Chem. 2018, 83, 3177–3187. [Google Scholar] [CrossRef] [PubMed]
- Auricchio, S.; Bini, A.; Pastormerlo, E.; Truscello, A.M. Iron dichloride induced isomerization or reductive cleavage of isoxazoles: A facile synthesis of 2-carboxy-azirines. Tetrahedron 1997, 53, 10911–10920. [Google Scholar] [CrossRef]
- Rostovskii, N.V.; Agafonova, A.V.; Smetanin, I.A.; Novikov, M.S.; Khlebnikov, A.F.; Ruvinskaya, J.O.; Starova, G.L. Metal-Catalyzed Isomerization of 5-Heteroatom-Substituted Isoxazoles as a New Route to 2-Halo-2H-azirines. Synthesis 2017, 49, 4478–4488. [Google Scholar]
- Chen, Y.; Yang, W.; Wu, J.; Sun, W.; Loh, T.-P.; Jiang, Y. 2H-Azirines as Potential Bifunctional Chemical Linkers of Cysteine Residues in Bioconjugate Technology. Org. Lett. 2020, 22, 2038–2043. [Google Scholar] [CrossRef]
- Nishiwaki, T.; Fujiyama, F.J. Studies on heterocyclic chemistry. Part XIII. Cleavage of 5-benzyl-amino-oxazoles, photoproducts of N-benzyl-2H-azirine-2-carboxamides, by dialkyl phosphite. J. Chem. Soc. Perkin Trans. 1972, 1, 1456–1459. [Google Scholar] [CrossRef]
- Ge, Y.; Sun, W.; Pei, B.; Ding, J.; Jiang, Y.; Loh, T.-P. Hoveyda–Grubbs II Catalyst: A Useful Catalyst for One-Pot Visible-Light-Promoted Ring Contraction and Olefin Metathesis Reactions. Org. Lett. 2018, 20, 2774–2777. [Google Scholar] [CrossRef]
- Fernandes, A.A.G.; da Silva, A.F.; Okada, C.Y., Jr.; Suzukawa, V.; Cormanich, R.A.; Jurberg, I.D. General Platform for the Conversion of Isoxazol-5-ones to 3, 5-Disubstituted Isoxazoles via Nucleophilic Substitutions and Palladium Catalyzed Cross-Coupling Strategies. Eur. J. Org. Chem. 2019, 2019, 3022–3034. [Google Scholar] [CrossRef]
- Galenko, E.E.; Shakirova, F.M.; Bodunov, V.A.; Novikov, M.S.; Khlebnikov, A.F. 1-(2H-Azirine-2-carbonyl)benzotriazoles: Building blocks for the synthesis of pyrrole-containing heterocycles. Org. Biomol. Chem. 2020, 18, 2283–2296. [Google Scholar] [CrossRef]
- Funt, L.D.; Krivolapova, Y.V.; Khoroshilova, O.V.; Novikov, M.S.; Khlebnikov, A.F. 2H-Azirine-2-carbonyl Azides: Preparation and Use as N-Heterocyclic Building Blocks. J. Org. Chem. 2020, 85, 4182–4194. [Google Scholar] [CrossRef]
- Efimenko, N.I.; Tomashenko, O.A.; Spiridonova, D.V.; Novikov, M.S.; Khlebnikov, A.F. Nucleophile-Induced Rearrangement of 2H-Azirine-2-carbonyl Azides to 2-(1H-Tetrazol-1-yl)acetic Acid Derivatives. Org. Lett. 2021, 23, 6362–6366. [Google Scholar] [CrossRef] [PubMed]
- Kanzian, T.; Nigst, T.A.; Maier, A.; Pichl, S.; Mayr, H. Nucleophilic reactivities of primary and secondary amines in acetonitrile. Eur. J. Org. Chem. 2009, 2009, 6379–6385. [Google Scholar] [CrossRef]
- Hall Jr, H.K. Correlation of the Base Strengths of Amines. J. Am. Chem. Soc. 1957, 79, 5441–5444. [Google Scholar] [CrossRef]
- Nantermet, P.G.; Barrow, J.C.; Lundell, G.F.; Pellicore, J.M.; Rittle, K.E.; Young, M.; Freidinger, R.M.; Connolly, T.M.; Condra, C.; Karczewski, J.; et al. Discovery of a nonpeptidic small molecule antagonist of the human platelet thrombin receptor (PAR-1). Bioorg. Med. Chem. Lett. 2002, 12, 319–323. [Google Scholar] [CrossRef]
- Okamoto, K.; Nanya, A.; Eguchi, A.; Ohe, K. Asymmetric Synthesis of 2H-Azirines with a Tetrasubstituted Stereocenter by Enantioselective Ring Contraction of Isoxazoles. Angew. Chem. Int. Ed. 2018, 57, 1039–1043. [Google Scholar] [CrossRef]
- Zhang, G.; Wang, Y.; Xu, J.; Sun, J.; Sun, F.; Zhang, Y.; Zhang, C.; Du, Y. A new hypervalent iodine (iii/v) oxidant and its application to the synthesis of 2H-azirines. Chem. Sci. 2020, 11, 947–953. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT-Integrated space-group and crystal-structure determination. Acta Cryst. 2015, A71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. 2015, C71, 3–8. [Google Scholar]

























Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).