Mitochondrial Iron Metabolism: The Crucial Actors in Diseases

 , ,
, ,
Abstract
:1. Introduction
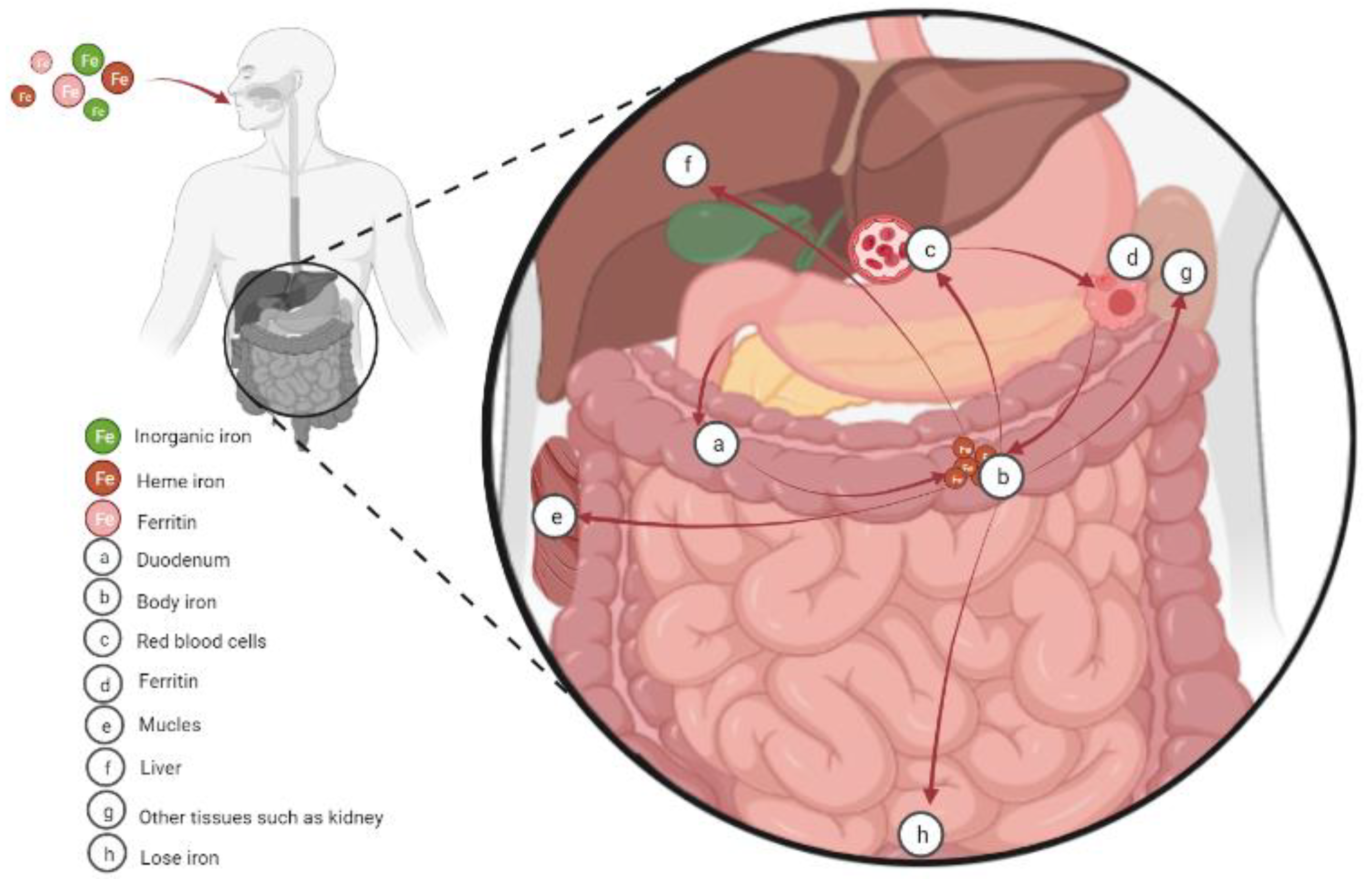
2. Cellular Iron Absorption, Utilization, and Homeostasis
3. Iron and Energy Metabolism
4. Mitochondrial Iron and Diseases
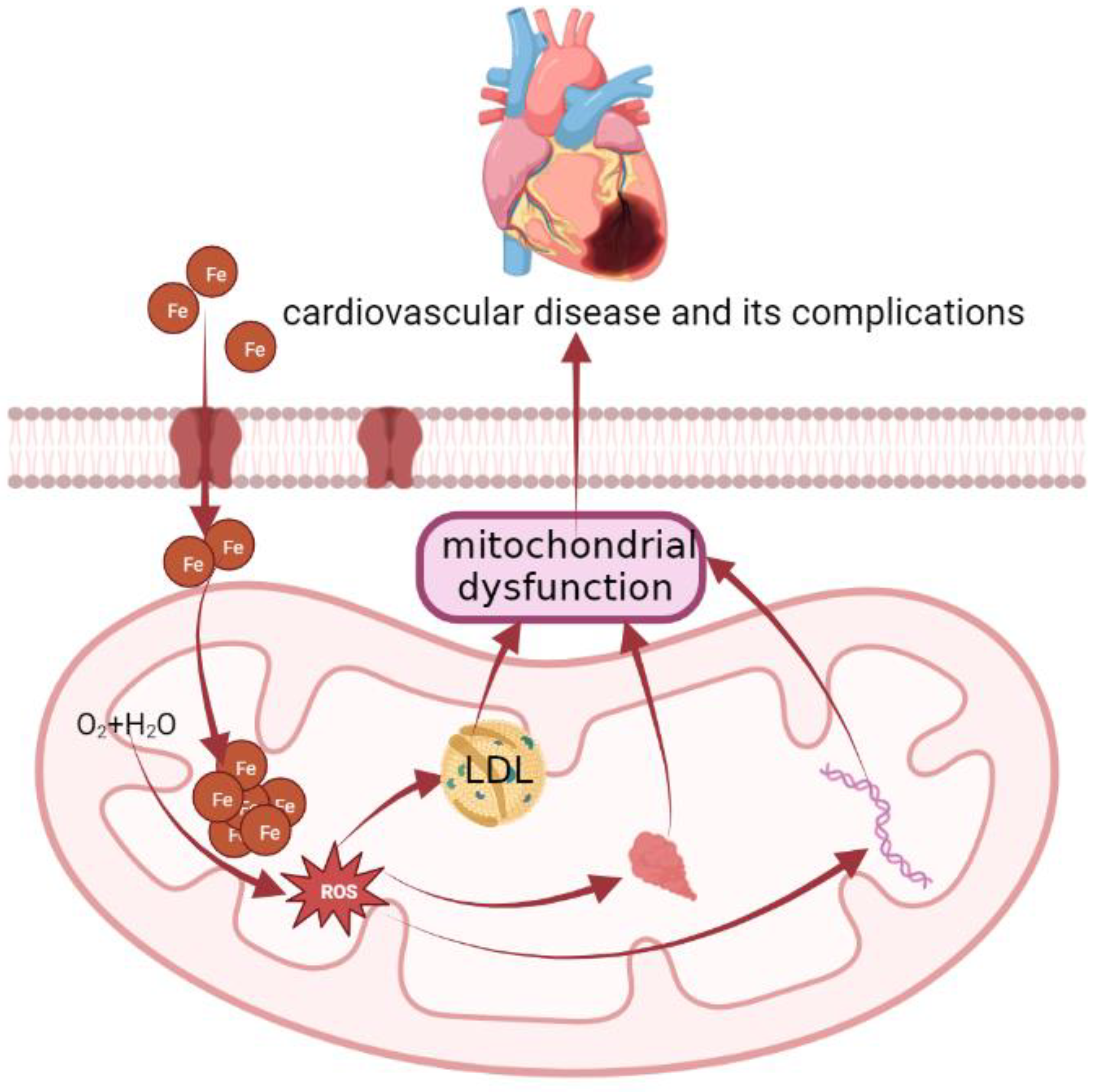
4.1. Cardiovascular Disease
4.2. Liver Disease
4.3. Muscle Atrophy
4.4. Obesity and Diabetes
4.5. Kidney Disease
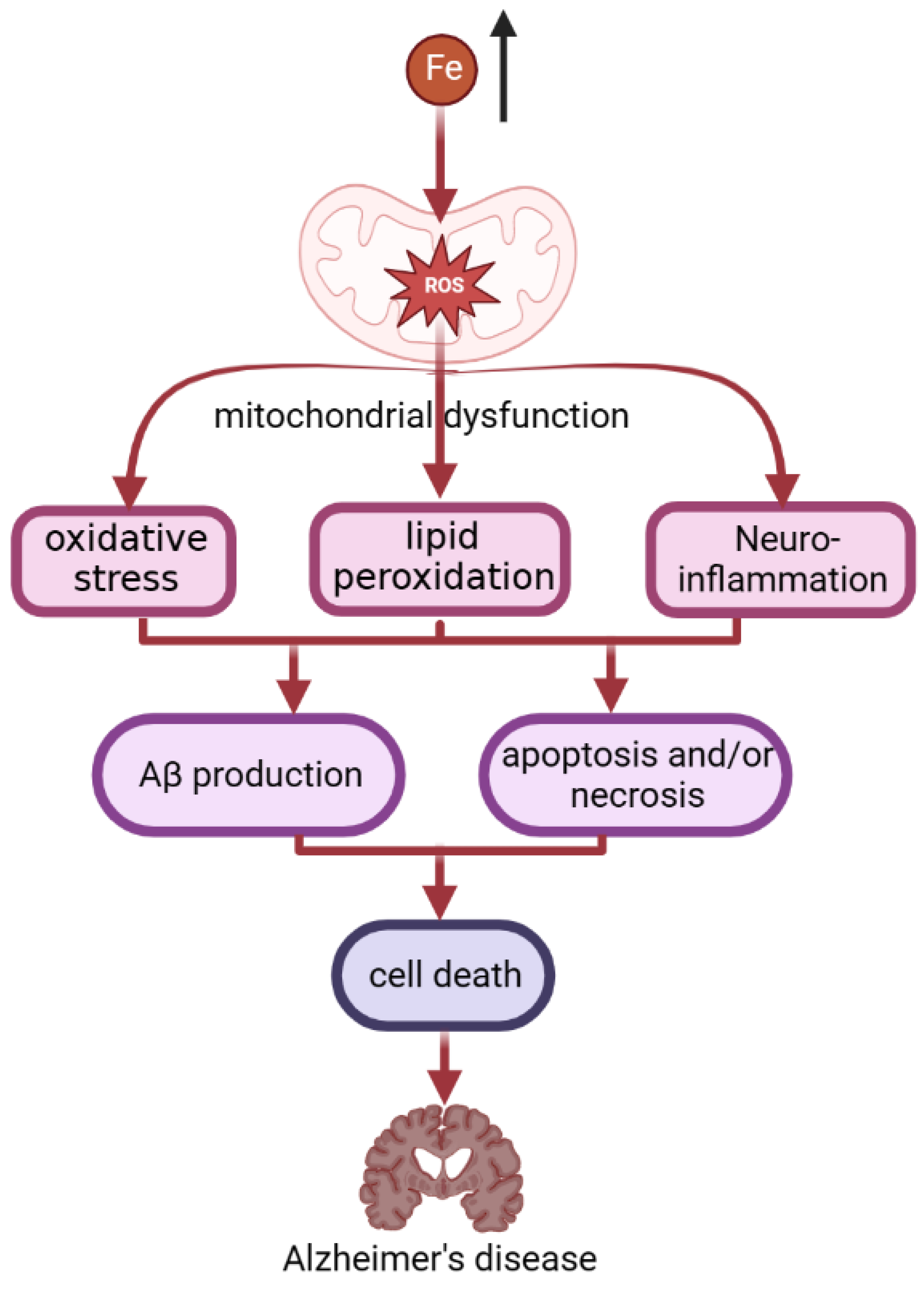
4.6. Neurodegenerative Disease (NDDs)
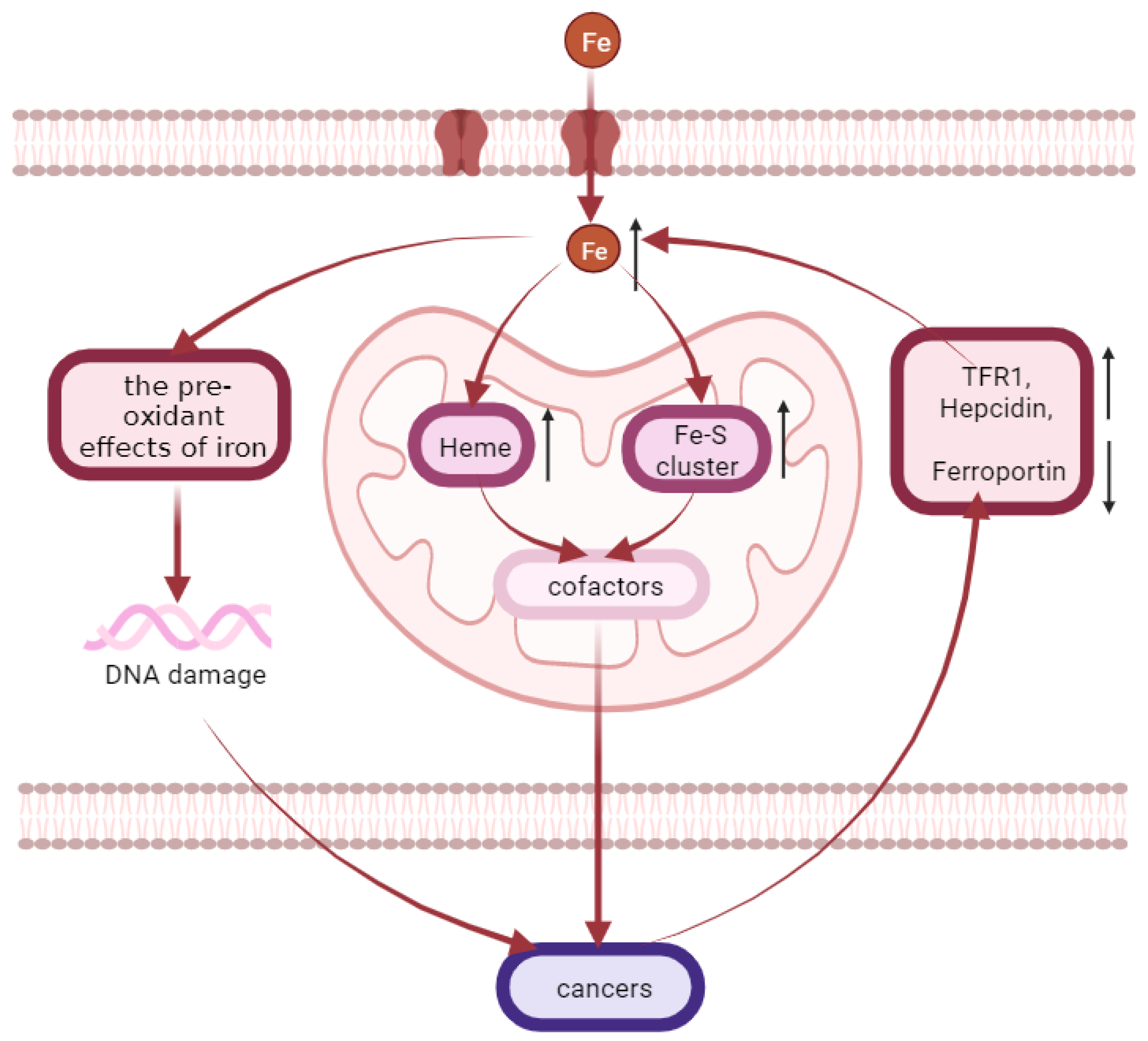
4.7. Cancers
5. Summary
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Dev, S.; Babitt, J.L. Overview of iron metabolism in health and disease. Hemodial. Int. 2017, 21 (Suppl. S1), S6–S20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galaris, D.; Pantopoulos, K. Oxidative stress and iron homeostasis: Mechanistic and health aspects. Crit. Rev. Clin. Lab. Sci. 2008, 45, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Stockwell, B.R. The role of iron and reactive oxygen species in cell death. Nat. Chem. Biol. 2014, 10, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Galy, B.; Ferring-Appel, D.; Sauer, S.W.; Kaden, S.; Lyoumi, S.; Puy, H.; Kolker, S.; Grone, H.J.; Hentze, M.W. Iron regulatory proteins secure mitochondrial iron sufficiency and function. Cell Metab. 2010, 12, 194–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kell, D.B. Iron behaving badly: Inappropriate iron chelation as a major contributor to the aetiology of vascular and other progressive inflammatory and degenerative diseases. BMC Med. Genom. 2009, 2, 2. [Google Scholar] [CrossRef] [PubMed]
- Swaminathan, S. Iron Homeostasis Pathways as Therapeutic Targets in Acute Kidney Injury. Nephron 2018, 140, 156–159. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Zhou, Q.; Wu, D.; Chen, L. Mitochondrial iron metabolism and its role in diseases. Clin. Chim. Acta 2021, 513, 6–12. [Google Scholar] [CrossRef]
- Levi, S.; Rovida, E. The role of iron in mitochondrial function. Biochim. Biophys. Acta 2009, 1790, 629–636. [Google Scholar] [CrossRef]
- McKie, A.T.; Barrow, D.; Latunde-Dada, G.O.; Rolfs, A.; Sager, G.; Mudaly, E.; Mudaly, M.; Richardson, C.; Barlow, D.; Bomford, A.; et al. An iron-regulated ferric reductase associated with the absorption of dietary iron. Science 2001, 291, 1755–1759. [Google Scholar] [CrossRef]
- Papanikolaou, G.; Pantopoulos, K. Iron metabolism and toxicity. Toxicol. Appl. Pharmacol. 2005, 202, 199–211. [Google Scholar] [CrossRef]
- Andrews, N.C. Disorders of iron metabolism. N. Engl. J. Med. 1999, 341, 1986–1995. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Pantopoulos, K. Regulation of cellular iron metabolism. Biochem. J. 2011, 434, 365–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, J.; Masaratana, P.; Latunde-Dada, G.O.; Arno, M.; Simpson, R.J.; McKie, A.T. Duodenal reductase activity and spleen iron stores are reduced and erythropoiesis is abnormal in Dcytb knockout mice exposed to hypoxic conditions. J. Nutr. 2012, 142, 1929–1934. [Google Scholar] [CrossRef] [Green Version]
- Muir, A.; Hopfer, U. Regional specificity of iron uptake by small intestinal brush-border membranes from normal and iron-deficient mice. Am. J. Physiol. 1985, 248 Pt 1, G376–G379. [Google Scholar] [CrossRef] [PubMed]
- Xue, X.; Ramakrishnan, S.K.; Weisz, K.; Triner, D.; Xie, L.; Attili, D.; Pant, A.; Gyorffy, B.; Zhan, M.; Carter-Su, C.; et al. Iron Uptake via DMT1 Integrates Cell Cycle with JAK-STAT3 Signaling to Promote Colorectal Tumorigenesis. Cell Metab. 2016, 24, 447–461. [Google Scholar] [CrossRef] [Green Version]
- Theil, E.C.; Chen, H.; Miranda, C.; Janser, H.; Elsenhans, B.; Nunez, M.T.; Pizarro, F.; Schumann, K. Absorption of iron from ferritin is independent of heme iron and ferrous salts in women and rat intestinal segments. J. Nutr. 2012, 142, 478–483. [Google Scholar] [CrossRef] [Green Version]
- Troadec, M.B.; Ward, D.M.; Lo, E.; Kaplan, J.; De Domenico, I. Induction of FPN1 transcription by MTF-1 reveals a role for ferroportin in transition metal efflux. Blood 2010, 116, 4657–4664. [Google Scholar] [CrossRef] [Green Version]
- Donovan, A.; Lima, C.A.; Pinkus, J.L.; Pinkus, G.S.; Zon, L.I.; Robine, S.; Andrews, N.C. The iron exporter ferroportin/Slc40a1 is essential for iron homeostasis. Cell Metab. 2005, 1, 191–200. [Google Scholar] [CrossRef] [Green Version]
- Donovan, A.; Brownlie, A.; Zhou, Y.; Shepard, J.; Pratt, S.J.; Moynihan, J.; Paw, B.H.; Drejer, A.; Barut, B.; Zapata, A.; et al. Positional cloning of zebrafish ferroportin1 identifies a conserved vertebrate iron exporter. Nature 2000, 403, 776–781. [Google Scholar] [CrossRef]
- Strzyz, P. Iron expulsion by exosomes drives ferroptosis resistance. Nat. Rev. Mol. Cell Biol. 2020, 21, 4–5. [Google Scholar] [CrossRef]
- Harrison, P.M.; Arosio, P. The ferritins: Molecular properties, iron storage function and cellular regulation. Biochim. Biophys. Acta 1996, 1275, 161–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pantopoulos, K.; Porwal, S.K.; Tartakoff, A.; Devireddy, L. Mechanisms of mammalian iron homeostasis. Biochemistry 2012, 51, 5705–5724. [Google Scholar] [CrossRef] [PubMed]
- Sohn, Y.S.; Ghoti, H.; Breuer, W.; Rachmilewitz, E.; Attar, S.; Weiss, G.; Cabantchik, Z.I. The role of endocytic pathways in cellular uptake of plasma non-transferrin iron. Haematologica 2012, 97, 670–678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wood, J.C. Guidelines for quantifying iron overload. Hematol. Am. Soc. Hematol. Educ. Program 2014, 2014, 210–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torti, S.V.; Torti, F.M. Iron and cancer: More ore to be mined. Nat. Rev. Cancer 2013, 13, 342–355. [Google Scholar] [CrossRef] [Green Version]
- Muckenthaler, M.U.; Galy, B.; Hentze, M.W. Systemic iron homeostasis and the iron-responsive element/iron-regulatory protein (IRE/IRP) regulatory network. Annu. Rev. Nutr. 2008, 28, 197–213. [Google Scholar] [CrossRef]
- Rouault, T.A. The role of iron regulatory proteins in mammalian iron homeostasis and disease. Nat. Chem. Biol. 2006, 2, 406–414. [Google Scholar] [CrossRef]
- Hentze, M.W.; Muckenthaler, M.U.; Galy, B.; Camaschella, C. Two to tango: Regulation of Mammalian iron metabolism. Cell 2010, 142, 24–38. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Pan, X.; Pan, G.; Song, Z.; He, Y.; Zhang, S.; Ye, X.; Yang, X.; Xie, E.; Wang, X.; et al. Transferrin Receptor 1 Regulates Thermogenic Capacity and Cell Fate in Brown/Beige Adipocytes. Adv. Sci. 2020, 7, 1903366. [Google Scholar] [CrossRef] [Green Version]
- Rensvold, J.W.; Krautkramer, K.A.; Dowell, J.A.; Denu, J.M.; Pagliarini, D.J. Iron Deprivation Induces Transcriptional Regulation of Mitochondrial Biogenesis. J. Biol. Chem. 2016, 291, 20827–20837. [Google Scholar] [CrossRef]
- Swenson, S.A.; Moore, C.M.; Marcero, J.R.; Medlock, A.E.; Reddi, A.R.; Khalimonchuk, O. From Synthesis to Utilization: The Ins and Outs of Mitochondrial Heme. Cells 2020, 9, 579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braymer, J.J.; Lill, R. Iron-sulfur cluster biogenesis and trafficking in mitochondria. J. Biol. Chem. 2017, 292, 12754–12763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pandey, A.K.; Pain, J.; Dancis, A.; Pain, D. Mitochondria export iron-sulfur and sulfur intermediates to the cytoplasm for iron-sulfur cluster assembly and tRNA thiolation in yeast. J. Biol. Chem. 2019, 294, 9489–9502. [Google Scholar] [CrossRef]
- Lill, R.; Muhlenhoff, U. Iron-sulfur protein biogenesis in eukaryotes: Components and mechanisms. Annu. Rev. Cell Dev. Biol. 2006, 22, 457–486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zorov, D.B.; Krasnikov, B.F.; Kuzminova, A.E.; Vysokikh, M.; Zorova, L.D. Mitochondria revisited. Alternative functions of mitochondria. Biosci. Rep. 1997, 17, 507–520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paul, B.T.; Manz, D.H.; Torti, F.M.; Torti, S.V. Mitochondria and Iron: Current questions. Expert Rev. Hematol. 2017, 10, 65–79. [Google Scholar] [CrossRef] [Green Version]
- Ward, D.M.; Cloonan, S.M. Mitochondrial Iron in Human Health and Disease. Annu. Rev. Physiol. 2019, 81, 453–482. [Google Scholar] [CrossRef]
- Santambrogio, P.; Dusi, S.; Guaraldo, M.; Rotundo, L.I.; Broccoli, V.; Garavaglia, B.; Tiranti, V.; Levi, S. Mitochondrial iron and energetic dysfunction distinguish fibroblasts and induced neurons from pantothenate kinase-associated neurodegeneration patients. Neurobiol. Dis. 2015, 81, 144–153. [Google Scholar] [CrossRef] [Green Version]
- Tavsan, Z.; Ayar Kayali, H. The Variations of Glycolysis and TCA Cycle Intermediate Levels Grown in Iron and Copper Mediums of Trichoderma harzianum. Appl. Biochem. Biotechnol. 2015, 176, 76–85. [Google Scholar] [CrossRef]
- Manceau, H.; Ausseil, J.; Masson, D.; Feugeas, J.P.; Sablonniere, B.; Guieu, R.; Puy, H.; Peoc’h, K. Neglected Comorbidity of Chronic Heart Failure: Iron Deficiency. Nutrients 2022, 14, 3214. [Google Scholar] [CrossRef]
- Corradi, F.; Fischetti, I.; De Caterina, R. Addenda online Ferro e cardiopatia ischemica stabile-lezioni dallo scompenso cardiaco. G. Ital. Cardiol. 2019, 20. [Google Scholar] [CrossRef]
- Payne, R.M. The Heart in Friedreich’s Ataxia: Basic Findings and Clinical Implications. Prog. Pediatr. Cardiol. 2011, 31, 103–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pietrangelo, A. Iron and the liver. Liver Int. 2016, 36 (Suppl. S1), 116–123. [Google Scholar] [CrossRef]
- DeRuisseau, K.C.; Park, Y.M.; DeRuisseau, L.R.; Cowley, P.M.; Fazen, C.H.; Doyle, R.P. Aging-related changes in the iron status of skeletal muscle. Exp. Gerontol. 2013, 48, 1294–1302. [Google Scholar] [CrossRef] [Green Version]
- Kusminski, C.M.; Ghaben, A.L.; Morley, T.S.; Samms, R.J.; Adams, A.C.; An, Y.; Johnson, J.A.; Joffin, N.; Onodera, T.; Crewe, C.; et al. A Novel Model of Diabetic Complications: Adipocyte Mitochondrial Dysfunction Triggers Massive beta-Cell Hyperplasia. Diabetes 2020, 69, 313–330. [Google Scholar] [CrossRef]
- Nankivell, B.J.; Boadle, R.A.; Harris, D.C. Iron accumulation in human chronic renal disease. Am. J. Kidney Dis. 1992, 20, 580–584. [Google Scholar] [CrossRef] [PubMed]
- Mezzaroba, L.; Alfieri, D.F.; Colado Simao, A.N.; Vissoci Reiche, E.M. The role of zinc, copper, manganese and iron in neurodegenerative diseases. Neurotoxicology 2019, 74, 230–241. [Google Scholar] [CrossRef] [PubMed]
- Mostile, G.; Cicero, C.E.; Giuliano, L.; Zappia, M.; Nicoletti, A. Iron and Parkinson’s disease: A systematic review and meta-analysis. Mol. Med. Rep. 2017, 15, 3383–3389. [Google Scholar] [CrossRef]
- Bajbouj, K.; Shafarin, J.; Abdalla, M.Y.; Ahmad, I.M.; Hamad, M. Estrogen-induced disruption of intracellular iron metabolism leads to oxidative stress, membrane damage, and cell cycle arrest in MCF-7 cells. Tumour Biol. 2017, 39, 1010428317726184. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.R.; Zweier, J.L. Cardiac mitochondria and reactive oxygen species generation. Circ. Res. 2014, 114, 524–537. [Google Scholar] [CrossRef]
- Gammella, E.; Recalcati, S.; Rybinska, I.; Buratti, P.; Cairo, G. Iron-induced damage in cardiomyopathy: Oxidative-dependent and independent mechanisms. Oxid. Med. Cell. Longev. 2015, 2015, 230182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melenovsky, V.; Petrak, J.; Mracek, T.; Benes, J.; Borlaug, B.A.; Nuskova, H.; Pluhacek, T.; Spatenka, J.; Kovalcikova, J.; Drahota, Z.; et al. Myocardial iron content and mitochondrial function in human heart failure: A direct tissue analysis. Eur. J. Heart Fail. 2017, 19, 522–530. [Google Scholar] [CrossRef] [PubMed]
- Loncar, G.; Obradovic, D.; Thiele, H.; von Haehling, S.; Lainscak, M. Iron deficiency in heart failure. ESC Heart Fail. 2021, 8, 2368–2379. [Google Scholar] [CrossRef] [PubMed]
- von Haehling, S.; Ebner, N.; Evertz, R.; Ponikowski, P.; Anker, S.D. Iron Deficiency in Heart Failure: An Overview. JACC Heart Fail. 2019, 7, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Chopra, V.K.; Anker, S.D. Anaemia, iron deficiency and heart failure in 2020: Facts and numbers. ESC Heart Fail. 2020, 7, 2007–2011. [Google Scholar] [CrossRef] [PubMed]
- Hoes, M.F.; Grote Beverborg, N.; Kijlstra, J.D.; Kuipers, J.; Swinkels, D.W.; Giepmans, B.N.G.; Rodenburg, R.J.; van Veldhuisen, D.J.; de Boer, R.A.; van der Meer, P. Iron deficiency impairs contractility of human cardiomyocytes through decreased mitochondrial function. Eur. J. Heart Fail. 2018, 20, 910–919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rineau, E.; Gaillard, T.; Gueguen, N.; Procaccio, V.; Henrion, D.; Prunier, F.; Lasocki, S. Iron deficiency without anemia is responsible for decreased left ventricular function and reduced mitochondrial complex I activity in a mouse model. Int. J. Cardiol. 2018, 266, 206–212. [Google Scholar] [CrossRef] [PubMed]
- Ascherio, A.; Willett, W.C.; Rimm, E.B.; Giovannucci, E.L.; Stampfer, M.J. Dietary iron intake and risk of coronary disease among men. Circulation 1994, 89, 969–974. [Google Scholar] [CrossRef] [Green Version]
- Van der, A.D.; Peeters, P.H.; Grobbee, D.E.; Marx, J.J.; van der Schouw, Y.T. Dietary haem iron and coronary heart disease in women. Eur. Heart J. 2005, 26, 257–262. [Google Scholar] [CrossRef] [Green Version]
- Kiechl, S.; Willeit, J.; Egger, G.; Poewe, W.; Oberhollenzer, F. Body iron stores and the risk of carotid atherosclerosis: Prospective results from the Bruneck study. Circulation 1997, 96, 3300–3307. [Google Scholar] [CrossRef]
- Vinchi, F.; Porto, G.; Simmelbauer, A.; Altamura, S.; Passos, S.T.; Garbowski, M.; Silva, A.M.N.; Spaich, S.; Seide, S.E.; Sparla, R.; et al. Atherosclerosis is aggravated by iron overload and ameliorated by dietary and pharmacological iron restriction. Eur. Heart J. 2020, 41, 2681–2695. [Google Scholar] [CrossRef]
- Brissot, P.; Ropert, M.; Le Lan, C.; Loreal, O. Non-transferrin bound iron: A key role in iron overload and iron toxicity. Biochim. Biophys. Acta 2012, 1820, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Michael, S.; Petrocine, S.V.; Qian, J.; Lamarche, J.B.; Knutson, M.D.; Garrick, M.D.; Koeppen, A.H. Iron and iron-responsive proteins in the cardiomyopathy of Friedreich’s ataxia. Cerebellum 2006, 5, 257–267. [Google Scholar] [CrossRef] [PubMed]
- Whitnall, M.; Suryo Rahmanto, Y.; Sutak, R.; Xu, X.; Becker, E.M.; Mikhael, M.R.; Ponka, P.; Richardson, D.R. The MCK mouse heart model of Friedreich’s ataxia: Alterations in iron-regulated proteins and cardiac hypertrophy are limited by iron chelation. Proc. Natl. Acad. Sci. USA 2008, 105, 9757–9762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puccio, H.; Simon, D.; Cossee, M.; Criqui-Filipe, P.; Tiziano, F.; Melki, J.; Hindelang, C.; Matyas, R.; Rustin, P.; Koenig, M. Mouse models for Friedreich ataxia exhibit cardiomyopathy, sensory nerve defect and Fe-S enzyme deficiency followed by intramitochondrial iron deposits. Nat. Genet. 2001, 27, 181–186. [Google Scholar] [CrossRef]
- Gao, X.; Qian, M.; Campian, J.L.; Marshall, J.; Zhou, Z.; Roberts, A.M.; Kang, Y.J.; Prabhu, S.D.; Sun, X.F.; Eaton, J.W. Mitochondrial dysfunction may explain the cardiomyopathy of chronic iron overload. Free Radic. Biol. Med. 2010, 49, 401–407. [Google Scholar] [CrossRef] [Green Version]
- Chang, H.C.; Wu, R.; Shang, M.; Sato, T.; Chen, C.; Shapiro, J.S.; Liu, T.; Thakur, A.; Sawicki, K.T.; Prasad, S.V.; et al. Reduction in mitochondrial iron alleviates cardiac damage during injury. EMBO Mol. Med. 2016, 8, 247–267. [Google Scholar] [CrossRef]
- Velasco-Sanchez, D.; Aracil, A.; Montero, R.; Mas, A.; Jimenez, L.; O’Callaghan, M.; Tondo, M.; Capdevila, A.; Blanch, J.; Artuch, R.; et al. Combined therapy with idebenone and deferiprone in patients with Friedreich’s ataxia. Cerebellum 2011, 10, 1–8. [Google Scholar] [CrossRef]
- Sohn, Y.S.; Breuer, W.; Munnich, A.; Cabantchik, Z.I. Redistribution of accumulated cell iron: A modality of chelation with therapeutic implications. Blood 2008, 111, 1690–1699. [Google Scholar] [CrossRef] [Green Version]
- Ichikawa, Y.; Bayeva, M.; Ghanefar, M.; Potini, V.; Sun, L.; Mutharasan, R.K.; Wu, R.; Khechaduri, A.; Jairaj Naik, T.; Ardehali, H. Disruption of ATP-binding cassette B8 in mice leads to cardiomyopathy through a decrease in mitochondrial iron export. Proc. Natl. Acad. Sci. USA 2012, 109, 4152–4157. [Google Scholar] [CrossRef]
- Elas, M.; Bielanska, J.; Pustelny, K.; Plonka, P.M.; Drelicharz, L.; Skorka, T.; Tyrankiewicz, U.; Wozniak, M.; Heinze-Paluchowska, S.; Walski, M.; et al. Detection of mitochondrial dysfunction by EPR technique in mouse model of dilated cardiomyopathy. Free Radic. Biol. Med. 2008, 45, 321–328. [Google Scholar] [CrossRef] [PubMed]
- Di Renzo, L.; Galvano, F.; Orlandi, C.; Bianchi, A.; Di Giacomo, C.; La Fauci, L.; Acquaviva, R.; De Lorenzo, A. Oxidative stress in normal-weight obese syndrome. Obesity 2010, 18, 2125–2130. [Google Scholar] [CrossRef] [PubMed]
- Grattagliano, I.; Palmieri, V.O.; Portincasa, P.; Moschetta, A.; Palasciano, G. Oxidative stress-induced risk factors associated with the metabolic syndrome: A unifying hypothesis. J. Nutr. Biochem. 2008, 19, 491–504. [Google Scholar] [CrossRef]
- Itabe, H.; Obama, T.; Kato, R. The Dynamics of Oxidized LDL during Atherogenesis. J. Lipids 2011, 2011, 418313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fantuzzi, G.; Mazzone, T. Adipose tissue and atherosclerosis: Exploring the connection. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 996–1003. [Google Scholar] [CrossRef] [PubMed]
- Burgos Alves, M.I.; Aviles Plaza, F.; Martinez-Tomas, R.; Sanchez-Campillo, M.; Larque, E.; Perez-Llamas, F.; Martinez Hernandez, P.; Parra Pallares, S. Oxidized LDL and its correlation with lipid profile and oxidative stress biomarkers in young healthy Spanish subjects. J. Physiol. Biochem. 2010, 66, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Di Renzo, L.; Carraro, A.; Valente, R.; Iacopino, L.; Colica, C.; De Lorenzo, A. Intake of red wine in different meals modulates oxidized LDL level, oxidative and inflammatory gene expression in healthy people: A randomized crossover trial. Oxid. Med. Cell. Longev. 2014, 2014, 681318. [Google Scholar] [CrossRef] [Green Version]
- Klip, I.T.; Comin-Colet, J.; Voors, A.A.; Ponikowski, P.; Enjuanes, C.; Banasiak, W.; Lok, D.J.; Rosentryt, P.; Torrens, A.; Polonski, L.; et al. Iron deficiency in chronic heart failure: An international pooled analysis. Am. Heart J. 2013, 165, 575–582.e3. [Google Scholar] [CrossRef]
- Anker, S.D.; Comin Colet, J.; Filippatos, G.; Willenheimer, R.; Dickstein, K.; Drexler, H.; Luscher, T.F.; Bart, B.; Banasiak, W.; Niegowska, J.; et al. Ferric carboxymaltose in patients with heart failure and iron deficiency. N. Engl. J. Med. 2009, 361, 2436–2448. [Google Scholar] [CrossRef] [Green Version]
- Ponikowski, P.; van Veldhuisen, D.J.; Comin-Colet, J.; Ertl, G.; Komajda, M.; Mareev, V.; McDonagh, T.; Parkhomenko, A.; Tavazzi, L.; Levesque, V.; et al. Beneficial effects of long-term intravenous iron therapy with ferric carboxymaltose in patients with symptomatic heart failure and iron deficiencydagger. Eur. Heart J. 2015, 36, 657–668. [Google Scholar] [CrossRef]
- Camaschella, C.; Nai, A. Ineffective erythropoiesis and regulation of iron status in iron loading anaemias. Br. J. Haematol. 2016, 172, 512–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valenti, L.; Fracanzani, A.L.; Bugianesi, E.; Dongiovanni, P.; Galmozzi, E.; Vanni, E.; Canavesi, E.; Lattuada, E.; Roviaro, G.; Marchesini, G.; et al. HFE genotype, parenchymal iron accumulation, and liver fibrosis in patients with nonalcoholic fatty liver disease. Gastroenterology 2010, 138, 905–912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cabantchik, Z.I. Labile iron in cells and body fluids: Physiology, pathology, and pharmacology. Front. Pharmacol. 2014, 5, 45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pietrangelo, A. Metals, oxidative stress, and hepatic fibrogenesis. Semin. Liver Dis. 1996, 16, 13–30. [Google Scholar] [CrossRef] [PubMed]
- Pietrangelo, A. Pathogens, Metabolic Adaptation, and Human Diseases—An Iron-Thrifty Genetic Model. Gastroenterology 2015, 149, 834–838. [Google Scholar] [CrossRef]
- Ikeda, Y.; Imao, M.; Satoh, A.; Watanabe, H.; Hamano, H.; Horinouchi, Y.; Izawa-Ishizawa, Y.; Kihira, Y.; Miyamoto, L.; Ishizawa, K.; et al. Iron-induced skeletal muscle atrophy involves an Akt-forkhead box O3-E3 ubiquitin ligase-dependent pathway. J. Trace Elem. Med. Biol. 2016, 35, 66–76. [Google Scholar] [CrossRef]
- Evans, W.J. What is sarcopenia? J. Gerontol. A Biol. Sci. Med. Sci. 1995, 50, 5–8. [Google Scholar] [CrossRef]
- Fulster, S.; Tacke, M.; Sandek, A.; Ebner, N.; Tschope, C.; Doehner, W.; Anker, S.D.; von Haehling, S. Muscle wasting in patients with chronic heart failure: Results from the studies investigating co-morbidities aggravating heart failure (SICA-HF). Eur. Heart J. 2013, 34, 512–519. [Google Scholar] [CrossRef] [Green Version]
- Mak, R.H.; Ikizler, A.T.; Kovesdy, C.P.; Raj, D.S.; Stenvinkel, P.; Kalantar-Zadeh, K. Wasting in chronic kidney disease. J. Cachexia Sarcopenia Muscle 2011, 2, 9–25. [Google Scholar] [CrossRef] [Green Version]
- Marzetti, E.; Hwang, J.C.; Lees, H.A.; Wohlgemuth, S.E.; Dupont-Versteegden, E.E.; Carter, C.S.; Bernabei, R.; Leeuwenburgh, C. Mitochondrial death effectors: Relevance to sarcopenia and disuse muscle atrophy. Biochim. Biophys. Acta 2010, 1800, 235–244. [Google Scholar] [CrossRef]
- Kramer, I.F.; Snijders, T.; Smeets, J.S.J.; Leenders, M.; van Kranenburg, J.; den Hoed, M.; Verdijk, L.B.; Poeze, M.; van Loon, L.J.C. Extensive Type II Muscle Fiber Atrophy in Elderly Female Hip Fracture Patients. J. Gerontol. A Biol. Sci. Med. Sci. 2017, 72, 1369–1375. [Google Scholar] [CrossRef] [Green Version]
- Sayed, R.K.; de Leonardis, E.C.; Guerrero-Martinez, J.A.; Rahim, I.; Mokhtar, D.M.; Saleh, A.M.; Abdalla, K.E.; Pozo, M.J.; Escames, G.; Lopez, L.C.; et al. Identification of morphological markers of sarcopenia at early stage of aging in skeletal muscle of mice. Exp. Gerontol. 2016, 83, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Altun, M.; Edstrom, E.; Spooner, E.; Flores-Moralez, A.; Bergman, E.; Tollet-Egnell, P.; Norstedt, G.; Kessler, B.M.; Ulfhake, B. Iron load and redox stress in skeletal muscle of aged rats. Muscle Nerve 2007, 36, 223–233. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Knutson, M.D.; Carter, C.S.; Leeuwenburgh, C. Iron accumulation with age, oxidative stress and functional decline. PLoS ONE 2008, 3, e2865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kasztura, M.; Dziegala, M.; Kobak, K.; Bania, J.; Mazur, G.; Banasiak, W.; Ponikowski, P.; Jankowska, E.A. Both iron excess and iron depletion impair viability of rat H9C2 cardiomyocytes and L6G8C5 myocytes. Kardiol. Pol. 2017, 75, 267–275. [Google Scholar] [CrossRef]
- Ding, H.; Chen, S.; Pan, X.; Dai, X.; Pan, G.; Li, Z.; Mai, X.; Tian, Y.; Zhang, S.; Liu, B.; et al. Transferrin receptor 1 ablation in satellite cells impedes skeletal muscle regeneration through activation of ferroptosis. J. Cachexia Sarcopenia Muscle 2021, 12, 746–768. [Google Scholar] [CrossRef]
- Chung, J.Y.; Kang, H.T.; Lee, D.C.; Lee, H.R.; Lee, Y.J. Body composition and its association with cardiometabolic risk factors in the elderly: A focus on sarcopenic obesity. Arch. Gerontol. Geriatr. 2013, 56, 270–278. [Google Scholar] [CrossRef]
- Perna, S.; Peroni, G.; Faliva, M.A.; Bartolo, A.; Naso, M.; Miccono, A.; Rondanelli, M. Sarcopenia and sarcopenic obesity in comparison: Prevalence, metabolic profile, and key differences. A cross-sectional study in Italian hospitalized elderly. Aging Clin. Exp. Res. 2017, 29, 1249–1258. [Google Scholar] [CrossRef]
- Kim, T.H.; Hwang, H.J.; Kim, S.H. Relationship between Serum Ferritin Levels and Sarcopenia in Korean Females Aged 60 Years and Older Using the Fourth Korea National Health and Nutrition Examination Survey (KNHANES IV-2, 3), 2008–2009. PLoS ONE 2014, 9, e90105. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Hwang, J.C.; Lees, H.A.; Wohlgemuth, S.E.; Knutson, M.D.; Judge, A.R.; Dupont-Versteegden, E.E.; Marzetti, E.; Leeuwenburgh, C. Long-term perturbation of muscle iron homeostasis following hindlimb suspension in old rats is associated with high levels of oxidative stress and impaired recovery from atrophy. Exp. Gerontol. 2012, 47, 100–108. [Google Scholar] [CrossRef]
- Argiles, J.M.; Fontes-Oliveira, C.C.; Toledo, M.; Lopez-Soriano, F.J.; Busquets, S. Cachexia: A problem of energetic inefficiency. J. Cachexia Sarcopenia Muscle 2014, 5, 279–286. [Google Scholar] [CrossRef] [PubMed]
- Halon-Golabek, M.; Borkowska, A.; Herman-Antosiewicz, A.; Antosiewicz, J. Iron Metabolism of the Skeletal Muscle and Neurodegeneration. Front. Neurosci. 2019, 13, 165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reardon, T.F.; Allen, D.G. Iron injections in mice increase skeletal muscle iron content, induce oxidative stress and reduce exercise performance. Exp. Physiol. 2009, 94, 720–730. [Google Scholar] [CrossRef]
- Kobak, K.; Kasztura, M.; Dziegala, M.; Bania, J.; Kapusniak, V.; Banasiak, W.; Ponikowski, P.; Jankowska, E.A. Iron limitation promotes the atrophy of skeletal myocytes, whereas iron supplementation prevents this process in the hypoxic conditions. Int. J. Mol. Med. 2018, 41, 2678–2686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, P.; He, J.; Wang, F.; Gong, J.; Wang, L.; Wu, Q.; Li, W.; Liu, H.; Wang, J.; Zhang, K.; et al. Hemojuvelin is a novel suppressor for Duchenne muscular dystrophy and age-related muscle wasting. J. Cachexia Sarcopenia Muscle 2019, 10, 557–573. [Google Scholar] [CrossRef] [Green Version]
- Ikeda, Y.; Funamoto, M.; Tsuchiya, K. The role of iron in obesity and diabetes. J. Med. Investig. 2022, 69, 1–7. [Google Scholar] [CrossRef]
- Yan, H.F.; Liu, Z.Y.; Guan, Z.A.; Guo, C. Deferoxamine ameliorates adipocyte dysfunction by modulating iron metabolism in ob/ob mice. Endocr. Connect. 2018, 7, 604–616. [Google Scholar] [CrossRef] [Green Version]
- Fernandez-Real, J.M.; Lopez-Bermejo, A.; Ricart, W. Cross-talk between iron metabolism and diabetes. Diabetes 2002, 51, 2348–2354. [Google Scholar] [CrossRef] [Green Version]
- Orr, J.S.; Kennedy, A.; Anderson-Baucum, E.K.; Webb, C.D.; Fordahl, S.C.; Erikson, K.M.; Zhang, Y.; Etzerodt, A.; Moestrup, S.K.; Hasty, A.H. Obesity alters adipose tissue macrophage iron content and tissue iron distribution. Diabetes 2014, 63, 421–432. [Google Scholar] [CrossRef] [Green Version]
- Tajima, S.; Ikeda, Y.; Sawada, K.; Yamano, N.; Horinouchi, Y.; Kihira, Y.; Ishizawa, K.; Izawa-Ishizawa, Y.; Kawazoe, K.; Tomita, S.; et al. Iron reduction by deferoxamine leads to amelioration of adiposity via the regulation of oxidative stress and inflammation in obese and type 2 diabetes KKAy mice. Am. J. Physiol. Endocrinol. Metab. 2012, 302, E77–E86. [Google Scholar] [CrossRef]
- Xue, H.; Chen, D.; Zhong, Y.K.; Zhou, Z.D.; Fang, S.X.; Li, M.Y.; Guo, C. Deferoxamine ameliorates hepatosteatosis via several mechanisms in ob/ob mice. Ann. N. Y. Acad. Sci. 2016, 1375, 52–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horinouchi, Y.; Ikeda, Y.; Tamaki, T. Body iron accumulation in obesity, diabetes and its complications, and the possibility of therapeutic application by iron regulation. Nihon Yakurigaku Zasshi 2019, 154, 316–321. [Google Scholar] [CrossRef] [PubMed]
- Blesia, V.; Patel, V.B.; Al-Obaidi, H.; Renshaw, D.; Zariwala, M.G. Excessive Iron Induces Oxidative Stress Promoting Cellular Perturbations and Insulin Secretory Dysfunction in MIN6 Beta Cells. Cells 2021, 10, 1141. [Google Scholar] [CrossRef] [PubMed]
- Qiu, F.; Wu, L.; Yang, G.; Zhang, C.; Liu, X.; Sun, X.; Chen, X.; Wang, N. The role of iron metabolism in chronic diseases related to obesity. Mol. Med. 2022, 28, 130. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Navarrete, J.M.; Ortega, F.; Moreno, M.; Ricart, W.; Fernandez-Real, J.M. Fine-tuned iron availability is essential to achieve optimal adipocyte differentiation and mitochondrial biogenesis. Diabetologia 2014, 57, 1957–1967. [Google Scholar] [CrossRef] [PubMed]
- Medina-Gomez, G. Mitochondria and endocrine function of adipose tissue. Best Pract. Res. Clin. Endocrinol. Metab. 2012, 26, 791–804. [Google Scholar] [CrossRef]
- Rumberger, J.M.; Peters, T., Jr.; Burrington, C.; Green, A. Transferrin and iron contribute to the lipolytic effect of serum in isolated adipocytes. Diabetes 2004, 53, 2535–2541. [Google Scholar] [CrossRef] [Green Version]
- Rajpathak, S.N.; Wylie-Rosett, J.; Gunter, M.J.; Negassa, A.; Kabat, G.C.; Rohan, T.E.; Crandall, J.; Diabetes Prevention Program Research Group. Biomarkers of body iron stores and risk of developing type 2 diabetes. Diabetes Obes. Metab. 2009, 11, 472–479. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.C.; Tang, T.T.; Lv, L.L.; Lan, H.Y. Renal tubule injury: A driving force toward chronic kidney disease. Kidney Int. 2018, 93, 568–579. [Google Scholar] [CrossRef]
- Baliga, R.; Ueda, N.; Shah, S.V. Increase in bleomycin-detectable iron in ischaemia/reperfusion injury to rat kidneys. Biochem. J. 1993, 291 Pt 3, 901–905. [Google Scholar] [CrossRef]
- Baliga, R.; Zhang, Z.; Baliga, M.; Ueda, N.; Shah, S.V. In vitro and in vivo evidence suggesting a role for iron in cisplatin-induced nephrotoxicity. Kidney Int. 1998, 53, 394–401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wish, J.B.; Aronoff, G.R.; Bacon, B.R.; Brugnara, C.; Eckardt, K.U.; Ganz, T.; Macdougall, I.C.; Nunez, J.; Perahia, A.J.; Wood, J.C. Positive Iron Balance in Chronic Kidney Disease: How Much is Too Much and How to Tell? Am. J. Nephrol. 2018, 47, 72–83. [Google Scholar] [CrossRef] [PubMed]
- van Swelm, R.P.L.; Wetzels, J.F.M.; Swinkels, D.W. The multifaceted role of iron in renal health and disease. Nat. Rev. Nephrol. 2020, 16, 77–98. [Google Scholar] [CrossRef] [PubMed]
- ElAlfy, M.S.; Khalil Elsherif, N.H.; Ebeid, F.S.E.; Ismail, E.A.R.; Ahmed, K.A.; Darwish, Y.W.; Ibrahim, A.S.; Elghamry, I.R.F.; Shokrey, N.A.; Alajeil, D.N. Renal iron deposition by magnetic resonance imaging in pediatric beta-thalassemia major patients: Relation to renal biomarkers, total body iron and chelation therapy. Eur. J. Radiol. 2018, 103, 65–70. [Google Scholar] [CrossRef]
- Linkermann, A.; Skouta, R.; Himmerkus, N.; Mulay, S.R.; Dewitz, C.; De Zen, F.; Prokai, A.; Zuchtriegel, G.; Krombach, F.; Welz, P.S.; et al. Synchronized renal tubular cell death involves ferroptosis. Proc. Natl. Acad. Sci. USA 2014, 111, 16836–16841. [Google Scholar] [CrossRef] [Green Version]
- Tang, S.; Xiao, X. Ferroptosis and kidney diseases. Int. Urol. Nephrol. 2020, 52, 497–503. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Liu, C.; Zhao, Y.; Gao, G. Mitochondria regulation in ferroptosis. Eur. J. Cell Biol. 2020, 99, 151058. [Google Scholar] [CrossRef] [PubMed]
- Deng, H.F.; Yue, L.X.; Wang, N.N.; Zhou, Y.Q.; Zhou, W.; Liu, X.; Ni, Y.H.; Huang, C.S.; Qiu, L.Z.; Liu, H.; et al. Mitochondrial Iron Overload-Mediated Inhibition of Nrf2-HO-1/GPX4 Assisted ALI-Induced Nephrotoxicity. Front. Pharmacol. 2020, 11, 624529. [Google Scholar] [CrossRef]
- Bhat, A.H.; Dar, K.B.; Anees, S.; Zargar, M.A.; Masood, A.; Sofi, M.A.; Ganie, S.A. Oxidative stress, mitochondrial dysfunction and neurodegenerative diseases; a mechanistic insight. Biomed. Pharmacother. 2015, 74, 101–110. [Google Scholar] [CrossRef]
- Moos, T.; Morgan, E.H. The metabolism of neuronal iron and its pathogenic role in neurological disease: Review. Ann. N. Y. Acad. Sci. 2004, 1012, 14–26. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.T. Oxidative stress and mitochondrial dysfunction-linked neurodegenerative disorders. Neurol. Res. 2017, 39, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Federico, A.; Cardaioli, E.; Da Pozzo, P.; Formichi, P.; Gallus, G.N.; Radi, E. Mitochondria, oxidative stress and neurodegeneration. J. Neurol. Sci. 2012, 322, 254–262. [Google Scholar] [CrossRef]
- Ward, R.J.; Dexter, D.T.; Crichton, R.R. Ageing, neuroinflammation and neurodegeneration. Front. Biosci. Schol. Ed. 2015, 7, 189–204. [Google Scholar] [CrossRef] [PubMed]
- Garza-Lombo, C.; Posadas, Y.; Quintanar, L.; Gonsebatt, M.E.; Franco, R. Neurotoxicity Linked to Dysfunctional Metal Ion Homeostasis and Xenobiotic Metal Exposure: Redox Signaling and Oxidative Stress. Antioxid. Redox Signal. 2018, 28, 1669–1703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winklhofer, K.F.; Haass, C. Mitochondrial dysfunction in Parkinson’s disease. Biochim. Biophys. Acta 2010, 1802, 29–44. [Google Scholar] [CrossRef]
- Quintana, C.; Cowley, J.M.; Marhic, C. Electron nanodiffraction and high-resolution electron microscopy studies of the structure and composition of physiological and pathological ferritin. J. Struct. Biol. 2004, 147, 166–178. [Google Scholar] [CrossRef]
- Cheng, R.; Dhorajia, V.V.; Kim, J.; Kim, Y. Mitochondrial iron metabolism and neurodegenerative diseases. Neurotoxicology 2022, 88, 88–101. [Google Scholar] [CrossRef]
- Worwood, M. Ferritin in human tissues and serum. Clin. Haematol. 1982, 11, 275–307. [Google Scholar] [CrossRef]
- Powell, L.W.; Alpert, E.; Isselbacher, K.J.; Drysdale, J.W. Human isoferritins: Organ specific iron and apoferritin distribution. Br. J. Haematol. 1975, 30, 47–55. [Google Scholar] [CrossRef]
- Zhang, N.; Yu, X.; Xie, J.; Xu, H. New Insights into the Role of Ferritin in Iron Homeostasis and Neurodegenerative Diseases. Mol. Neurobiol. 2021, 58, 2812–2823. [Google Scholar] [CrossRef] [PubMed]
- Bossoni, L.; Grand Moursel, L.; Bulk, M.; Simon, B.G.; Webb, A.; van der Weerd, L.; Huber, M.; Carretta, P.; Lascialfari, A.; Oosterkamp, T.H. Human-brain ferritin studied by muon spin rotation: A pilot study. J. Phys. Condens. Matter 2017, 29, 415801. [Google Scholar] [CrossRef] [PubMed]
- Strbak, O.; Balejcikova, L.; Mihalikova, M.; Kopcansky, P.; Dobrota, D. Quantitative Analysis of Magnetoferritin-Induced Relaxivity Enhancement in MRI. Acta Phys. Pol. A 2020, 137, 720–722. [Google Scholar] [CrossRef]
- Meraz-Rios, M.A.; Franco-Bocanegra, D.; Toral Rios, D.; Campos-Pena, V. Early onset Alzheimer’s disease and oxidative stress. Oxid. Med. Cell. Longev. 2014, 2014, 375968. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.; Atwood, C.S.; Hartshorn, M.A.; Multhaup, G.; Goldstein, L.E.; Scarpa, R.C.; Cuajungco, M.P.; Gray, D.N.; Lim, J.; Moir, R.D.; et al. The A beta peptide of Alzheimer’s disease directly produces hydrogen peroxide through metal ion reduction. Biochemistry 1999, 38, 7609–7616. [Google Scholar] [CrossRef]
- Pratico, D.; Zhukareva, V.; Yao, Y.; Uryu, K.; Funk, C.D.; Lawson, J.A.; Trojanowski, J.Q.; Lee, V.M. 12/15-lipoxygenase is increased in Alzheimer’s disease: Possible involvement in brain oxidative stress. Am. J. Pathol. 2004, 164, 1655–1662. [Google Scholar] [CrossRef]
- Yang, H.; Zhuo, J.M.; Chu, J.; Chinnici, C.; Pratico, D. Amelioration of the Alzheimer’s disease phenotype by absence of 12/15-lipoxygenase. Biol. Psychiatry 2010, 68, 922–929. [Google Scholar] [CrossRef]
- Feeney, C.J.; Frantseva, M.V.; Carlen, P.L.; Pennefather, P.S.; Shulyakova, N.; Shniffer, C.; Mills, L.R. Vulnerability of glial cells to hydrogen peroxide in cultured hippocampal slices. Brain Res. 2008, 1198, 1–15. [Google Scholar] [CrossRef]
- Rubinsztein, D.C.; Carmichael, J. Huntington’s disease: Molecular basis of neurodegeneration. Expert Rev. Mol. Med. 2003, 5, 1–21. [Google Scholar] [CrossRef]
- Shi, Z.H.; Nie, G.; Duan, X.L.; Rouault, T.; Wu, W.S.; Ning, B.; Zhang, N.; Chang, Y.Z.; Zhao, B.L. Neuroprotective mechanism of mitochondrial ferritin on 6-hydroxydopamine-induced dopaminergic cell damage: Implication for neuroprotection in Parkinson’s disease. Antioxid. Redox Signal. 2010, 13, 783–796. [Google Scholar] [CrossRef]
- Guan, H.; Yang, H.; Yang, M.; Yanagisawa, D.; Bellier, J.P.; Mori, M.; Takahata, S.; Nonaka, T.; Zhao, S.; Tooyama, I. Mitochondrial ferritin protects SH-SY5Y cells against H2O2-induced oxidative stress and modulates alpha-synuclein expression. Exp. Neurol. 2017, 291, 51–61. [Google Scholar] [CrossRef]
- Mizuno, Y.; Ikebe, S.; Hattori, N.; Nakagawa-Hattori, Y.; Mochizuki, H.; Tanaka, M.; Ozawa, T. Role of mitochondria in the etiology and pathogenesis of Parkinson’s disease. Biochim. Biophys. Acta 1995, 1271, 265–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munoz, Y.; Carrasco, C.M.; Campos, J.D.; Aguirre, P.; Nunez, M.T. Parkinson’s Disease: The Mitochondria-Iron Link. Park. Dis. 2016, 2016, 7049108. [Google Scholar]
- Carroll, C.B.; Zeissler, M.L.; Chadborn, N.; Gibson, K.; Williams, G.; Zajicek, J.P.; Morrison, K.E.; Hanemann, C.O. Changes in iron-regulatory gene expression occur in human cell culture models of Parkinson’s disease. Neurochem. Int. 2011, 59, 73–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mena, N.P.; Garcia-Beltran, O.; Lourido, F.; Urrutia, P.J.; Mena, R.; Castro-Castillo, V.; Cassels, B.K.; Nunez, M.T. The novel mitochondrial iron chelator 5-((methylamino)methyl)-8-hydroxyquinoline protects against mitochondrial-induced oxidative damage and neuronal death. Biochem. Biophys. Res. Commun. 2015, 463, 787–792. [Google Scholar] [CrossRef]
- Mastroberardino, P.G.; Hoffman, E.K.; Horowitz, M.P.; Betarbet, R.; Taylor, G.; Cheng, D.; Na, H.M.; Gutekunst, C.A.; Gearing, M.; Trojanowski, J.Q.; et al. A novel transferrin/TfR2-mediated mitochondrial iron transport system is disrupted in Parkinson’s disease. Neurobiol. Dis. 2009, 34, 417–431. [Google Scholar] [CrossRef] [Green Version]
- Toyokuni, S.; Kong, Y.; Cheng, Z.; Sato, K.; Hayashi, S.; Ito, F.; Jiang, L.; Yanatori, I.; Okazaki, Y.; Akatsuka, S. Carcinogenesis as Side Effects of Iron and Oxygen Utilization: From the Unveiled Truth toward Ultimate Bioengineering. Cancers 2020, 12, 3320. [Google Scholar] [CrossRef]
- Torti, S.V.; Manz, D.H.; Paul, B.T.; Blanchette-Farra, N.; Torti, F.M. Iron and Cancer. Annu. Rev. Nutr. 2018, 38, 97–125. [Google Scholar] [CrossRef]
- Ludwig, H.; Evstatiev, R.; Kornek, G.; Aapro, M.; Bauernhofer, T.; Buxhofer-Ausch, V.; Fridrik, M.; Geissler, D.; Geissler, K.; Gisslinger, H.; et al. Iron metabolism and iron supplementation in cancer patients. Wien. Klin. Wochenschr. 2015, 127, 907–919. [Google Scholar] [CrossRef] [Green Version]
- Manz, D.H.; Blanchette, N.L.; Paul, B.T.; Torti, F.M.; Torti, S.V. Iron and cancer: Recent insights. Ann. N. Y. Acad. Sci. 2016, 1368, 149–161. [Google Scholar] [CrossRef] [Green Version]
- Bystrom, L.M.; Rivella, S. Cancer cells with irons in the fire. Free Radic. Biol. Med. 2015, 79, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Quiros Roldan, E.; Biasiotto, G.; Magro, P.; Zanella, I. The possible mechanisms of action of 4-aminoquinolines (chloroquine/hydroxychloroquine) against SARS-CoV-2 infection (COVID-19): A role for iron homeostasis? Pharmacol. Res. 2020, 158, 104904. [Google Scholar] [CrossRef] [PubMed]
- Cerqueira, N.M.; Fernandes, P.A.; Ramos, M.J. Ribonucleotide reductase: A critical enzyme for cancer chemotherapy and antiviral agents. Recent Pat. Anticancer Drug Discov. 2007, 2, 11–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, Y.; Akatsuka, S.; Motooka, Y.; Zheng, H.; Cheng, Z.; Shiraki, Y.; Mashimo, T.; Imaoka, T.; Toyokuni, S. BRCA1 haploinsufficiency promotes chromosomal amplification under Fenton reaction-based carcinogenesis through ferroptosis-resistance. Redox Biol. 2022, 54, 102356. [Google Scholar] [CrossRef] [PubMed]
- Hooda, J.; Cadinu, D.; Alam, M.M.; Shah, A.; Cao, T.M.; Sullivan, L.A.; Brekken, R.; Zhang, L. Enhanced heme function and mitochondrial respiration promote the progression of lung cancer cells. PLoS ONE 2013, 8, e63402. [Google Scholar] [CrossRef] [Green Version]
- Basuli, D.; Tesfay, L.; Deng, Z.; Paul, B.; Yamamoto, Y.; Ning, G.; Xian, W.; McKeon, F.; Lynch, M.; Crum, C.P.; et al. Iron addiction: A novel therapeutic target in ovarian cancer. Oncogene 2017, 36, 4089–4099. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Chang, W.; Wu, H.; Wang, Y.H.; Gong, Y.W.; Zhao, Y.L.; Liu, S.H.; Wang, H.Z.; Svatek, R.S.; Rodriguez, R.; et al. Pan-cancer analysis of iron metabolic landscape across the Cancer Genome Atlas. J. Cell Physiol. 2020, 235, 1013–1024. [Google Scholar] [CrossRef]
- Xue, D.; Zhou, C.X.; Shi, Y.B.; Lu, H.; He, X.Z. Decreased expression of ferroportin in prostate cancer. Oncol. Lett. 2015, 10, 913–916. [Google Scholar] [CrossRef] [Green Version]
- Pinnix, Z.K.; Miller, L.D.; Wang, W.; D’Agostino, R., Jr.; Kute, T.; Willingham, M.C.; Hatcher, H.; Tesfay, L.; Sui, G.; Di, X.; et al. Ferroportin and iron regulation in breast cancer progression and prognosis. Sci. Transl. Med. 2010, 2, 43ra56. [Google Scholar] [CrossRef]
- Guo, W.; Zhang, S.; Chen, Y.; Zhang, D.; Yuan, L.; Cong, H.; Liu, S. An important role of the hepcidin-ferroportin signaling in affecting tumor growth and metastasis. Acta Biochim. Biophys. Sin. 2015, 47, 703–715. [Google Scholar] [CrossRef] [Green Version]
- Shan, Z.; Wei, Z.; Shaikh, Z.A. Suppression of ferroportin expression by cadmium stimulates proliferation, EMT, and migration in triple-negative breast cancer cells. Toxicol. Appl. Pharmacol. 2018, 356, 36–43. [Google Scholar] [CrossRef] [PubMed]
- Hung, H.I.; Schwartz, J.M.; Maldonado, E.N.; Lemasters, J.J.; Nieminen, A.L. Mitoferrin-2-dependent mitochondrial iron uptake sensitizes human head and neck squamous carcinoma cells to photodynamic therapy. J. Biol. Chem. 2013, 288, 677–686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lui, G.Y.; Kovacevic, Z.; Richardson, V.; Merlot, A.M.; Kalinowski, D.S.; Richardson, D.R. Targeting cancer by binding iron: Dissecting cellular signaling pathways. Oncotarget 2015, 6, 18748–18779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Estrov, Z.; Tawa, A.; Wang, X.H.; Dube, I.D.; Sulh, H.; Cohen, A.; Gelfand, E.W.; Freedman, M.H. In vitro and in vivo effects of deferoxamine in neonatal acute leukemia. Blood 1987, 69, 757–761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdelaal, G.; Veuger, S. Reversing oncogenic transformation with iron chelation. Oncotarget 2021, 12, 106–124. [Google Scholar] [CrossRef] [PubMed]
- Crielaard, B.J.; Lammers, T.; Rivella, S. Targeting iron metabolism in drug discovery and delivery. Nat. Rev. Drug Discov. 2017, 16, 400–423. [Google Scholar] [CrossRef] [Green Version]
- De Domenico, I.; Ward, D.M.; Kaplan, J. Specific iron chelators determine the route of ferritin degradation. Blood 2009, 114, 4546–4551. [Google Scholar] [CrossRef] [Green Version]
- Lesjak, M.; Balesaria, S.; Skinner, V.; Debnam, E.S.; Srai, S.K.S. Quercetin inhibits intestinal non-haem iron absorption by regulating iron metabolism genes in the tissues. Eur. J. Nutr. 2019, 58, 743–753. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Yuan, X.; Li, Z.; Lu, Z.; Kong, S.; Jiang, H.; Du, H.; Pan, X.; Nandi, M.; Kong, X.; et al. CN128: A New Orally Active Hydroxypyridinone Iron Chelator. J. Med. Chem. 2020, 63, 4215–4226. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disease Examples or Models | Changes in Iron Level | Major Consequences |
|---|---|---|
| Patients [40] and mice [41] with heart failure | Intracellular iron is deficient | Severe heart failure |
| Cardiomyopathy of Friedreich’s ataxia [42] | Mitochondrial iron levels increased | Cardiomyocyte death and fibrosis, impaired systolic and diastolic function. |
| β-thalassemia and hereditary hemochromatosis [43] | Iron overload | Liver fibrosis, cirrhosis, and even hepatoma |
| Aged rats muscle atrophy [44] | Mitochondrial iron accumulation | Muscle mass decreased |
| Patients with obesity and diabetes [45] | Iron accumulation | Mitochondrial dysfunction in adipocytes causes toxic effects on β cells leading to defects in insulin synthesis and secretion |
| Patients with chronic kidney disease [46] | Tubular cell lysosomal iron accumulation | Renal cell damage |
| AD [47] | Diffuse accumulation of iron in the cerebral cortex and hippocampus, and the content of iron in senile plaques increases slightly | Apoptosis and/or necrosis, thus leading to cell death |
| PD [48] | Focal accumulation of iron in the substantia nigra | The formation of α-synuclein leaded to synaptic dysfunction and disruption of ax-onal transport |
| Breast cancer [49] | Iron overload1 | Promoting cancer cell proliferation |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Duan, G.; Li, J.; Duan, Y.; Zheng, C.; Guo, Q.; Li, F.; Zheng, J.; Yu, J.; Zhang, P.; Wan, M.; et al. Mitochondrial Iron Metabolism: The Crucial Actors in Diseases. Molecules 2023, 28, 29. https://doi.org/10.3390/molecules28010029
Duan G, Li J, Duan Y, Zheng C, Guo Q, Li F, Zheng J, Yu J, Zhang P, Wan M, et al. Mitochondrial Iron Metabolism: The Crucial Actors in Diseases. Molecules. 2023; 28(1):29. https://doi.org/10.3390/molecules28010029
Chicago/Turabian StyleDuan, Geyan, Jianjun Li, Yehui Duan, Changbing Zheng, Qiuping Guo, Fengna Li, Jie Zheng, Jiayi Yu, Peiwen Zhang, Mengliao Wan, and et al. 2023. "Mitochondrial Iron Metabolism: The Crucial Actors in Diseases" Molecules 28, no. 1: 29. https://doi.org/10.3390/molecules28010029