

Inhibition of Cancer Cell Proliferation and Bacterial Growth by Silver(I) Complexes Bearing a CH3-Substituted Thiadiazole-Based Thioamide
 , ,
, ,  , ,
, ,  and
and
Abstract
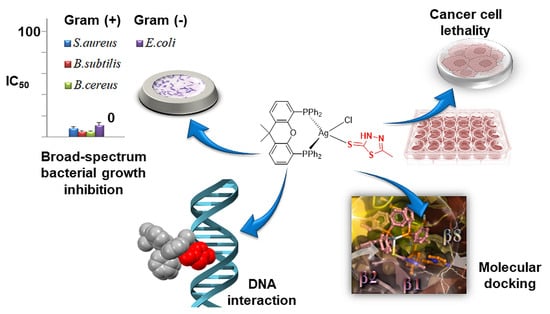
:
1. Introduction
2. Results and Discussion
2.1. Synthesis and Characterization
2.2. Photophysical Properties
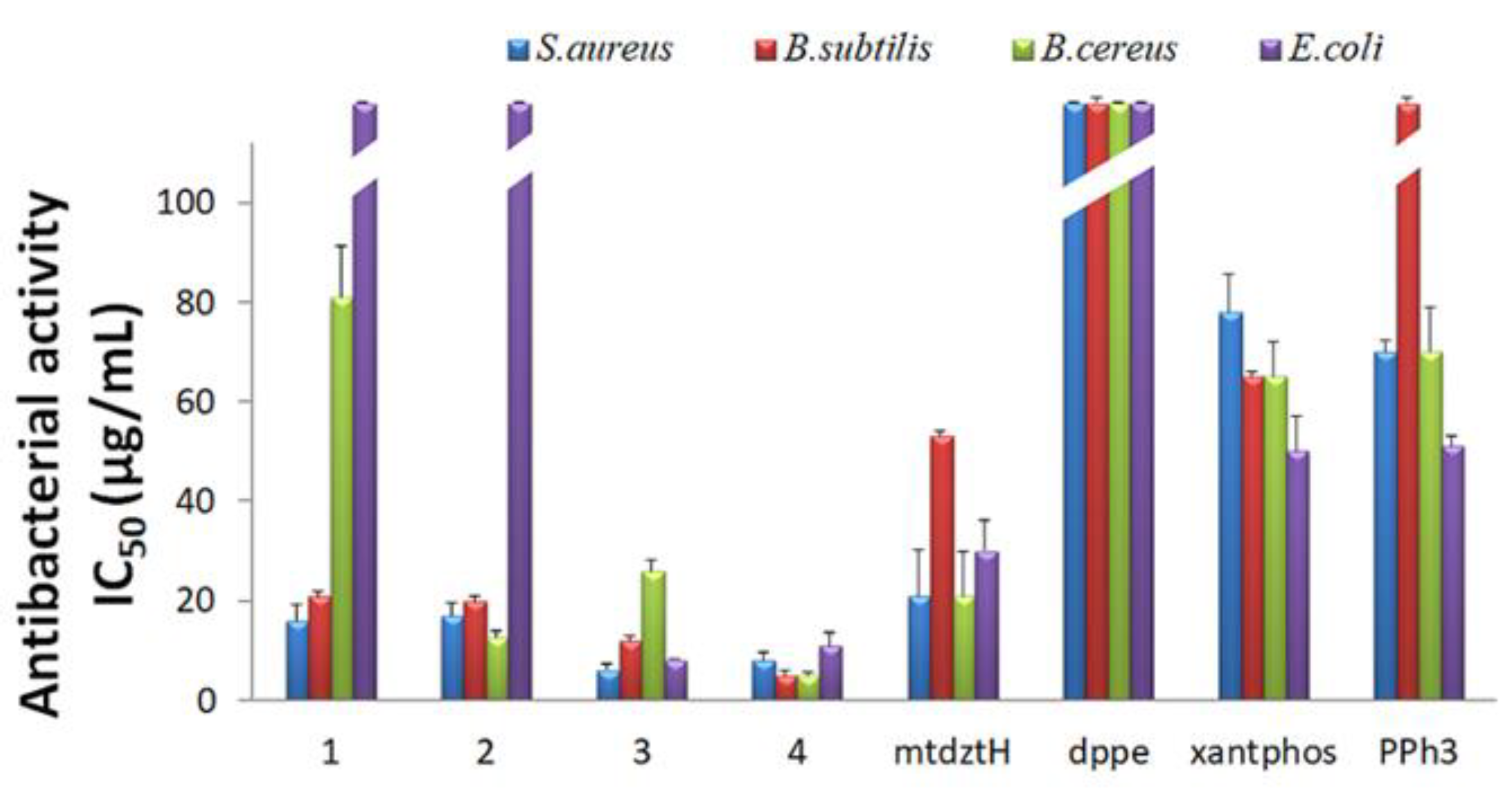
2.3. In Vitro Antibacterial Activity
2.4. In Vitro Anti-Cancer Activity
2.5. In Vitro Antioxidant Activity
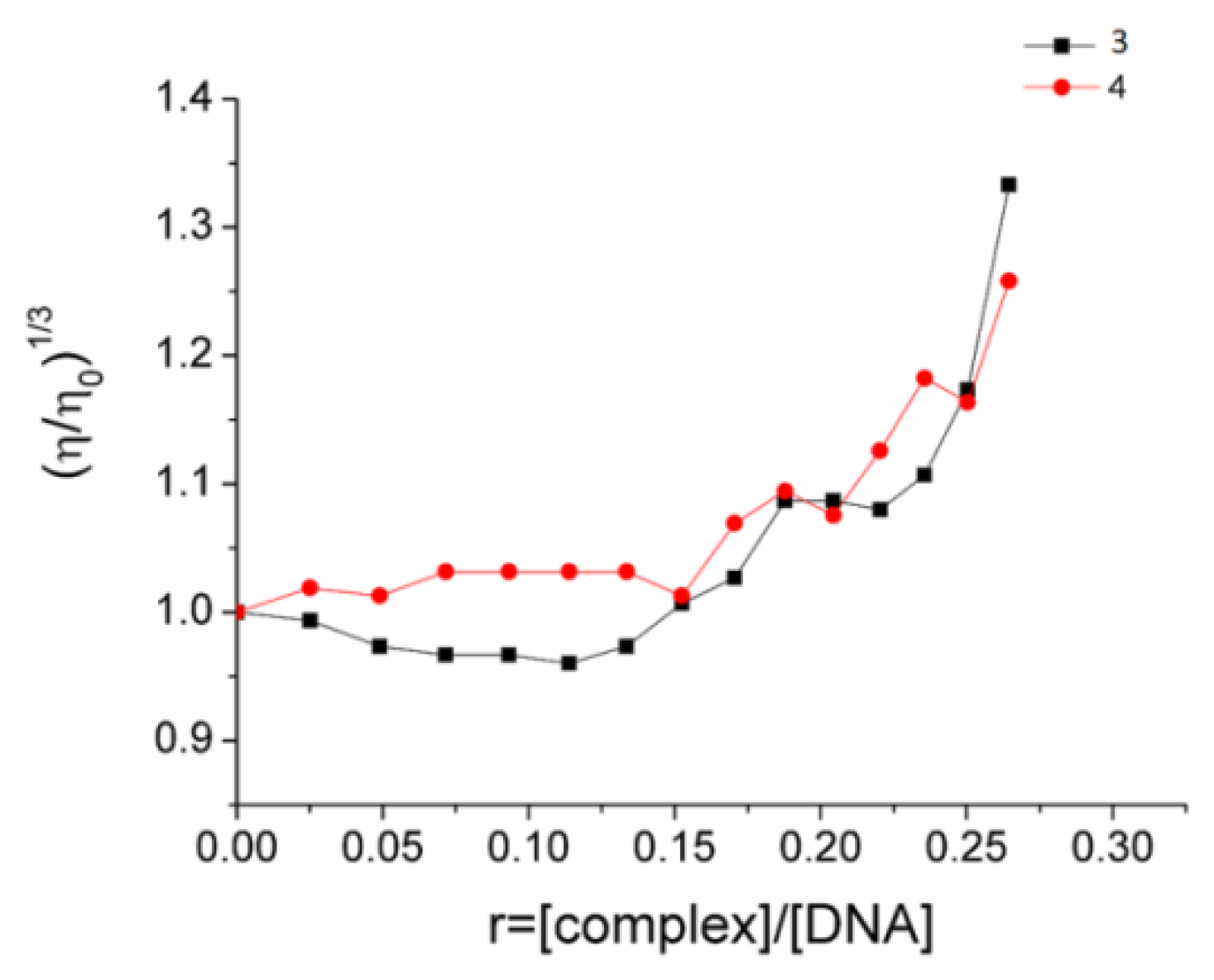
2.6. CT DNA Interaction
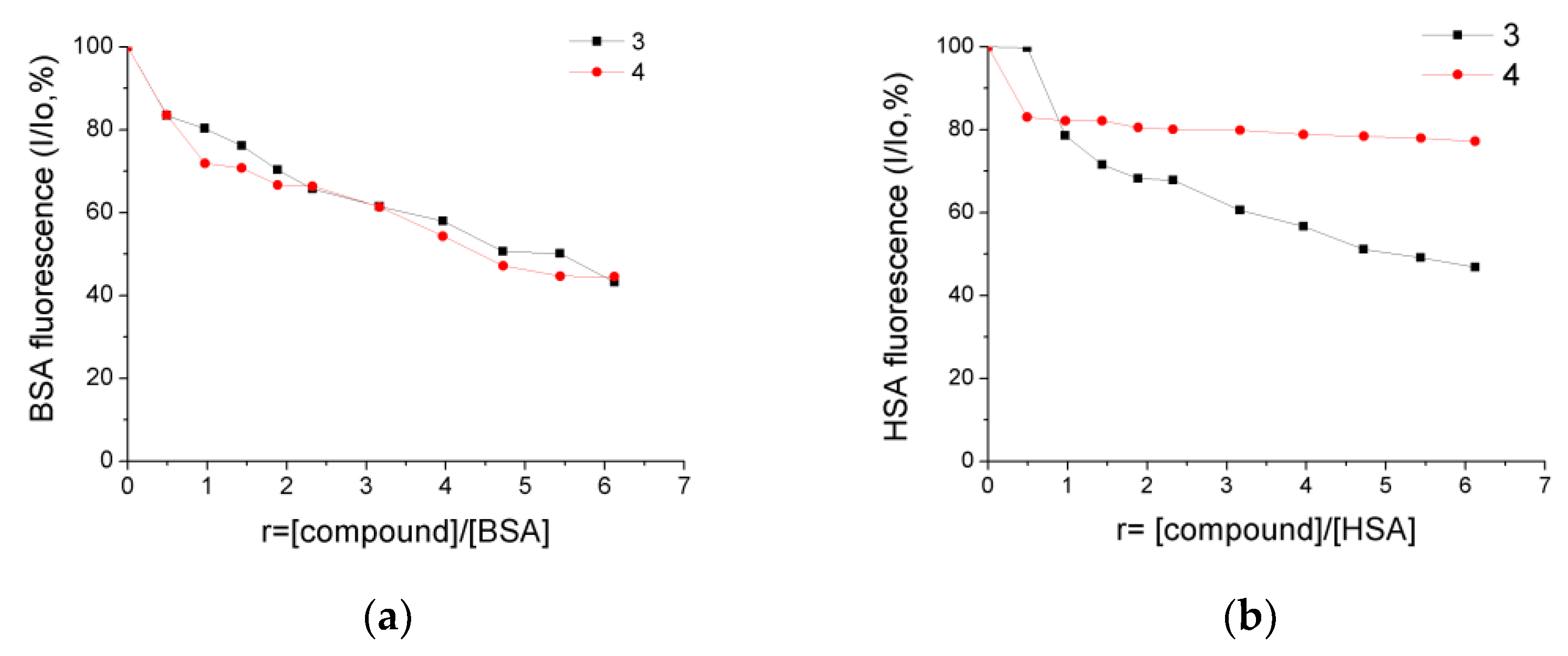
2.7. Albumin-Binding Studies
2.8. Molecular Docking Calculations
3. Conclusions
4. Materials and Methods
4.1. General Procedures and Chemicals
4.2. Syntheses
4.2.1. [AgCl(mtdztH)(PPh3)2] (1)
4.2.2. [Ag(mtdzt)(PPh3)3] (2)
4.2.3. [AgCl(mtdztH)(xantphos)] (3)
4.2.4. [Ag(mtdztH)(dppe)2(NO3)]n (4)
4.3. Instrumentation
4.4. X-ray Crystal Structure Determinations
4.5. In Vitro Antibacterial Activity
4.6. In Vitro Anticancer Activity
4.6.1. Cell Culture
4.6.2. In Vitro Cytotoxicity Assay
4.7. In Vitro Antioxidant Activity
4.7.1. Reduction of Hydrogen Peroxide
4.7.2. Determination of the Reducing Activity of DPPH Radical
4.7.3. Assay of Radical Cation ABTS Scavenging Activity
4.8. CT DNA Interaction
4.9. Serum Albumins Binding
4.10. Molecular Docking Calculations
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Bray, F.; Jemal, A.; Grey, N.; Ferlay, J.; Forman, D. Global cancer transitions according to the Human Development Index (2008–2030): A population-based study. Lancet Oncol. 2012, 13, 790–801. [Google Scholar] [CrossRef]
- Kuehl, R.; Brunetto, P.S.; Woischnig, A.K.; Varisco, M.; Rajacic, Z.; Vosbeck, J.; Terracciano, L.; Fromm, K.M.; Khanna, N. Preventing implant-associated infections by silver coating. Antimicrob. Agents Chemother. 2016, 60, 2467–2475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.; Zhou, L.; Xie, B.; Wang, Y.; Ren, L.; Chen, X.; Cen, B.; Lv, H.; Wang, H. Novel fast-acting pyrazole/pyridine-functionalized N-heterocyclic carbene silver complexes assembled with nanoparticles show enhanced safety and efficacy as anticancer therapeutics. Dalton Trans. 2020, 49, 2505–2516. [Google Scholar] [CrossRef]
- Mármol, I.; Montanel-Perez, S.; Carlos Royo, J.; Gimeno, M.C.; Villacampa, M.D.; Rodríguez-Yoldi, M.J.; Cerrada, E. Gold(I) and silver(I) complexes with 2-anilinopyridine-based heterocycles as multitarget drugs against colon cancer. Inorg. Chem. 2020, 59, 17732–17745. [Google Scholar] [CrossRef]
- Quintana, M.; Rodriguez-Rius, A.; Vellé, A.; Vives, S.; Sanz Miguel, P.J.; Triola, G. Dinuclear silver and gold bisNHC complexes as drug candidates for cancer therapy. Bioorg. Med. Chem. 2022, 67, 116814:1–116814:8. [Google Scholar] [CrossRef]
- Raper, E.S. Complexes of heterocyclic thionates. Part 1. Complexes of monodentate and chelating ligands. Coord. Chem. Rev. 1996, 153, 199–255. [Google Scholar] [CrossRef]
- Lobana, T.S. Heterocyclic-2-thione derivatives of group 10–12 metals: Coordination versatility, activation of C=S (thione) bonds and biochemical potential. Coord. Chem. Rev. 2021, 441, 213884:1–213884:84. [Google Scholar] [CrossRef]
- Janowska, S.; Paneth, A.; Wujec, M. Cytotoxic properties of 1,3,4-thiadiazole derivatives—A review. Molecules 2020, 25, 4309. [Google Scholar] [CrossRef]
- Li, Y.; Geng, J.; Liu, Y.; Yu, S.; Zhao, G. Thiadiazole-a promising structure in medicinal chemistry. ChemMedChem 2013, 8, 27–41. [Google Scholar] [CrossRef]
- Asbury, R.F.; Blessing, J.A.; DiSaia, P.J.; Malfetano, J. Aminothiadiazole (NSC 4728) in patients with advanced nonsquamous carcinoma of the cervix. A phase II study of the Gynecologic Oncology Group. Am. J. Clin. Oncol. 1989, 12, 375–377. [Google Scholar] [CrossRef]
- Szeliga, M. Thiadiazole derivatives as anticancer agents. Pharmacol. Rep. 2020, 72, 1079–1100. [Google Scholar] [CrossRef]
- Kyros, L.; Kourkoumelis, N.; Kubicki, M.; Male, L.; Hursthouse, M.B.; Verginadis, I.I.; Gouma, E.; Karkabounas, S.; Char-alabopoulos, K.; Hadjikakou, S.K. Structural properties, cytotoxicity, and anti-inflammatory activity of silver(I) complexes with tris(p-tolyl)phosphine and 5-chloro-2-mercaptobenzothiazole. Bioinorg. Chem. Appl. 2010, 2010, 386860:1–386860:12. [Google Scholar] [CrossRef] [PubMed]
- Banti, C.N.; Kyros, L.; Geromichalos, G.D.; Kourkoumelis, N.; Kubicki, M.; Hadjikakou, S.K. A novel silver iodide meta-lo-drug: Experimental and computational modelling assessment of its interaction with intracellular DNA, lipoxygenase and glutathione. Eur. J. Med. Chem. 2014, 77, 388–399. [Google Scholar] [CrossRef] [PubMed]
- Aulakh, J.K.; Lobana, T.S.; Sood, H.; Arora, D.S.; Kaur, R.; Singh, J.; Garcia-Santos, I.; Kaur, M.; Jasinski, J.P. Silver derivatives of multi-donor heterocyclic thioamides as antimicrobial/anticancer agents: Unusual bioactivity against methicillin resistant S. aureus, S. epidermidis, and E. faecalis and human bone cancer MG63 cell line. RSC Adv. 2019, 9, 15470–15487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nawaz, S.; Isab, A.A.; Merz, K.; Vasylyeva, V.; Metzler-Nolte, N.; Saleem, M.; Ahmad, S. Synthesis, characterization and anti-microbial studies of mixed ligand silver(I) complexes of triphenylphosphine and heterocyclic thiones: Crystal structure of bis[{(l 2-diazinane-2-thione)(diazinane-2-thione)(triphenylphosphine)silver(I) nitrate. Polyhedron 2011, 30, 1502–1506. [Google Scholar] [CrossRef]
- Aulakh, J.K.; Lobana, T.S.; Sood, H.; Arora, D.S.; Smolinski, V.A.; Duff, C.E.; Jasinski, J.P. Synthesis, structures, and ESI-mass studies of silver(I) derivatives of imidazolidine-2-thiones: Antimicrobial potential and biosafety evaluation. J. Inorg. Biochem. 2018, 178, 18–31. [Google Scholar] [CrossRef]
- Altaf, M.; Stoeckli-Evans, H.; Cuin, A.; Sato, D.N.; Pavan, F.R.; Leite, C.Q.F.; Ahmad, S.; Bouakka, M.; Mimouni, M.; Khardli, F.Z.; et al. Synthesis, crystal structures, antimicrobial, antifungal and antituberculosis activities of mixed ligand silver(I) complexes. Polyhedron 2013, 62, 138–147. [Google Scholar] [CrossRef]
- Anastasiadou, D.; Geromichalou, E.; Tsavea, E.; Psomas, G.; Hatzidimitriou, A.G.; Kalogiannis, S.; Geromichalos, G.; Trafalis, D.; Dalezis, P.; Aslanidis, P. Silver complexes with heterocyclic thioamide and tertiary arylphosphane ligands: Synthesis, crystal structures, in vitro and in silico antibacterial and cytotoxic activity, and interaction with DNA. J. Inorg. Biochem. 2020, 210, 111167:1–111167:17. [Google Scholar] [CrossRef]
- Varna, D.; Zainuddin, D.I.; Hatzidimitriou, A.G.; Psomas, G.; Pantazaki, A.; Papi, R.; Angaridis, P.; Aslanidis, P. Homoleptic and heteroleptic silver(I) complexes bearing diphosphane and thioamide ligands: Synthesis, structures, DNA interactions and antibacterial activity studies. Mater. Sci. Eng. C 2019, 99, 450–459. [Google Scholar] [CrossRef]
- Varna, D.; Kapetanaki, E.; Koutsari, A.; Hatzidimitriou, A.G.; Psomas, G.; Angaridis, P.; Papi, R.; Pantazaki, A.A.; Aslanidis, P. Heterocyclic thioamide/phosphine mixed-ligand silver(I) complexes: Synthesis, molecular structures, DNA-binding proper-ties and antibacterial activity. Polyhedron 2018, 151, 131–140. [Google Scholar] [CrossRef]
- Varna, D.; Geromichalou, E.; Hatzidimitriou, A.G.; Papi, R.; Psomas, G.; Dalezis, P.; Aslanidis, P.; Choli-Papadopoulou, T.; Trafalis, D.T.; Angaridis, P.A. Silver(I) complexes bearing heterocyclic thioamide ligands with NH2 and CF3 substituents: Effect of ligand group substitution on antibacterial and anticancer properties. Dalton Trans. 2022, 51, 9412–9431. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Powell, D.R.; Houser, R.P. Structural variation in copper(I) complexes with pyridylmethylamide ligands: Structural analysis with a new four-coordinate geometry index, τ4. Dalt. Trans. 2007, 955–964. [Google Scholar] [CrossRef]
- Brandys, M.; Puddephatt, R.J. Polymeric Complexes of silver(I) with diphosphine ligands: Self-assembly of a puckered sheet network structure. J. Am. Chem. Soc. 2002, 124, 3946–3950. [Google Scholar] [CrossRef] [PubMed]
- Huheey, J.E. Inorganic Chemistry, Principles of Structure and Reactivity, 2nd ed.; Harper & Row: New York, NY, USA, 1978; pp. 230–257. ISBN 978-0060429867. [Google Scholar]
- Fabretti, A.C.; Franchini, G.C.; Peyronel, G. Tin (IV) tetrahalide complexes of 2,5-disubstituted 1,3,4-thiadiazoles. Spectrochim. Acta Part A 1980, 36, 517–520. [Google Scholar] [CrossRef]
- Fabretti, A.C.; Giusti, A. 2-Mercapto-5-methyl-1,3,4-thiadiazole as a ligand in Ru(III), Rh(III), Os(III) and Ir(III) trichloride complexes. Polyhedron 1986, 5, 1927–1930. [Google Scholar] [CrossRef]
- Hu, Y.; Li, C.-Y.; Wang, X.-M.; Yang, Y.-H.; Zhu, H.-L. 1,3,4-Thiadiazole: Synthesis, reactions, and applications in medicinal, agricultural, and materials chemistry. Chem. Rev. 2014, 114, 5572–5610. [Google Scholar] [CrossRef] [PubMed]
- Richter, M.F.; Drown, B.S.; Riley, A.P.; Garcia, A.; Shirai, T.; Svec, R.L.; Hergenrother, P.J. Predictive compound accumulation rules yield a broad-spectrum antibiotic. Nature 2017, 545, 299–304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varna, D.; Geromichalou, E.; Papachristou, E.; Papi, R.; Hatzidimitriou, A.G.; Panteris, E.; Psomas, G.; Geromichalos, G.D.; Aslanidis, P.; Choli-Papadopoulou, T.; et al. Biocompatible silver(I) complexes with heterocyclic thioamide ligands for selective killing of cancer cells and high antimicrobial activity—A combined in vitro and in silico study. J. Inorg. Biochem. 2022, 228, 111695:1–111695:18. [Google Scholar] [CrossRef]
- Cressier, D.; Prouillac, C.; Hernandez, P.; Amourette, C.; Diserbo, M.; Lion, C.; Rima, G. Synthesis, antioxidant properties and radioprotective effects of new benzothiazoles and thiadiazoles. Bioorg. Med. Chem. 2009, 17, 5275–5284. [Google Scholar] [CrossRef] [PubMed]
- Wettasinghe, M.; Shahidi, F. Scavenging of reactive-oxygen species and DPPH free radicals by extracts of borage and evening primrose meals. Food Chem. 2000, 70, 17–26. [Google Scholar] [CrossRef]
- Gurova, K. New hopes from old drugs: Revisiting DNA-binding small molecules as anticancer agents. Future Oncol. 2009, 5, 1685–1704. [Google Scholar] [CrossRef] [Green Version]
- Sirajuddin, M.; Ali, S.; Badshah, A. Drug–DNA interactions and their study by UV–visible, fluorescence spectroscopies and cyclic voltammetry. J. Photochem. Photobiol. B 2013, 124, 1–19. [Google Scholar] [CrossRef]
- Mjos, K.D.; Orvig, C. Metallodrugs in Medicinal Inorganic Chemistry. Chem. Rev. 2014, 114, 4540–4563. [Google Scholar] [CrossRef]
- Lakowicz, J.R. Principles of Fluorescence Spectroscopy, 3rd ed.; Plenum Press: New York, NY, USA, 2006; ISBN 978-0-387-46312-4. [Google Scholar]
- Zhang, Y.-Z.; Zhou, B.; Liu, Y.-X.; Zhou, C.-X.; Ding, X.-L.; Liu, Y. Fluorescence study on the interaction of bovine serum al-bumin with p-Aminoazobenzene. J. Fluoresc. 2008, 18, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, E.; Rabbani, G.; Zaidi, N.; Khan, M.A.; Qadeer, A.; Ishtikhar, M.; Singh, S.; Khan, R.H. Revisiting ligand-induced con-formational changes in proteins: Essence, advancements, implications and future challenges. J. Biomol. Struct. Dyn. 2013, 31, 630–648. [Google Scholar] [CrossRef] [PubMed]
- Qi, H.; Wang, Y.; Wang, X.; Su, L.; Wang, Y.; Wang, S. The different interactions of two anticancer drugs with bovine serum albumin based on multi-spectrum method combined with molecular dynamics simulations. Spectrochim. Acta Part A 2021, 259, 119809:1–119809:10. [Google Scholar] [CrossRef]
- Turner, N.; Grose, R. Fibroblast growth factor signalling: From development to cancer. Nat. Rev. Cancer 2010, 10, 116–129. [Google Scholar] [CrossRef] [PubMed]
- Ornitz, D.M.; Itoh, N. The Fibroblast Growth Factor signaling pathway. Wiley Interdiscip. Rev. Dev. Biol. 2015, 4, 215–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Babina, I.S.; Turner, N.C. Advances and challenges in targeting FGFR signalling in cancer. Nat. Rev. Cancer 2017, 17, 318–332. [Google Scholar] [CrossRef] [PubMed]
- Katoh, M.; Nakagama, H. FGF receptors: Cancer biology and therapeutics. Med. Res. Rev. 2014, 34, 280–300. [Google Scholar] [CrossRef] [PubMed]
- Datta, J.; Damodaran, S.; Parks, H.; Ocrainiciuc, C.; Miya, J.; Yu, L.; Gardner, E.P.; Samorodnitsky, E.; Wing, M.R.; Bhatt, D.; et al. Akt activation mediates acquired resistance to Fibroblast Growth Factor Receptor Inhibitor BGJ398. Mol. Cancer Ther. 2017, 16, 614–624. [Google Scholar] [CrossRef] [Green Version]
- Pardo, O.E.; Latigo, J.; Jeffery, R.E.; Nye, E.; Poulsom, R.; Spencer-Dene, B.; Lemoine, N.R.; Stamp, G.W.; Aboagye, E.O.; Seckl, M.J. The fibroblast growth factor receptor inhibitor PD173074 blocks small cell lung cancer growth in vitro and in vivo. Cancer Res. 2009, 69, 8645–8651. [Google Scholar] [CrossRef] [Green Version]
- Cole, C.; Lau, S.; Backen, A.; Clamp, A.; Rushton, G.; Dive, C.; Hodgkinson, C.; McVey, R.; Kitchener, H.; Jayson, G.C. Inhibi-tion of FGFR2 and FGFR1 increases cisplatin sensitivity in ovarian cancer. Cancer Biol. Ther. 2010, 10, 495–504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haq, F.; Sung, Y.-N.; Park, I.; Kayani, M.A.; Yousuf, F.; Hong, S.-M.; Ahn, S.-M. FGFR1 expression defines clinically distinct subtypes in pancreatic cancer. J. Transl. Med. 2018, 16, 374:1–374:7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quan, M.-Y.; Guo, Q.; Liu, J.; Yang, R.; Bai, J.; Wang, W.; Cai, Y.; Han, R.; Lv, Y.-Q.; Ding, L.; et al. An FGFR/AKT/SOX2 signaling axis controls pancreatic cancer stemness. Front. Cell Dev. Biol. 2020, 8, 287:1–287:15. [Google Scholar] [CrossRef]
- Dianat-Moghadam, H.; Teimoori-Toolabi, L. Implications of fibroblast growth factors (FGFs) in cancer: From prognostic to therapeutic applications. Curr. Drug Targets 2019, 20, 852–870. [Google Scholar] [CrossRef]
- Coleman, S.J.; Chioni, A.M.; Ghallab, M.; Anderson, R.K.; Lemoine, N.R.; Kocher, H.M.; Grose, R.P. Nuclear translocation of FGFR1 and FGF2 in pancreatic stellate cells facilitates pancreatic cancer cell invasion. EMBO Mol. Med. 2014, 6, 467–481. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, Y.; Yoshimura, H.; Suzuki, T.; Uchida, E.; Naito, Z.; Ishiwata, T. Inhibition of fibroblast growth factor receptor 2 at-tenuates proliferation and invasion of pancreatic cancer. Cancer Sci. 2014, 105, 1212–1219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koinis, F.; Corn, P.; Parikh, N.; Song, J.; Vardaki, I.; Mourkioti, I.; Lin, S.-H.; Logothetis, C.; Panaretakis, T.; Gallick, G. Resistance to MET/VEGFR2 inhibition by cabozantinib is mediated by YAP/TBX5-dependent induction of FGFR1 in castration-resistant prostate cancer. Cancers 2020, 12, 244. [Google Scholar] [CrossRef] [Green Version]
- Labanca, E.; Yang, J.; Shepherd, P.D.A.; Wan, X.; Starbuck, M.W.; Guerra, L.D.; Anselmino, N.; Bizzotto, J.A.; Dong, J.; Chin-naiyan, A.M.; et al. Fibroblast Growth Factor Receptor 1 drives the metastatic progression of prostate cancer. Eur. Urol. Oncol. 2022, 5, 164–175. [Google Scholar] [CrossRef]
- Wan, X.; Corn, P.G.; Yang, J.; Palanisamy, N.; Starbuck, M.W.; Efstathiou, E.; Li Ning Tapia, E.M.; Zurita, A.J.; Aparicio, A.; Ravoori, M.K.; et al. Prostate cancer cell-stromal cell crosstalk via FGFR1 mediates antitumor activity of dovitinib in bone metastases. Sci. Transl. Med. 2014, 6, 252ra122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tucker, J.A.; Klein, T.; Breed, J.; Breeze, A.L.; Overman, R.; Phillips, C.; Norman, R.A. Structural insights into FGFR kinase isoform selectivity: Diverse binding modes of AZD4547 and ponatinib in complex with FGFR1 and FGFR4. Structure 2014, 22, 1764–1774. [Google Scholar] [CrossRef] [Green Version]
- Bruker Analytical X-ray Systems, Inc. Apex2, Version 2 User Manual, M86-E01078; Bruker Analytical X-ray Systems, Inc.: Madison, WI, USA, 2006. [Google Scholar]
- Betteridge, P.W.; Carruthers, J.R.; Cooper, R.I.; Prout, K.; Watkin, D.J.J. Software for guided crystal structure analysis. J. Appl. Cryst. 2003, 36, 1487. [Google Scholar] [CrossRef]
- Macrae, C.F.P.; Edgington, R.; McCabe, P.; Pidcock, E.; Shields, G.P.; Taylor, R.; Towler, M.; van de Streek, J. Mercury: Visualization and analysis of crystal structures. J. Appl. Cryst. 2006, 39, 453–457. [Google Scholar] [CrossRef] [Green Version]
- Andrews, J.M. Determination of minimum inhibitory concentrations. J. Antimicrob. Chemother. 2001, 48, 5–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsiaggali, M.A.; Andreadou, E.G.; Hatzidimitriou, A.G.; Pantazaki, A.A.; Aslanidis, P. Copper(I) halide complexes of N-methylbenzothiazole-2-thione: Synthesis, structure, luminescence, antibacterial activity and interaction with DNA. J. Inorg. Biochem. 2013, 121, 121–128. [Google Scholar] [CrossRef]
- Finlay, G.J.; Wilson, W.R.; Baguley, B.C. Comparison of the in vitro activity of the cytotoxic drugs towards human carcinoma and leukemia cell lines. Eur. J. Cancer Clin. Oncol. 1986, 22, 655–662. [Google Scholar] [CrossRef]
- Alley, M.C.; Scudiero, D.A.; Monks, A.; Hursey, M.L.; Czerwinski, M.J.; Fine, D.L.; Abbott, B.J.; Mayo, J.G.; Shoemaker, R.H.; Boyd, M.R. Feasibility of drug screening with panels of human tumor cell lines using a microculture tetrazolium assay. Cancer Res. 1988, 48, 589–601. [Google Scholar] [PubMed]
- Trafalis, D.T.; Camoutsis, C.; Karamanakos, P.; Arvanitis, A.; Tegou, E.; Ziras, N.; Athanassiou, A.E. Preclinical evaluation of the homo-aza-steroid ester 13-hydroxy-13-amino-13,17- seco-5-androstan-17-oic-13,17-lactam-p-bis(2-chloroethyl)aminophenoxy acetate for the treatment of malignant melanoma. J. BUON 2003, 8, 333–339. [Google Scholar] [PubMed]
- Ruch, R.J.; Cheng, C.; Klaunig, J.E. Prevention of cytotoxicity and inhibition of intercellular communication by antioxidant catechins isolated from chinese green tea. Carcinogenesis 1989, 10, 1003–1008. [Google Scholar] [CrossRef]
- Kontogiorgis, C.; Hadjipavlou-Litina, D. Biological evaluation of several coumarin derivatives designed as possible anti-inflammatory/antioxidant Agents. J. Enz. Inhib. Med. Chem. 2003, 18, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Li, D.D.; Tian, J.; Gu, W.; Liu, X.; Yan, S. A novel 1,2,4-triazole-based copper(II) complex: Synthesis, characterization, magnetic property and nuclease activity. J. Inorg. Biochem. 2010, 104, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.; Lin, H.; Zhu, S.; Sun, H.; Chen, Y. Dinuclear palladium(II) complexes containing two monofunctional [Pd(en)(pyridine)Cl]+ units bridged by Se or S. Synthesis, characterization, cytotoxicity and kinetic studies of DNA-binding. J. Inorg. Biochem. 1998, 70, 219–226. [Google Scholar] [CrossRef]
- Heller, D.P.; Greenstock, C.L. Fluorescence lifetime analysis of DNA intercalated ethidium bromide and quenching by free dye. Biophys. Chem. 1994, 50, 305–312. [Google Scholar] [CrossRef] [PubMed]
- Hehre, W.; Ohlinger, S. Spartan’14 for Windows, Macintosh and Linux, Tutorial and User’s Guide; Wavefunction Inc.: Irvine, CA, USA, 2014. [Google Scholar]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berman, H.M.; Henrick, K.; Nakamura, H. Announcing the worldwide Protein Data Bank. Nat. Struct. Mol. Biol. 2003, 10, 980. [Google Scholar] [CrossRef]
- Bernstein, F.C.; Koetzle, T.F.; Williams, G.J.; Meyer, E.F., Jr.; Brice, M.D.; Rodgers, J.R.; Kennard, O.; Shimanouchi, T.; Tasumi, M. The Protein Data Bank: A computer-based archival file for macromolecular structures. J. Mol. Biol. 1977, 112, 535–542. [Google Scholar] [CrossRef]
- Fosgerau, K.; Hoffmann, T. Peptide therapeutics: Current status and future directions. Drug Discov. Today 2015, 20, 122–128. [Google Scholar] [CrossRef] [Green Version]
- Shivakumar, D.; Williams, J.; Wu, Y.; Damm, W.; Shelley, J.; Sherman, W. Prediction of absolute solvation free energies using molecular dynamics free energy perturbation and the OPLS force field. J. Chem. Theory Comput. 2010, 6, 1509–1519. [Google Scholar] [CrossRef]
- Sastry, G.M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, M.P.; Pincus, D.L.; Rapp, C.S.; Day, T.J.F.; Honig, B.; Shaw, D.E.; Friesner, R.A. A hierarchical approach to all-atom protein loop prediction. Proteins Struct. Funct. Bioinform. 2004, 55, 351–367. [Google Scholar] [CrossRef] [Green Version]
- Jacobson, M.P.; Friesner, R.A.; Xiang, Z.; Honig, B. On the role of the crystal environment in determining protein side-chain conformations. J. Mol. Biol. 2002, 320, 597–608. [Google Scholar] [CrossRef] [PubMed]
- Halgren, T. New method for fast and accurate binding-site identification and analysis. Chem. Biol. Drug Des. 2007, 69, 146–148. [Google Scholar] [CrossRef] [PubMed]
- Halgren, T. Identifying and characterizing binding sites and assessing druggability. J. Chem. Inf. Model. 2009, 49, 377–389. [Google Scholar] [CrossRef]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shaw, D.E.; Shelley, M.; et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Farid, R.; Day, T.; Friesner, R.A.; Pearlstein, R.A. New insights about HERG blockade obtained from protein modeling, poten-tial energy mapping, and docking studies. Bioorg. Med. Chem. 2006, 14, 3160–3173. [Google Scholar] [CrossRef]
- Sherman, W.; Day, T.; Jacobson, M.P.; Friesner, R.A.; Farid, R. Novel procedure for modeling ligand/receptor induced fit ef-fects. J. Med. Chem. 2006, 49, 534–553. [Google Scholar] [CrossRef]
- Geromichalos, G.D.; Alifieris, C.E.; Geromichalou, E.G.; Trafalis, D.T. Overview on the current status of virtual high-throughput screening and combinatorial chemistry approaches in multi-target anticancer drug discovery; Part I. J. BUON 2016, 21, 764–779. [Google Scholar]
- DeLano, W.L. The PyMOL Molecular Graphics System 0.99; DeLano Scientific: San Carlos, CA, USA, 2006. [Google Scholar]
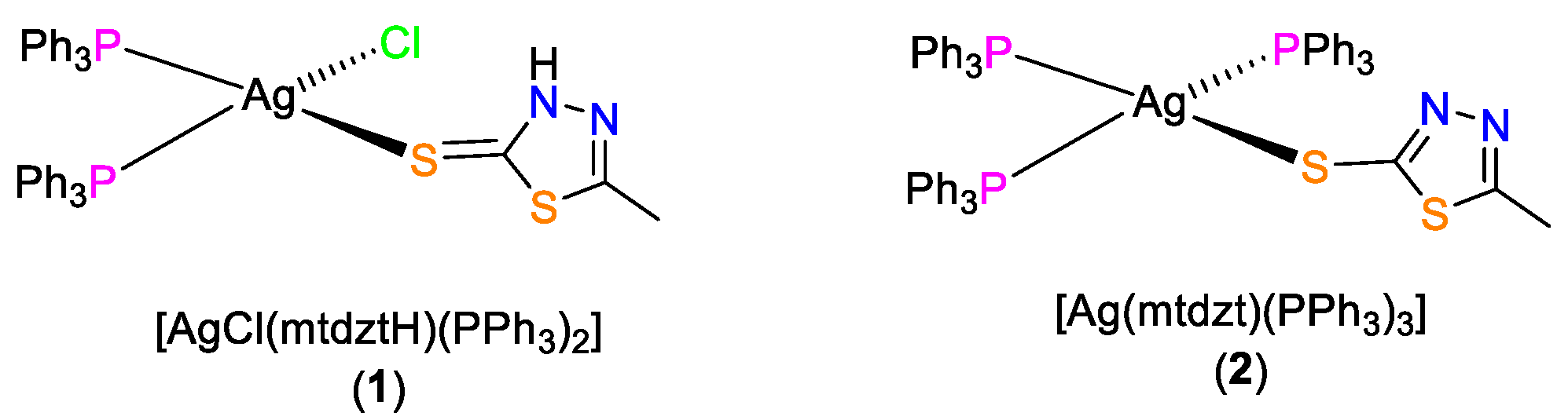








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 3 | 4 | |
|---|---|---|
| Bond lengths (Å) | ||
| Ag1–P1 | 2.513(1) | 2.451(1) |
| Ag1–P2 | 2.471(1) | 2.449(1) |
| Ag1–S1 | 2.699(2) | 2.654(1) |
| Ag1–Cl1 | 2.561(1) | |
| Ag1···O1 | 2.768(3) | |
| Bond angles (°) | ||
| P1–Ag1–P2 | 109.68(3) | 130.69(3) |
| P2–Ag1–S1 | 103.31(3) | 112.24(3) |
| P1–Ag1–S1 | 108.38(4) | 102.89(3) |
| Ag1–S1–C1 | 99.98(14) | 111.7(2) |
| P1–Ag–Cl1 | 113.00(3) | |
| P2–Ag–Cl1 | 121.74(3) | |
| S1–Ag–Cl1 | 98.88(3) | |
| Compound | λmax(abs)/nm (ε/M−1cm−1) | λmax(em)/nm |
|---|---|---|
| 1 | 256 (26,500), 310 (3300) | 428 |
| 2 | 257 (37,000) | 460 |
| 3 | 254 (16,300), 307 (10,300) | 432 |
| 4 | 265 (30,700) | 400, 419, 434 |
| Compound | λmax(nm) (ΔA/Aο (%) a, Δλ (nm) b) | Kb (M−1) |
|---|---|---|
| 3 | 275 nm (+15, +2) | 2.48 (±0.32) × 104 |
| 4 | 272 nm (–13, 0 ) | 6.63 (±0.12) × 105 |
| mtdztH | 313 (–12,0) | 7.34 (±0.02) × 107 |
| Compound | ΔI/Iο (%) | KSV (M−1) | kq (M−1s−1) |
|---|---|---|---|
| 3 | 25.3 | 2.40 (±0.08) × 104 | 1.40 (±0.03) × 1012 |
| 4 | 27.6 | 5.68 (±0.20) × 104 | 2.47 (±0.10) × 1012 |
| Compound | BSA | HSA | ||
|---|---|---|---|---|
| kq (M−1s−1) | K (M−1) | kq (M−1s−1) | K (M−1) | |
| 3 | 6.62 (±0.29) × 1012 | 7.89 (±0.8) × 104 | 5.91 (±0.25) × 1012 | 1.45 (±0.11) × 105 |
| 4 | 7.15 (±0.40) × 1012 | 1.29 (±0.09) × 105 | 5.11 (±0.37) × 1011 | 7.21 (±0.53) × 104 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Varna, D.; Geromichalou, E.; Karlioti, G.; Papi, R.; Dalezis, P.; Hatzidimitriou, A.G.; Psomas, G.; Choli-Papadopoulou, T.; Trafalis, D.T.; Angaridis, P.A. Inhibition of Cancer Cell Proliferation and Bacterial Growth by Silver(I) Complexes Bearing a CH3-Substituted Thiadiazole-Based Thioamide. Molecules 2023, 28, 336. https://doi.org/10.3390/molecules28010336
Varna D, Geromichalou E, Karlioti G, Papi R, Dalezis P, Hatzidimitriou AG, Psomas G, Choli-Papadopoulou T, Trafalis DT, Angaridis PA. Inhibition of Cancer Cell Proliferation and Bacterial Growth by Silver(I) Complexes Bearing a CH3-Substituted Thiadiazole-Based Thioamide. Molecules. 2023; 28(1):336. https://doi.org/10.3390/molecules28010336
Chicago/Turabian StyleVarna, Despoina, Elena Geromichalou, Georgia Karlioti, Rigini Papi, Panagiotis Dalezis, Antonios G. Hatzidimitriou, George Psomas, Theodora Choli-Papadopoulou, Dimitrios T. Trafalis, and Panagiotis A. Angaridis. 2023. "Inhibition of Cancer Cell Proliferation and Bacterial Growth by Silver(I) Complexes Bearing a CH3-Substituted Thiadiazole-Based Thioamide" Molecules 28, no. 1: 336. https://doi.org/10.3390/molecules28010336
APA StyleVarna, D., Geromichalou, E., Karlioti, G., Papi, R., Dalezis, P., Hatzidimitriou, A. G., Psomas, G., Choli-Papadopoulou, T., Trafalis, D. T., & Angaridis, P. A. (2023). Inhibition of Cancer Cell Proliferation and Bacterial Growth by Silver(I) Complexes Bearing a CH3-Substituted Thiadiazole-Based Thioamide. Molecules, 28(1), 336. https://doi.org/10.3390/molecules28010336