
Design of κ-Opioid Receptor Agonists for the Development of Potential Treatments of Pain with Reduced Side Effects


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. KOR Agonists
2.1. Small Molecules
2.1.1. Morphinans
2.1.2. Benzomorphans
2.1.3. Arylacetamides
2.1.4. Triazoles
2.1.5. Diphenethylamines
2.1.6. Salvinorins
2.2. Peptides and Peptidomimetics
2.2.1. Dynorphins
2.2.2. Difelikefalin (CR845)
2.2.3. Conorphins
2.2.4. Helianorphins
2.2.5. H-Tyr-Amo-Trp-PheNH2
2.2.6. CJ-15,209 and Derivatives
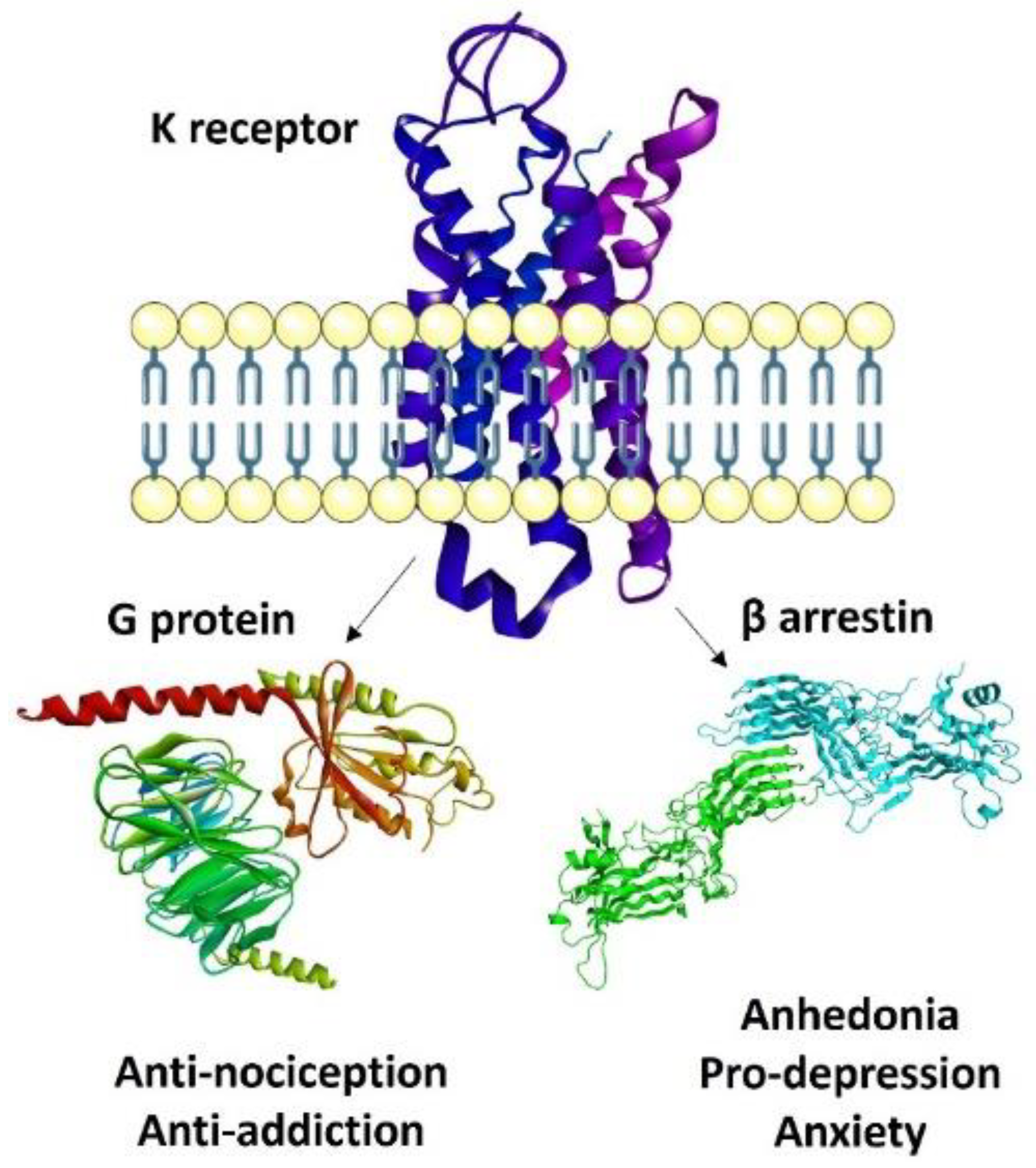
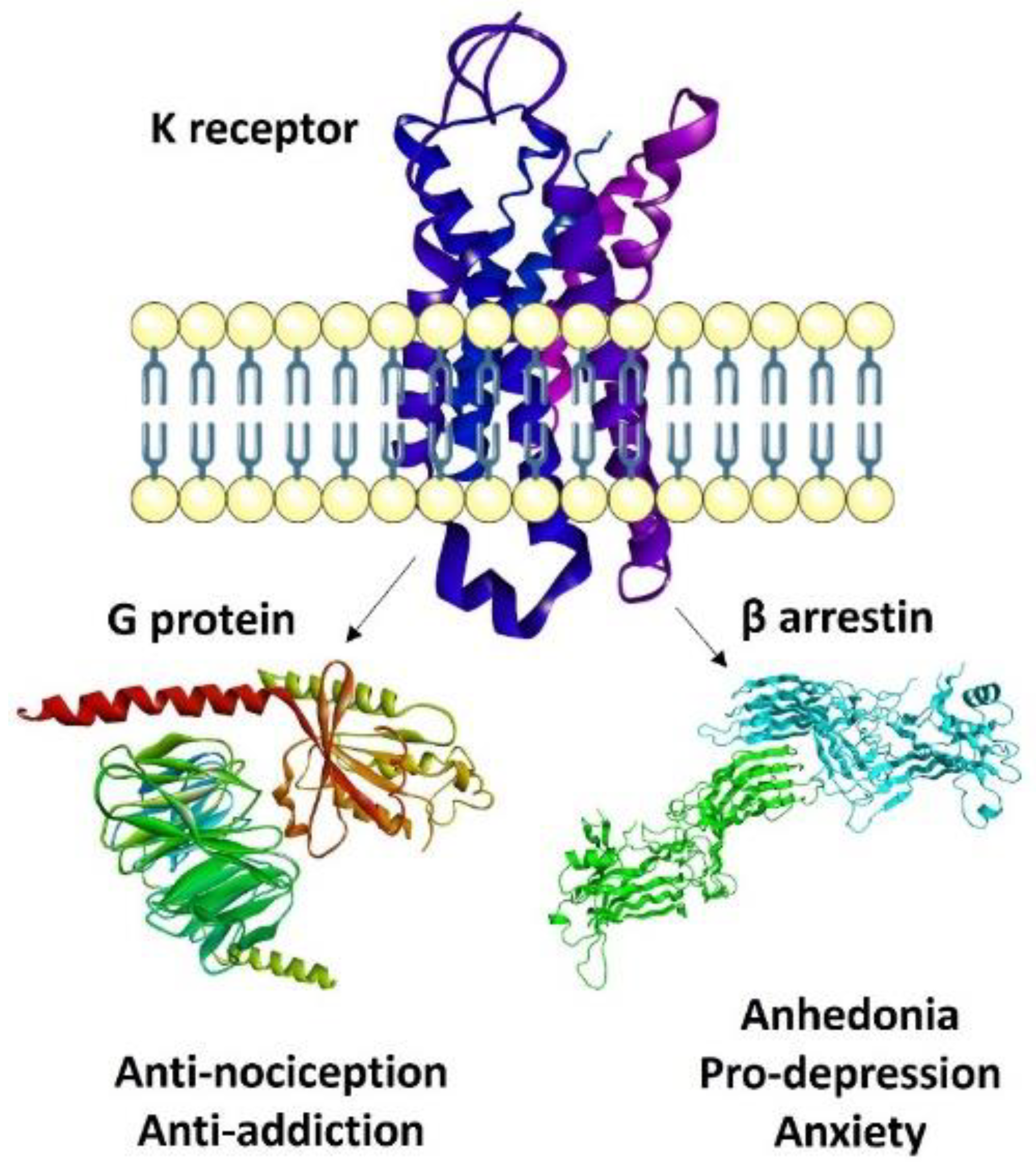
3. Recent Paradigms in KOR Agonism: Biased, Partial, Peripheral
3.1. Biased Ligands
Biased Ligands at KOR
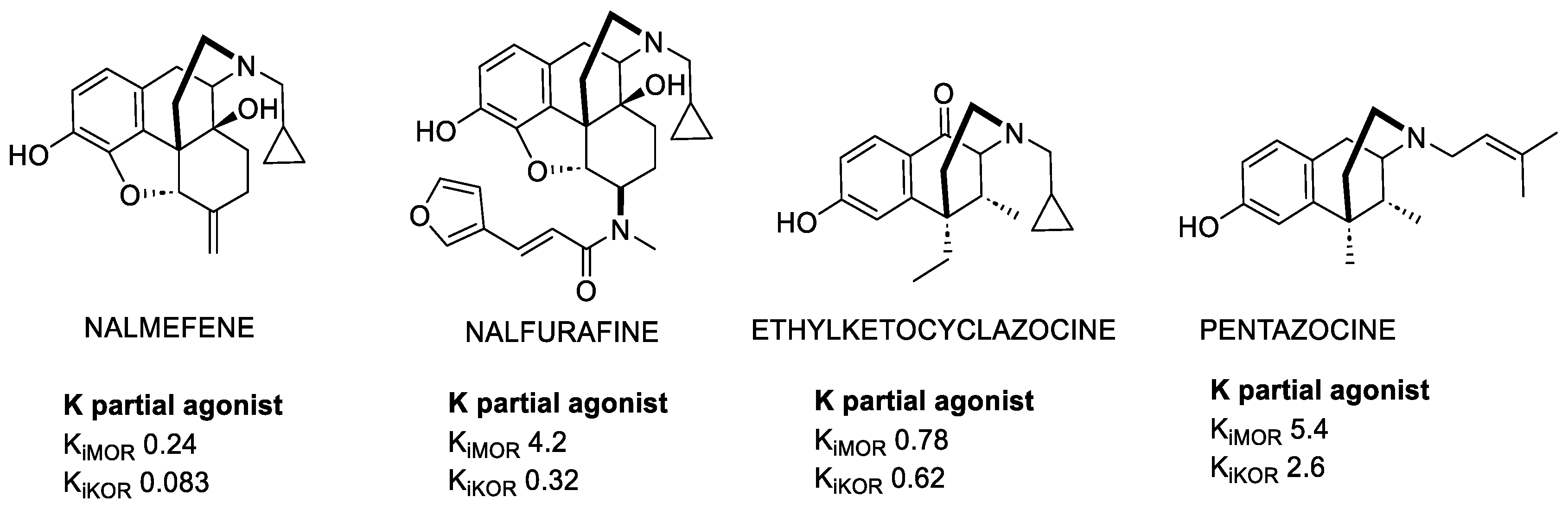
3.2. KOR-Selective Partial Agonists
3.3. Central vs. Peripheral Activity
4. Structural Insights in Ligand-Receptor Interactions for KOR Agonists
4.1. Crystal Structure of KOR–JDTic
4.2. Crystal Structure of KOR-MP1104
4.3. Docking Simulations
4.3.1. Docking of Salvinorins
4.3.2. Docking of Dynorphin
4.3.3. Docking of Conorphin
4.3.4. Docking of Tyr-Amo-Trp-PheNH2
4.4. Design of Biased KOR Ligands
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lemos, C.J.; Chavkin, C. Kappa opioid receptor function. In The Opiate Receptors, 2nd ed.; Pasternak, C.W., Ed.; Humana Press: Totowa, NJ, USA, 2011; pp. 265–305. [Google Scholar]
- Liu-Chen, L.-Y.; Inan, S. The Kappa Opioid Receptor, Handbook of Experimental Pharmacology, 1st ed.; Springer: Cham, Switzerland, 2022; Volume 271, p. 577. [Google Scholar]
- Janecka, A.; Fichna, J.; Janecki, T. Opioid receptors and their ligands. Curr. Top. Med. Chem. 2004, 4, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carlezon, W.A., Jr.; Béguin, C.; Knoll, A.T.; Cohen, B.M. Kappa-opioid ligands in the study and treatment of mood disorders. Pharm. Ther. 2009, 123, 334–343. [Google Scholar]
- Clark, S.D. The Role of Dynorphin and the Kappa Opioid Receptor in Schizophrenia and Major Depressive Disorder: A Translational Approach. In The Kappa Opioid Receptor. Handbook of Experimental Pharmacology; Liu-Chen, L.Y., Inan, S., Eds.; Springer: Cham, Switzerland, 2020; Volume 271, pp. 525–546. [Google Scholar]
- Chavkin, C.; James, I.; Goldstein, A. Dynorphin is a specific endogenous ligand of the kappa opioid receptor. Science 1982, 215, 413–415. [Google Scholar] [CrossRef] [PubMed]
- Lazenka, M.F. Antinociceptive Effects of Kappa-Opioid Receptor Agonists. Handb. Exp. Pharmacol. 2022, 271, 293–313. [Google Scholar] [PubMed]
- Beck, T.C.; Hapstack, M.A.; Beck, K.R.; Dix, T.A. Therapeutic Potential of Kappa Opioid Agonists. Pharmaceuticals 2019, 12, 95. [Google Scholar] [CrossRef] [Green Version]
- Dalefield, M.L.; Scouller, B.; Bibi, R.; Kivell, B.M. The Kappa Opioid Receptor: A Promising Therapeutic Target for Multiple Pathologies. Front. Pharmacol. 2022, 13, 837671. [Google Scholar] [CrossRef]
- Karkhanis, A.; Holleran, K.M.; Jones, S.R. Dynorphin/Kappa Opioid Receptor Signaling in Preclinical Models of Alcohol, Drug, and Food Addiction. Int. Rev. Neurobiol. 2017, 136, 53–88. [Google Scholar] [PubMed]
- Estave, P.M.; Spodnick, M.B.; Karkhanis, A.N. KOR Control over addiction processing: An exploration of the mesolimbic dopamine pathway. In The Kappa Opioid Receptor. Handbook of Experimental Pharmacology; Liu-Chen, L.Y., Inan, S., Eds.; Springer: Cham, Switzerland, 2020; Volume 271. [Google Scholar]
- Del Pilar Escobar, A.; Casanova, J.P.; Andrés, M.E.; Fuentealba, J.A. Crosstalk Between Kappa Opioid and Dopamine Systems in Compulsive Behaviors. Front. Pharmacol. 2020, 11, 57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brust, T.F.; Morgenweck, J.; Kim, S.A.; Rose, J.H.; Locke, J.L.; Schmid, C.L.; Zhou, L.; Stahl, E.L.; Cameron, M.D.; Scarry, S.M.; et al. Biased Agonists of the Kappa Opioid Receptor Suppress Pain and Itch without Causing Sedation or Dysphoria. Sci. Signal. 2016, 9, ra117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faouzi, A.; Varga, B.R.; Majumdar, S. Biased Opioid Ligands. Molecules 2020, 25, 4257. [Google Scholar] [CrossRef]
- Neto, J.A.; Costanzini, A.; De Giorgio, R.; Lambert, D.G.; Ruzza, C.; Calò, G. Biased versus partial agonism in the search for safer opioid analgesics. Molecules 2020, 25, 3870. [Google Scholar] [CrossRef] [PubMed]
- Albert-Vartanian, A.; Boyd, M.R.; Hall, A.L.; Morgado, S.J.; Nguyen, E.; Nguyen, V.P.H.; Patel, S.P.; Russo, L.J.; Shao, A.J.; Raffa, R.B. Will peripherally restricted kappa-opioid receptor agonists (pKORAs) relieve pain with less opioid adverse effects and abuse potential? J. Clin.Pharm. Ther. 2016, 41, 371–382. [Google Scholar] [CrossRef] [Green Version]
- Inui, S. Nalfurafine hydrochloride to treat pruritus: A review. Clin. Cosmet. Investig. Dermatol. 2015, 8, 249–255. [Google Scholar] [CrossRef] [Green Version]
- Keating, G.M. Nalmefene: A review of its use in the treatment of alcohol dependence. CNS Drugs 2013, 27, 761–772. [Google Scholar] [CrossRef] [PubMed]
- Martin, W.R.; Eades, C.G.; Thompson, J.A.; Huppler, R.E.; Gilbert, P.E. The effects of morphine- and nalorphine-like drugs in the nondependent and morphine-dependent chronic spinal dog. J. Pharmacol. Exp. Ther. 1976, 197, 517–532. [Google Scholar] [PubMed]
- Schattauer, S.S.; Kuhar, J.R.; Song, A.; Chavkin, C. Nalfurafine is a G-protein biased agonist having significantly greater bias at the human than rodent form of the kappa opioid receptor. Cell. Signal. 2017, 32, 59–65. [Google Scholar] [CrossRef] [Green Version]
- Dortch-Carnes, J.; Potter, D.E. Bremazocine: A kappa-opioid agonist with potent analgesic and other pharmacologic properties. CNS Drug Rev. 2005, 11, 195–212. [Google Scholar] [CrossRef]
- Szmuszkovicz, J.; VonVoigtlander, P.F. Benzeneacetamide amines: Structurally novel non-m.mu. opioids. J. Med. Chem. 1982, 25, 1125–1126. [Google Scholar] [CrossRef] [PubMed]
- Mangel, A.W.; Williams, V.S.L. Asimadoline in the treatment of irritable bowel syndrome. Expert Opin. Investig. Drugs 2010, 19, 1257–1264. [Google Scholar] [CrossRef]
- Spetea, M.; Schmidhammer, H. Kappa Opioid Receptor Ligands and Pharmacology: Diphenethylamines, a Class of Structurally Distinct, Selective Kappa Opioid Ligands. Handb. Exp. Pharmacol. 2022, 271, 163–195. [Google Scholar]
- Zhou, L.; Lovell, K.M.; Frankowski, K.J.; Slauson, S.R.; Phillips, A.M.; Streicher, J.M.; Stahl, E.; Schmid, C.L.; Hodder, P.; Madoux, F.; et al. Development of functionally selective, small molecule agonists at kappa opioid receptors. J. Biol. Chem. 2013, 288, 36703–36716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nedelec, L.; Dumont, C.; Oberlander, C.; Frechet, D.; Laurent, J.; Boissier, J.R. Synthèse et étude de l’activité dopaminergique de dérivés de la di(phénéthyl)amine. Eur. J. Med. Chem. Chim. Ther. 1978, 13, 553–563. [Google Scholar]
- Schmidhammer, H.; Erli, F.; Guerrieri, E.; Spetea, M. Development of Diphenethylamines as Selective Kappa Opioid Receptor Ligands and Their Pharmacological Activities. Molecules 2020, 25, 5092. [Google Scholar] [CrossRef] [PubMed]
- Spetea, M.; Berzetei-Gurske, I.P.; Guerrieri, E.; Schmidhammer, H. Discovery and pharmacological evaluation of a diphenethylamine derivative (HS665), a highly potent and selective κ opioid receptor agonist. J. Med. Chem. 2012, 55, 10302–10306. [Google Scholar] [CrossRef] [PubMed]
- Spetea, M.; Eans, S.O.; Ganno, M.L.; Lantero, A.; Mairegger, M.; Toll, L.; Schmidhammer, H.; McLaughlin, J.P. Selective k receptor partial agonist HS666 produces potent antinociception without inducing aversion after i.c.v. administration in mice. Br. J. Pharmacol. 2017, 174, 2444–2456. [Google Scholar] [CrossRef] [Green Version]
- Erli, F.; Guerrieri, E.; Haddou, B.T.; Lantero, A.; Mairegger, M.; Schmidhammer, H.; Spetea, M. Highly Potent and Selective New Diphenethylamines Interacting with the κ-Opioid Receptor: Synthesis, Pharmacology, and Structure-Activity Relationships. J. Med. Chem. 2017, 60, 7579–7590. [Google Scholar] [CrossRef] [Green Version]
- Ortega, A.; Blount, J.F.; Manchand, P.S. Salvinorin, a new trans-neoclerodane diterpene from Salvia divinorum (Labiatae). J. Chem. Soc. Perkin Trans. 1 1982, 2505–2508. [Google Scholar] [CrossRef]
- Valdes, L.J.; Butler, W.B.; Hatfield, G.M.; Paul, A.G.; Koreeda, M. Divinorin A, a Psychotropic Terpenoid, and Divinorin B from the Hallucinogenic Mexican Mint Salvia divinorum. J. Org. Chem. 1984, 49, 4716–4720. [Google Scholar] [CrossRef]
- Valdes, L.J.; Chang, H.M.; Visger, D.C.; Koreeda, M. Salvinorin C, a New Neoclerodane Diterpene from a Bioactive Fraction of the Hallucinogenic Mexican Mint Salvia divinorum. Org. Lett. 2001, 3, 3935–3937. [Google Scholar] [CrossRef]
- Roach, J.J.; Shenvi, R.A. A Review of Salvinorin Analogs and their Kappa-Opioid Receptor Activity. Bioorg. Med. Chem. Lett. 2018, 28, 1436–1445. [Google Scholar] [CrossRef]
- Morani, A.S.; Ewald, A.; Prevatt-Smith, K.M.; Prisinzano, T.E.; Kivell, B.M. The 2-methoxy methyl analogue of salvinorin A attenuates cocaine-induced drug seeking and sucrose reinforcements in rats. Eur. J. Pharmacol. 2013, 720, 69–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kivell, B.M.; Ewald, A.W.M.; Prisinzano, T.E. Salvinorin A Analogs and Other Kappa-Opioid Receptor Compounds as Treatments for Cocaine Abuse. Adv. Pharmacol. 2014, 69, 481–511. [Google Scholar] [PubMed] [Green Version]
- Kivell, B.M.; Paton, K.F.; Kumar, N.; Morani, A.S.; Culverhouse, A.; Shepherd, A.; Welsh, S.A.; Biggerstaff, A.; Crowley, R.S.; Prisinzano, T.E. Kappa Opioid Receptor Agonist Mesyl Sal B Attenuates Behavioral Sensitization to Cocaine with Fewer Aversive Side-Effects than Salvinorin A in Rodents. Molecules 2018, 23, 2602. [Google Scholar] [CrossRef] [PubMed]
- White, K.L.; Robinson, J.E.; Zhu, H.; DiBerto, J.F.; Polepally, P.R.; Zjawiony, J.K.; Nichols, D.E.; Malanga, C.J.; Roth, B.L. The G protein-biased κ-opioid receptor agonist RB-64 is analgesic with a unique spectrum of activities in vivo. J. Pharmacol. Exp. Ther. 2015, 352, 98–109. [Google Scholar] [CrossRef] [Green Version]
- Bui, A.M.; Cavé, A.; Janot, M.M.; Parello, J.; Potier, P.; Scheidegger, U. Isolement et Analyse Structurale Du Collybolide, Nouveau Sesquiterpene Extrait de Collybia Maculata Alb. et Sch. Ex Fries (Basidiomycetes). Tetrahedron 1974, 30, 1327–1336. [Google Scholar] [CrossRef]
- Shevick, S.L.; Freeman, S.M.; Tong, G.; Russo, R.J.; Bohn, L.M.; Shenvi, R.A. Asymmetric Syntheses of (+)- and (−)-Collybolide Enable Reevaluation of kappa-Opioid Receptor Agonism. ACS Cent. Sci. 2022, 8, 948–954. [Google Scholar] [CrossRef]
- Aldrich, J.V.; McLaughlin, J.P. Peptide Kappa Opioid Receptor Ligands: Potential for Drug Development. AAPS J. 2009, 11, 312–322. [Google Scholar] [CrossRef] [Green Version]
- Gentilucci, L. New Trends in the Development of Opioid Peptide Analogues as Advanced Remedies for Pain Relief. Curr. Top. Med. Chem. 2004, 4, 19–38. [Google Scholar] [CrossRef]
- Gentilucci, L.; De Marco, R.; Cerisoli, L. Chemical Modifications Designed to Improve Peptide Stability: Incorporation of Non-Natural Amino Acids, Pseudo-Peptide Bonds, and Cyclization. Curr. Pharm. Des. 2010, 16, 3185–3203. [Google Scholar] [CrossRef]
- Chavkin, C. Dynorphin–Still an Extraordinarily Potent Opioid Peptide. Mol. Pharmacol. 2013, 83, 729–736. [Google Scholar] [CrossRef] [Green Version]
- Schlechtingen, G.; DeHaven, R.N.; Daubert, J.D.; Cassel, J.A.; Chung, N.N.; Schiller, P.W.; Taulane, J.P.; Goodman, M. Structureactivity relationships of dynorphin A analogues modified in the address sequence. J. Med. Chem. 2003, 46, 2104–2109. [Google Scholar] [CrossRef] [PubMed]
- Vanderah, T.W.; Schteingart, C.D.; Trojnar, J.; Junien, J.-L.; Lai, J.; Riviere, P.J.-M. FE200041 (D-Phe-D-Phe-D-Nle-D-Arg-NH2): A peripheral efficacious kappa opioid agonist with unprecedented selectivity. J. Pharmacol. Exp. Ther. 2004, 310, 326–333. [Google Scholar] [CrossRef] [PubMed]
- Vanderah, T.W.; Largent-Milnes, T.; Lai, J.; Porreca, F.; Houghten, R.A.; Menzaghi, F.; Wisniewski, K.; Stalewski, J.; Sueiras-Diaz, J.; Galyean, R.; et al. Novel D-amino acid tetrapeptides produce potent antinociception by selectively acting at peripheral kappa-opioid receptors. Eur. J. Pharmacol. 2008, 583, 62–72. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Gou, X.; Yu, X.; Bai, D.; Tan, B.; Cao, P.; Qian, M.; Zheng, X.; Wang, H.; Tang, P.; et al. Antinociceptive and Antipruritic Effects of HSK21542, a Peripherally-Restricted Kappa Opioid Receptor Agonist, in Animal Models of Pain and Itch. Front. Pharmacol. 2021, 12, 773204. [Google Scholar] [CrossRef] [PubMed]
- Beck, T.C.; Reichel, C.M.; Helke, K.L.; Bhadsavle, S.S.; Dix, T.A. Non-addictive orally-active kappa opioid agonists for the treatment of peripheral pain in rats. Eur. J. Pharmacol. 2019, 856, 172396. [Google Scholar] [CrossRef]
- Fugal, J.; Serpa, S.M. Difelikefalin: A New κ-Opioid Receptor Agonist for the Treatment of Hemodialysis-Dependent Chronic Kidney Disease–Associated Pruritus. Ann. Pharmacother. 2022; Online First. [Google Scholar] [CrossRef]
- Kutlu Yalcin, E.; Araujo-Duran, J.; Turan, A. Emerging drugs for the treatment of postsurgical pain. Expert Opin. Emerg. Drugs 2021, 26, 371–384. [Google Scholar] [CrossRef]
- Schmidtko, A.; Loetsch, J.; Freynhagen, R.; Geisslinger, G. Ziconotide for treatment of severe chronic pain. Lancet 2010, 375, 1569–1577. [Google Scholar] [CrossRef]
- Brust, A.; Croker, D.E.; Colless, B.; Ragnarsson, L.; Andersson, Å.; Jain, K.; Garcia-Caraballo, S.; Castro, J.; Brierley, S.M.; Alewood, P.F.; et al. Conopeptide-derived κ opioid agonists (Conorphins): Potent, selective, and metabolic stable dynorphin A mimetics with antinociceptive properties. J. Med. Chem. 2016, 59, 2381–2395. [Google Scholar] [CrossRef] [Green Version]
- Muratspahić, E.; Tomašević, N.; Koehbach, J.; Duerrauer, L.; Hadžić, S.; Castro, J.; Schober, G.; Sideromenos, S.; Clark, R.J.; Brierley, S.M.; et al. Design of a Stable Cyclic Peptide Analgesic Derived from Sunflower Seeds that Targets the κ-Opioid Receptor for the Treatment of Chronic Abdominal Pain. J. Med. Chem. 2021, 64, 9042–9055. [Google Scholar] [CrossRef]
- Zadina, J.E.; Hackler, L.; Ge, L.J.; Kastin, A.J. A potent and selective endogenous agonist for the mu-opiate receptor. Nature 1997, 386, 499–502. [Google Scholar] [CrossRef] [PubMed]
- Harrison, C.; McNulty, S.; Smart, D.; Rowbotham, D.J.; Grandy, D.K.; Devi, L.A.; Lambert, D.G. The effects of endomorphin-1 and endomorphin-2 in CHO cells expressing recombinant mu-opioid receptors and SH-SY5Y cells. Br. J. Pharmacol. 1999, 128, 472–478. [Google Scholar] [CrossRef] [Green Version]
- Fichna, J.; Janecka, A.; Costentin, J.; Do Rego, J.C. The endomorphin system and its evolving neurophysiological role. Pharmacol. Rev. 2007, 59, 88–123. [Google Scholar] [CrossRef] [PubMed]
- De Marco, R.; Bedini, A.; Spampinato, S.; Comellini, L.; Zhao, J.; Artali, R.; Gentilucci, L. Constraining endomorphin-1 by beta,alpha-hybrid dipeptide/heterocycle scaffolds: Identification of a novel kappa-opioid receptor selective partial agonist. J. Med. Chem. 2018, 61, 5751–5757. [Google Scholar] [CrossRef] [PubMed]
- Greco, A.; Tani, S.; De Marco, R.; Gentilucci, L. Synthesis and analysis of the conformational preferences of 5-aminomethyloxazolidine-2,4-dione scaffolds: First examples of β2- and β2,2-homo-Freidinger lactam analogues. Chem. Eur. J. 2014, 20, 13390–13404. [Google Scholar] [CrossRef] [PubMed]
- Saito, T.; Hirai, H.; Kim, Y.J.; Kojima, Y.; Matsunaga, Y.; Nishida, H.; Sakakibara, T.; Suga, O.; Sujaku, T.; Kojima, N. CJ-15,208, a novel kappa opioid receptor antagonist from a fungus, Ctenomyces serratus ATCC15502. J. Antibiot. 2002, 55, 847–854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dolle, R.E.; Michaut, M.; Martinez-Teipel, B.; Seida, P.R.; Ajello, C.W.; Muller, A.L.; DeHaven, R.N.; Carroll, P.J. Nascent structure-activity relationship study of a diastereomeric series of kappa opioid receptor antagonists derived from CJ-15,208. Bioorg. Med. Chem. Lett. 2009, 19, 3647–3650. [Google Scholar] [CrossRef]
- Aldrich, J.V.; Kulkarni, S.S.; Senadheera, S.N.; Ross, N.C.; Reilley, K.J.; Eans, S.O.; Ganno, M.L.; Murray, T.F.; McLaughlin, J.P. Unexpected opioid activity profiles of analogues of the novel peptide kappa opioid receptor ligand CJ-15,208. ChemMedChem 2011, 6, 1739–1745. [Google Scholar] [CrossRef]
- Ross, N.C.; Reilley, K.J.; Murray, T.F.; Aldrich, J.V.; McLaughlin, J.P. Novel opioid cyclic tetrapeptides: Trp isomers of CJ-15,208 exhibit distinct opioid receptor agonism and short-acting k opioid receptor antagonism. Br. J. Pharmacol. 2012, 165, 1097–1108. [Google Scholar] [CrossRef] [Green Version]
- Aldrich, J.V.; Senadheera, S.N.; Ross, N.C.; Reilley, K.A.; Ganno, M.L.; Eans, S.E.; Murray, T.F.; McLaughlin, J.P. Alanine analogues of [D-Trp] CJ-15,208: Novel opioid activity profiles and prevention of drug- and stress-induced reinstatement of cocaine seeking behavior. Br. J. Pharmacol. 2014, 171, 3212–3222. [Google Scholar] [CrossRef] [Green Version]
- Aldrich, J.V.; Senadheera, S.N.; Ross, N.C.; Ganno, M.L.; Eans, S.O.; McLaughlin, J.P. The macrocyclic peptide natural product CJ-15,208 is orally active and prevents reinstatement of extinguished cocaine-seeking behavior. J. Nat. Prod. 2013, 76, 433–438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eans, S.O.; Ganno, M.L.; Reilley, K.J.; Patkar, K.A.; Senadheera, S.N.; Aldrich, J.V.; McLaughlin, J.P. The macrocyclic tetrapeptide [D-Trp] CJ-15,208 produces short-acting k opioid receptor antagonism in the CNS after oral administration. Br. J. Pharmacol. 2013, 169, 426–436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brice-Tutt, A.C.; Senadheera, S.N.; Ganno, M.L.; Eans, S.O.; Khaliq, T.; Murray, T.F.; McLaughlin, J.P.; Aldrich, J.V. Phenylalanine Stereoisomers of CJ-15,208 and [d-Trp] CJ-15,208 Exhibit Distinctly Different Opioid Activity Profiles. Molecules 2020, 25, 3999. [Google Scholar] [CrossRef] [PubMed]
- Cardillo, G.; Gentilucci, L.; Tolomelli, A.; Spinosa, R.; Calienni, M.; Qasem, A.R.; Spampinato, S. Synthesis and evaluation of the affinity toward μ-opioid receptors of atypical, lipophilic ligands based on the sequence c[Tyr-Pro-Trp-Phe-Gly]. J. Med. Chem. 2004, 47, 5198–5203. [Google Scholar] [CrossRef] [PubMed]
- Bedini, A.; Baiula, M.; Gentilucci, L.; Tolomelli, A.; De Marco, R.; Spampinato, S. Peripheral antinociceptive effects of the cyclic endomorphin-1 analog c[YpwFG] in a mouse visceral pain model. Peptides 2010, 31, 2135–2140. [Google Scholar] [CrossRef] [PubMed]
- Gentilucci, L.; Tolomelli, A.; De Marco, R.; Spampinato, S.; Bedini, A.; Artali, R. The inverse type II beta-turn on D-Trp-Phe, a pharmacophoric motif for MOR agonists. ChemMedChem 2011, 6, 1640–1653. [Google Scholar] [CrossRef]
- De Marco, R.; Tolomelli, A.; Spampinato, S.; Bedini, A.; Gentilucci, L. Opioid activity profiles of oversimplified peptides lacking in the protonable N-terminus. J. Med. Chem. 2012, 55, 10292–10296. [Google Scholar] [CrossRef]
- De Marco, R.; Bedini, A.; Spampinato, S.; Gentilucci, L. Synthesis of tripeptides containing D-Trp substituted at the indole ring, assessment of opioid receptor binding and in vivo central antinociception. J. Med. Chem. 2014, 57, 6861–6866. [Google Scholar] [CrossRef]
- Gentilucci, L.; Squassabia, F.; Artali, R. Re-discussion of the importance of ionic interactions in stabilizing ligand-opioid receptor complex and in activating signal transduction. Curr. Drug Targets 2007, 8, 185–196. [Google Scholar] [CrossRef]
- Gentilucci, L.; Tolomelli, A.; De Marco, R.; Artali, R. Molecular docking of opiates and opioid peptides, a tool for the design of selective agonists and antagonists, and for the investigation of atypical ligand-receptor interactions. Curr. Med. Chem. 2012, 19, 1587–1601. [Google Scholar] [CrossRef]
- De Marco, R.; Bedini, A.; Spampinato, S.; Cavina, L.; Pirazzoli, E.; Gentilucci, L. Versatile Picklocks To Access All Opioid Receptors: Tuning the Selectivity and Functional Profile of the Cyclotetrapeptide c[Phe-D-Pro-Phe-Trp] (CJ-15,208). J. Med. Chem. 2016, 59, 9255–9261. [Google Scholar] [CrossRef]
- Bedini, A.; Di Cesare Mannelli, L.; Micheli, L.; Baiula, M.; Vaca, G.; De Marco, R.; Gentilucci, L.; Ghelardini, C.; Spampinato, S. Functional selectivity and antinociceptive effects of a novel KOR agonist. Front. Pharmacol. 2020, 11, 188. [Google Scholar] [CrossRef] [Green Version]
- De Marco, R.; Gentilucci, L. Tryptophan-Containing Non-Cationizable Opioid Peptides—A new chemotype with unusual structure and in vivo activity. Future Med. Chem. 2017, 9, 2099–2115. [Google Scholar] [CrossRef] [PubMed]
- Kenakin, T.; Christopoulos, A. Signalling bias in new drug discovery: Detection, quantification and therapeutic impact. Nat. Rev. Drug Discov. 2013, 12, 205–216. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.T.; Pitis, P.; Liu, G.; Yuan, C.; Gotchev, D.; Cowan, C.L.; Rominger, D.H.; Koblish, M.; Dewire, S.M.; Crombie, A.L.; et al. Structure-activity relationships and discovery of a G protein biased _ opioid receptor ligand, [(3-methoxythiophen-2-yl) methyl] ({2-[(9R)-9-(pyridin-2-yl)-6-oxaspiro-[4.5] decan-9-yl]ethyl})amine (TRV130), for the treatment of acute severe pain. J. Med. Chem. 2013, 56, 8019–8031. [Google Scholar] [CrossRef]
- DeWire, S.M.; Yamashita, D.S.; Rominger, D.H.; Liu, G.; Cowan, C.L.; Graczyk, T.M.; Chen, X.T.; Pitis, P.M.; Gotchev, D.; Yuan, C.; et al. A G protein-biased ligand at the κ-opioid receptor is potently analgesic with reduced gastrointestinal and respiratory dysfunction compared with morphine. J. Pharmacol. Exp. Ther. 2013, 344, 708–717. [Google Scholar] [CrossRef] [Green Version]
- Manglik, A.; Lin, H.; Aryal, D.K.; McCorvy, J.D.; Dengler, D.; Corder, G.; Levit, A.; Kling, R.C.; Bernat, V.; Hübner, H.; et al. Structure-based discovery of opioid analgesics with reduced side effects. Nature 2016, 537, 185–190. [Google Scholar] [CrossRef] [Green Version]
- Soergel, D.G.; Subach, R.A.; Sadler, B.; Connell, J.; Marion, A.S.; Cowan, C.L.; Violin, J.D.; Lark, M.W. First clinical experience with TRV130: Pharmacokinetics and pharmacodynamics in healthy volunteers. J. Clin. Pharmacol. 2014, 54, 351–357. [Google Scholar] [CrossRef]
- Lambert, D.; Calo, G. Approval of oliceridine (TRV130) for intravenous use in moderate to severe pain in adults. Br. J. Anaesth. 2020, 125, e473–e474. [Google Scholar] [CrossRef]
- Hill, R.; Disney, A.; Conibear, A.; Sutcliffe, K.; Dewey, W.; Husbands, S.; Bailey, C.; Kelly, E.; Henderson, G. The novel mu-opioid receptor agonist PZM21 depresses respiration and induces tolerance to antinociception. Br. J. Pharmacol. 2018, 175, 2653–2661. [Google Scholar] [CrossRef] [Green Version]
- Kliewer, A.; Schmiedel, F.; Sianati, S.; Bailey, A.; Bateman, J.T.; Levitt, E.S.; Williams, J.T.; Christie, M.J.; Schulz, S. Phosphorylationdeficient G-protein-biased _-opioid receptors improve analgesia and diminish tolerance but worsen opioid side effects. Nat. Commun. 2019, 21, 367. [Google Scholar] [CrossRef] [Green Version]
- Bohn, L.M.; Lefkowitz, R.J.; Gainetdinov, R.R.; Peppel, K.; Caron, M.G.; Lin, F.T. Enhanced morphine analgesia in mice lacking β-arrestin 2. Science 1999, 286, 2495–2498. [Google Scholar] [CrossRef] [PubMed]
- Conibear, A.E.; Kelly, E. A biased view of κ-opioid receptors? Mol. Pharmacol. 2019, 96, 542–549. [Google Scholar] [CrossRef] [PubMed]
- Gillis, A.; Gondin, A.B.; Kliewer, A.; Sanchez, J.; Lim, H.D.; Alamein, C.; Manandhar, P.; Santiago, M.; Fritzwanker, S.; Schmidel, F.; et al. Low intrinsic efficacy for G protein activation can explain the improved side effect profiles of new opioid agonists. Sci. Signal. 2020, 13, e3140. [Google Scholar] [CrossRef] [PubMed]
- Mores, K.L.; Cummins, B.R.; Cassell, R.J.; van Rijn, R.M. A Review of the Therapeutic Potential of Recently Developed G Protein-Biased Kappa Agonists. Front. Pharmacol. 2019, 10, 407. [Google Scholar] [CrossRef]
- Trevisan, G.; Rossato, M.F.; Walker, C.I.; Oliveira, S.M.; Rosa, F.; Tonello, R.; Silva, C.R.; Machado, P.; Boligon, A.A.; Martins, M.A.; et al. A novel, potent, oral active and safe antinociceptive pyrazole targeting kappa opioid receptors. Neuropharmacology 2013, 73, 261–273. [Google Scholar]
- Zheng, Z.; Huang, X.-P.; Mangano, T.J.; Zou, R.; Chen, X.; Zaidi, S.A.; Roth, B.L.; Stevens, R.C.; Katritch, V. Structure-based discovery of new antagonist and biased agonist chemotypes for the kappa opioid receptor. J. Med. Chem. 2017, 60, 3070–3081. [Google Scholar]
- Dunn, A.; Reed, B.; Erazo, J.; Ben-Ezra, A.; Kreek, M.J. Signaling properties of structurally diverse kappa opioid receptor ligands: Towards in vitro models of in vivo responses. ACS Chem. Neurosci. 2019, 10, 9b00195. [Google Scholar] [CrossRef]
- Kong, L.; Shu, X.; Tang, S.; Ye, R.; Sun, H.; Jiang, S.; Li, Z.; Chai, J.; Fang, Y.; Lan, Y.; et al. SLL-627 Is a Highly Selective and Potent κ Opioid Receptor (KOR) Agonist with an Unexpected Nonreduction in Locomotor Activity. J. Med. Chem. 2022, 65, 10377–10392. [Google Scholar] [CrossRef]
- Bidlack, J.M.; McLaughlin, J.P.; Wentland, M.P. Partial opioids: Medications for the treatment of pain and drug abuse. Ann. N. Y. Acad. Sci. 2000, 909, 1–11. [Google Scholar]
- Aldrich, J.V.; McLaughlin, J.P. Opioid Peptides: Potential for drug development. Drug Discov. Today Technol. 2012, 9, e23–e31. [Google Scholar] [CrossRef] [Green Version]
- Aldrich, J.V.; Patkar, K.A.; McLaughlin, J.P. Zyklophin, a systemically active selective kappa opioid receptor peptide antagonist with short duration of action. Proc. Natl. Acad. Sci. USA 2009, 106, 18396–18401. [Google Scholar] [CrossRef] [Green Version]
- Beck, T.C.; Dix, T.A. Targeting peripheral ϰ-opioid receptors for the non-addictive treatment of pain. Future Drug Discov. 2019, 1, FDD17. [Google Scholar] [CrossRef] [Green Version]
- Gentilucci, L.; Tolomelli, A.; Squassabia, F. Peptides and Peptidomimetics in Medicine, Surgery and Biotechnology. Curr. Med. Chem. 2006, 13, 2449–2466. [Google Scholar] [CrossRef]
- Rivière, P. J-M. Peripheral kappa-opioid agonists for visceral pain. Br. J. Pharmacol. 2004, 141, 1331–1334. [Google Scholar] [CrossRef] [Green Version]
- Zaidi, S.A.; Katritch, V. Structural Characterization of KOR Inactive and Active States for 3D Pharmacology and Drug Discovery. In The Kappa Opioid Receptor. Handbook of Experimental Pharmacology; Liu-Chen, L.Y., Inan, S., Eds.; Springer: Cham, Switzerland, 2021; Volume 271, pp. 41–64. [Google Scholar]
- Ferré, G.; Czaplicki, G.; Demange, P.; Milon, A. Structure and dynamics of dynorphin peptide and its receptor. Vitam Horm. 2019, 111, 17–47. [Google Scholar]
- Wu, H.; Wacker, D.; Mileni, M.; Katritch, V.; Han, G.W.; Vardy, E.; Liu, W.; Thompson, A.A.; Huang, X.-P.; Carroll, F.I.; et al. Structure of the human κ-opioid receptor in complex with JDTic. Nature 2012, 485, 327–332. [Google Scholar] [CrossRef]
- Carbone, J.; Ghidini, A.; Romano, A.; Gentilucci, L.; Musiani, F. PacDOCK: A Web Server for Positional Distance-Based and Interaction-Based Analysis of Docking Results. Molecules 2022, 27, 6884. [Google Scholar] [CrossRef]
- Che, T.; English, J.; Krumm, B.E.; Kim, K.; Pardon, E.; Olsen, R.H.J.; Wang, S.; Zhang, S.; Diberto, J.F.; Sciaky, N.; et al. Nanobody-enabled monitoring of kappa opioid receptor states. Nat. Commun. 2020, 11, 1145. [Google Scholar] [CrossRef] [Green Version]
- Che, T.; Majumdar, S.; Zaidi, S.A.; Ondachi, P.; McCorvy, J.D.; Wang, S.; Mosier, P.D.; Uprety, R.; Vardy, E.; Krumm, B.E.; et al. Structure of the nanobody-stabilized active state of the kappa opioid receptor. Cell 2018, 172, 55–67. [Google Scholar] [CrossRef] [Green Version]
- Puls, K.; Schmidhammer, H.; Wolber, G.; Spetea, M. Mechanistic Characterization of the Pharmacological Profile of HS-731, a Peripherally Acting Opioid Analgesic, at the µ-, δ-, κ-Opioid and Nociceptin Receptors. Molecules 2022, 27, 919. [Google Scholar] [CrossRef]
- Mehr-Un-Nisa; Munawar, M.A.; Rankin, D.; Hruby, V.J.; Porreca, F.; Lee, Y.S. C-terminal modified Enkephalin-like tetrapeptides with enhanced affinities at the kappa opioid receptor and monoamine transporters. Bioorg. Med. Chem. 2021, 51, 116509. [Google Scholar] [CrossRef]
- He, Q.; Wei, Y.; Liu, X.; Ye, R.; Kong, L.; Li, Z.; Jiang, S.; Yu, L.; Chai, J.; Xie, Q.; et al. Discovery of an M-Substituted N-Cyclopropylmethyl-7α-phenyl-6,14-endoethano tetrahydronorthebaine as a Selective, Potent, and Orally Active κ-Opioid Receptor Agonist with an Improved Central Nervous System Safety Profile. J. Med. Chem. 2021, 64, 12414–12433. [Google Scholar] [CrossRef]
- Yadav, V.D.; Kumar, L.; Kumari, P.; Kumar, S.; Singh, M.; Siddiqi, M.I.; Yadav, P.N.; Batra, S. Synthesis and Assessment of Fused β-Carboline Derivatives as Kappa Opioid Receptor Agonists. ChemMedChem 2021, 16, 1917–1926. [Google Scholar] [CrossRef]
- Stefanucci, A.; Iobbi, V.; Della Valle, A.; Scioli, G.; Pieretti, S.; Minosi, P.; Mirzaie, S.; Novellino, E.; Mollica, A. In Silico Identification of Tripeptides as Lead Compounds for the Design of KOR Ligands. Molecules 2021, 26, 4767. [Google Scholar] [CrossRef]
- Guerrieri, E.; Bermudez, M.; Wolber, G.; Berzetei-Gurske, I.P.; Schmidhammer, H.; Spetea, M. Structural determinants of diphenethylamines for interaction with the κ opioid receptor: Synthesis, pharmacology and molecular modeling studies. Bioorg. Med. Chem. Lett. 2016, 26, 4769–4774. [Google Scholar] [CrossRef]
- Wan, X.-H.; Huang, X.Q.; Zhou, D.H.; Jiang, H.L.; Chen, K.X.; Chi, Z.Q. Building 3D-structural model of kappa opioid receptor and studying its interaction mechanism with dynorphin A (1-8). Acta Pharmacol. Sin. 2000, 21, 701–708. [Google Scholar]
- Vardy, E.; Mosier, P.D.; Frankowski, K.J.; Wu, H.; Katritch, V.; Westkaemper, R.B.; Aube, J.; Stevens, R.C.; Roth, B.L. Chemotype-selective modes of action of kappa-opioid receptor agonists. J. Biol. Chem. 2013, 288, 34470–34483. [Google Scholar] [CrossRef]
- O’Connor, C.; White, K.L.; Doncescu, N.; Didenko, T.; Roth, B.L.; Czaplicki, G.; Stevens, R.C.; Wüthrich, K.; Milon, A. NMR structure and dynamics of the agonist dynorphin peptide bound to the human kappa opioid receptor. Proc. Natl. Acad. Sci. USA 2015, 112, 11852–11857. [Google Scholar] [CrossRef] [Green Version]
- Wtorek, K.; Ghidini, A.; Gentilucci, L.; Adamska-Bartłomiejczyk, A.; Piekielna-Ciesielska, J.; Ruzza, C.; Sturaro, C.; Calò, G.; Pieretti, S.; Kluczyk, A.; et al. Synthesis, Biological Activity and Molecular Docking of Chimeric Peptides Targeting Opioid and NOP Receptors. Int. J. Mol. Sci. 2022, 23, 12700. [Google Scholar] [CrossRef]
- Mafi, M.; Kim, S.-K.; Goddard, W.A., III. Mechanism of β-arrestin recruitment by the κ-opioid G protein-coupled receptor. Proc. Natl. Acad. Sci. USA 2020, 117, 16346–16355. [Google Scholar] [CrossRef] [PubMed]
- Piekielna-Ciesielska, J.; Artali, R.; Azzam, A.A.H.; Lambert, D.G.; Kluczyk, A.; Gentilucci, L.; Janecka, A. PharmacologicalCharacterization of μ-Opioid Receptor Agonists with Biased G Protein or β-Arrestin Signaling, and Computational Study of Conformational Changes during Receptor Activation. Molecules 2021, 26, 13. [Google Scholar] [CrossRef] [PubMed]
- Schneider, S.; Provasi, D.; Filizola, M. How oliceridine (TRV-130) binds and stabilizes a κ-opioid receptor. conformational state that selectively triggers G protein signaling pathways. Biochemistry 2016, 55, 6456–6466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uprety, R.; Che, T.; Zaidi, S.A.; Grinnell, S.G.; Varga, B.R.; Faouzi, A.; Slocum, S.T.; Allaoa, A.; Varadi, A.; Nelson, M.; et al. Controlling opioid receptor functional selectivity by targeting distinct subpockets of the orthosteric site. eLife 2021, 10, e56519. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Santino, F.; Gentilucci, L. Design of κ-Opioid Receptor Agonists for the Development of Potential Treatments of Pain with Reduced Side Effects. Molecules 2023, 28, 346. https://doi.org/10.3390/molecules28010346
Santino F, Gentilucci L. Design of κ-Opioid Receptor Agonists for the Development of Potential Treatments of Pain with Reduced Side Effects. Molecules. 2023; 28(1):346. https://doi.org/10.3390/molecules28010346
Chicago/Turabian StyleSantino, Federica, and Luca Gentilucci. 2023. "Design of κ-Opioid Receptor Agonists for the Development of Potential Treatments of Pain with Reduced Side Effects" Molecules 28, no. 1: 346. https://doi.org/10.3390/molecules28010346