2.1. Redox Potentials of the P-Cluster
We started the investigation with testing our methodology [
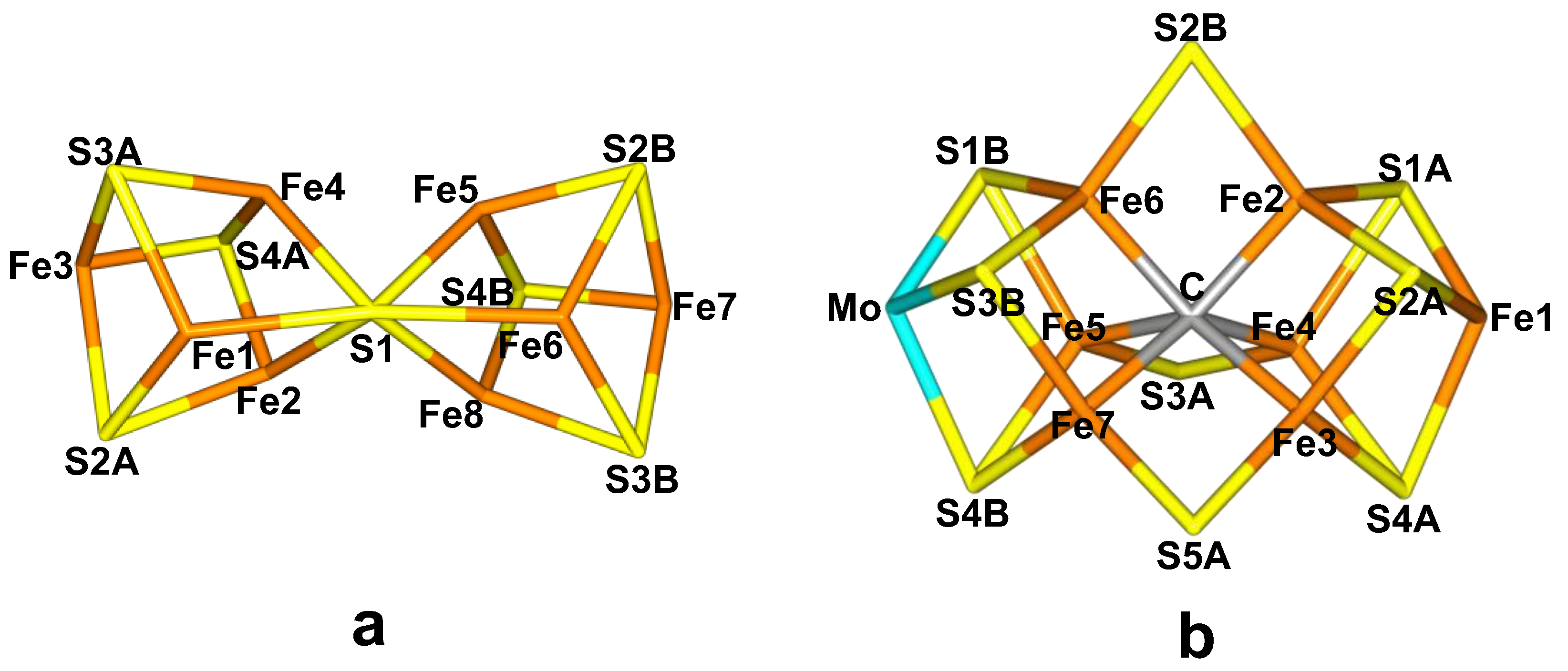
41] on the P-cluster to see if the results are reliable also for the large iron–sulfur clusters in nitrogenase and for redox reactions involving protonation of the clusters. The P-cluster (
Figure 1a) contains eight Fe ions and the resting P
N state has been shown to be the fully reduced
state [
44,
45,
46]. Three additional states have been experimentally observed, oxidised by 1–3 electrons [
44,
45,
46]. They are denoted P
1+–P
3+. Only the first two states are believed to be involved in the catalytic mechanism, although there are some evidence that also the P
2+ state may be used [
46,
47,
48,
49]. Crystallographic studies have shown that in the P
N state, the cluster is essentially two [4Fe–4S] clusters merged by the S1 sulfide ion that coordinates to six Fe ions (cf.
Figure 1a). In the P
2+ state, Ser-188D becomes deprotonated and coordinates to Fe6 [
48,
50,
51]. Likewise, the backbone N atom of Cys-88 (the sidechain of which is one of the ligands to the P-cluster) also becomes deprotonated and coordinates to Fe5 [
48,
50,
51]. This leads to cleavage of the Fe5–S1 and Fe6–S1 bonds. The same structure is expected for the P
3+ state. The structure of the P
1+ state is more uncertain, because redox titrations indicated that only the P
2+/P
1+ redox-couple is pH-dependent, whereas no evidence was found for a coupled electron- and proton-transfer for the P
1+/P
N couple [
52,
53]. However, a recent crystal structure was interpreted to contain deprotonated Ser-188D and protonated Cys-88, although it is probably a mixture of the P
1+ and the P
2+ states [
54,
55,
56].
Six states of the P-cluster were considered in this study as are described in
Table 1. Each state was first QM/MM optimised with the small QM system and then subjected to a single-point energy calculation with the large QM system in a COSMO continuum solvent with a dielectric constant of 80. The QM/MM structures and the best BS states were taken from our previous publication [
55]. The calculated redox potentials are listed in
Table 2 and they are compared to experimentally measured redox potentials [
5] in order to gauge the accuracy of the method when applied to the P-clusters of the nitrogenase. For the P
1+ state, we tested three different protonation states.
It can be seen that the redox couples P
2+ → P
3+ and P
NH
2 → P
1+H give errors of 0.05 and 0.33 V, respectively, compared to experimental potentials. These are within the range of errors observed in our previous study (maximum error 0.44 V) [
41]. This is quite satisfying, especially considering that one of the considered redox potentials for the P-cluster involves coupled redox and protonation reaction, whereas the calibration study involved only pure redox reactions. This gives us confidence to apply the method also to the FeMo cluster.
For the P
1+/P
N redox potential, the calculated result is essentially independent on whether we include the proton transfer or not in the calculations (the P
NH
2 → P
1+H
2 and P
NH
2 → P
1+H give calculated redox potentials that differ by only 0.02 V). This is in agreement with the experimental observation that the P
1+/P
N redox potential is pH-independent [
52,
53] and may solve the enigma why redox titrations did not observe any pH-dependence although crystal structures indicate that a deprotonation should be involved in this redox reaction [
54,
55,
56].
For the P
1+H → P
2+ transition, the comparison with experimental results is somewhat harder, because the measured redox potential changes with pH, from −0.224 V at pH 6.0 to −0.348 V at pH 8.5 [
52]. This has been interpreted to reflect the deprotonation of the backbone amide group of Cys-88: At very low pH, it is protonated in both the P
1+ and P
2+ states, whereas at high pH, it is deprotonated in both redox states. At intermediate pH (i.e., in the measured range), it is protonated in the P
1+ state and deprotonated in the P
2+ state [
52]. Our calculations confirm these suggestions: We obtain a more positive potential for the P
2+/P
1+H couple (
= 0.1 V) than for the P
2+/P
1+ couple (
= −1.1 V). However, the restricted pH range of the measured potentials makes it hard to make a more detailed judgement of the calculated potentials. The experiments indicate that the redox potential of the pure P
1+H → P
2+ transition is larger than −0.224 V, indicating an error of less than 0.36 V for our calculated potential. Likewise, the redox potential of the pure P
1+ → P
2+ transition is more negative than −0.348 V, indicating an error of less than 0.72 V. Thus, our calculations confirm the experimental observation that the P
1+ → P
2+ transition involves a proton transfer [
52,
53], but we cannot obtain any detailed estimate of the error for this redox couple.
2.2. Accuracy of the Redox Potential of the FeMo Cluster and Oxidation Level of the Cluster
As mentioned above, only two experimental redox
potentials have been reported for the FeMo cluster [
24,
25].
A potential of −0.042 V has been measured between the resting state and a
one-electron oxidised state [
26,
45] (not
involved in the normal reaction mechanism). Redox potentials for more reduced
states are harder to measure, because reduction starts substrate or proton
reduction. No firm measurement is available, but the potential between the
resting state and a steady-state reduced state (which my represent more than
one reduced state) has been estimated in four studies between −0.45 and −0.49 V
[
24,
25,
27,
28,
29]. In two cases, another potential
of −0.30 to −0.32 V was also reported, but it may come from the P-cluster [
24].
For the FeMo cluster, we wanted to study two related questions, viz. whether we can reproduce these potentials with our QM calculations, within the accuracy of the method, and whether we can identify the correct redox couple of the FeMo cluster. Recent Mössbauer, anomalous dispersion and QM investigations have suggested that the resting E
0 state of the FeMo cluster is in the
oxidation state [
13,
42,
57,
58]. However, this gives a large net negative charge of the cluster and its direct ligands (−5
e), which is only partly neutralised by two nearby arginine residues. Moreover, protonation energies of various groups of the FeMo cluster are unexpectedly large [
59,
60]. Therefore, an independent confirmation of the oxidation-state assignment is desirable.
To this end, twelve states of the FeMo cluster were considered, described in
Table 3. We studied the resting E
0 state, together with the one-electron oxidised and one-electron reduced states. For the latter, we considered both a structure with no protons added and a state with a proton added on the S2B µ
2 bridging sulfide ion, as has been suggested by several QM investigations [
18,
59] and is also supported by experimental studies [
61,
62]. For the standard oxidation-state assignment (
for E
0), these states are denoted E
0, E
−1, E
1 and E
1H. In addition, we considered two alternative charge states, either with two more or two electrons less (experimentally the resting state is a quartet [
2,
3], i.e., with an odd number of electrons, so electrons need to be added or removed in pairs). These states are called A
0, A
−1, A
1 and A
1H when two electrons were added (i.e., giving
for the A
0 state) and R
0, R
−1, R
1 and R
1H when two electrons were removed (i.e., giving
for the R
0 state; note that R
1 = E
−1 and A
−1 = E
1). The calculated redox potentials are listed in
Table 4.
It can be seen that with the standard charge state (
for E
0), our calculations reproduce the two experimental redox potentials with errors of 0.17 and 0.19 V, i.e., well within the error range observed in our previous study (maximum error 0.44 V) [
41] and also for the P-cluster. However, the good results are observed only if it is assumed that the reduction of E
0 is accompanied by the uptake of by a proton (i.e., E
0 → E
1H; for E
0 → E
1, the error is 0.6 V), showing that the calculations confirm that a proton transfer is involved in the redox reaction.
If we instead consider a FeMo cluster with two electrons less (i.e., with a
assignment for the resting state, here called R
0), the calculated potentials reproduce the experimental redox potentials worse: The R
−1 → R
0 transition gives and error of 2.0 V, much larger than the maximum error in our previous study [
41], whereas R
0 → R
1H gives an error of 0.43 V.
Likewise, if we instead add two extra electrons ( for the resting state), we get a very large error (over 6 V) for the A−1 → A0 potential. For the A0 → A1H transition, the error is smaller, −0.48 V, but it is still somewhat larger than the maximum error observed in our calibration study. Thus, our calculations confirm that is the proper redox assignment for E0. Apparently, the E−1/E0 redox potential is more sensitive to the involved redox couple than the E0/E1H potential. The calculations also confirm that the E1 state is protonated.
2.3. Redox Potentials of the E0–E4 States of the FeMo Cluster
Next, we studied also the E
0–E
4 states of the FeMo cluster from Lowe–Thorneley reaction cycle. The aim was to examine the suggestion that all the E
0–E
4 states should have similar redox potentials, because they use only two formal redox states,
and
[
42,
43]. This is suggested to be accomplished by the added protons, which bind to Fe ions in the E
2 and E
4 states, thereby formally becoming hydride ions and changing the oxidation state of the Fe ions by two. Thus, the five states E
0–E
4 would formally be
,
,
,
and
(leaving out Mo, which always is in the +III state).
We used mainly structures from previous studies [
59,
63,
64,
65,
66,
67] and included a few alternative structures for each state (except for E
0 and E
1H, for which there is a reasonable consensus), to see if we can discriminate between different possibilities using the redox potentials. The various structures are described in
Table 5 (they are also shown in
Figure S3) and the calculated redox potentials are listed in
Table 6. We use the redox potential of the E
0/E
1H couple as a reference (
to judge if all transitions have similar redox potentials.
In our previous studies, we found that with the TPSS functional, the best E
2H
2 structure has a proton on S2B and a hydride ion bridging Fe2 and Fe6 [
59,
64]. It can be seen that the estimated redox potential for the E
1H → E
2H
2 transition is similar to that of the E
0 → E
1H transition, only 0.08 V less negative. This confirms the conjecture that the FeMo cluster in the early E
n states should have a constant redox potential.
If we instead use another structure for the E
2H
2 state with the extra two protons bound to S2B and as a hydride ion bridging Fe2 and Fe6, but with S2B dissociated from Fe2 (but not from Fe6; called E
2H
2′), the redox potential for the E
1H → E
2H
2′ transition decreases by 0.25 V. This simply reflects that with TPSS-D4/def2-SV(P) and the 399-atom redox model, the E
2H
2′ structure is 0.25 eV (24 kJ/mol) less stable than the E
2H
2 structure. For the 184-atom QM/MM model, the difference is 15 kJ/mol. In our previous study, we showed that the relative stability of the structures with the protonated S2B group bridging Fe2 and Fe6 or dissociated from one of the two iron ions depends on what DFT functional is used [
64]. For example, with the TPSSh functional, the E
2H
2′ structure is instead 11 kJ/mol more stable. Consequently, the redox potentials will also depend on the QM method used, with differences of ~0.3 V.
If we instead use a structure for the E
2H
2 state with the two protons on S2B and S5A (E
2H
2″), the calculated redox potential is 0.08 V more negative than with the most stable E
2H
2 state. Again, this reflects that this protonation state is 9 kJ/mol less stable. However, it also shows that the formal oxidation states in the FeMo cluster have only a minor influence on the redox potential. The E
2H
2″ state involves two protons on sulfide groups and therefore represents a doubly reduced formal
state in contrast to the
state for E
2H
2 and E
2H
2′. This shows that as long as the various protonation states have similar relative energies, they will also have similar redox potentials, showing that there is no major difference between different formal oxidation states of the Fe ions. This is in line with suggestions by Dance, who has pointed out that it is misleading to make a sharp contrast between protons on sulfides and hydride ions on Fe ions, because there is only a small difference in the charge populations on the H atom [
18].
For the E
3H
3 state, we used a structure with the third proton bridging Fe3 and Fe7 (in addition a proton on S2B and a hydride ion bridging Fe2 and Fe6). The estimated redox potential for the E
2H
2 → E
3H
3 transition is 0.19 V more negative than that of the E
0 → E
1H transition. This is well within the maximum error in our calibration study 0.44 V [
41], and therefore still in agreement with the expectation that the electrons can be donated to the FeMo cluster at a constant redox potential. We tested also another structure, taken from our previous systematic study [
59], in which the third proton bound to Fe5 instead (E
3H
3′). It gave a 0.35 V more negative redox potential (i.e., further away from that of the E
0 → E
1H transition), reflecting that this structure is less stable. Interestingly, we found in contrast to our previous study that the broken-symmetry BS-14 state was more favourable for both these structures with the large QM model used in the redox calculations (but only for the best structure with the smaller QM/MM-optimised model).
For the E
3H
3 → E
4H
4 transition, our estimated redox potential is 0.41 V more positive than that of the E
0 → E
1H transition. This is within the maximum error in our calibration study [
41], but considering that the redox potential of the E
2H
2/E
3H
3 couple was a bit to negative and this redox potential is a bit too positive, it might indicate that we have not yet found the best structure for the E
3 state. For E
4H
4, we employed the best structure in our previous investigation of this state [
63], viz. a structure with two protons on S2B and S5A and two hydride ions bridging Fe2/6 and Fe3/7. There are several possible conformations of such a structure. The best one has all H atoms pointing towards S3A, except the Fe3/7 hydride, which points towards S2B (all structures are shown in
Figure S3). If we instead use a structure with the Fe2/6 hydride on the other side of S2B, i.e., the same face as the Fe2/6 hydride, the redox potential becomes 0.23 V more negative, reflecting that such a structure is 22 kJ/mol less stable. If we instead use the structure suggested by Hoffman and coworkers [
2,
21], i.e., with all four H atoms on the same face of the cluster (i.e., the two protons on S2B and S5A pointing in the opposite direction compared to E
4H
4 and E
4H
4′ structures), the redox potential becomes even more negative by 0.24 V. Likewise, if we use the best structure in our first investigation of the E
4H
4 state [
59] (with H atoms on S2B, Fe2/6, Fe5 and Fe6), the redox potential becomes 0.15 V even more negative, reflecting that this structure is 59 kJ/mol less stable than the best state.
In conclusion, we find that for all four calculated redox potentials for the E
0–E
4 states are similar, within 0.41 V, i.e., within the accuracy of our method. This confirm the expectation that the redox potentials should be similar [
42,
43] so that they can accept electrons from the same source. However, the results are very sensitive to which structures are employed and which QM method and broken-symmetry state is used. On the other hand, we show that the formal oxidation states of the Fe ions (the number of protons on sulfide ions or hydride ions on Fe) is less important for the redox potentials, contrary to the suggestion that the redox potential depends on the formal oxidation state of the Fe ions [
43,
68].
2.4. Redox Potentials of the E4–E8 States of the FeMo Cluster
Finally, we studied also redox potentials of the FeMo cluster in the later part of the reaction, after binding of N
2 and its protonation to N
2H
2. In previous studies, we have suggested thermodynamically stable structures for the E
4N
2H
2 to E
8NH
3 states, either with S2B bound or dissociated from the cluster [
65,
66,
67]. We use these structures also in this study. They are described in
Table 5 and are shown in
Figure S3. In
Table 6, the calculated redox potentials for both scenarios are presented.
It can be seen that both with and without S2B, the calculated redox potentials are all less negative than that for the E
0/E
1H couple, by 0.2–2.65 V. This reflects that once N
2H
2 has been formed, the following reactions are quite facile. Electrons tend to move towards sites with a more positive redox potential. Therefore, redox potentials that are more positive than those of the E
0/E
1H couple indicate that the electron transfer is more exothermic than in the E
0 → E
1 step. Thus, the electron transfers of the E
4–E
8 steps of the nitrogenase reaction are more downhill than those of the E
0–E
4 reactions. However, it might also indicate that the assumption that the bound N
2 directly is protonated may be incorrect. In fact, the first protonation of N
2 is the hardest step in the reduction of N
2 to ammonia [
69], and it is possible that it requires further reduction of the FeMo cluster before it is feasible (this part of the reaction was not studied in our previous studies). Our results indicate that this should be further studied.
The relative sizes of the four redox potentials for the E4–E8 states are also rather independent on whether S2B remains bound or is dissociated: The potentials of the first and third steps E4 → E5 and E6 → E7 are most positive, especially the latter, whereas the redox potentials of the other two steps are closer to that of the E0/E1H couple. For the four reductions with S2B still bound, our calculated redox potentials are 1.13, 0.41, 2.02 and 0.57 V, whereas with S2B dissociated, the four calculated redox potentials are 0.97, 0.20, 2.65 and 0.19 V (i.e., with a somewhat larger variation). The similarity of the trends for the two sets of potentials is conspicuous considering that the N–N bond is cleaved in the E5N2H3 → E6N2H4 transition with S2B bound, but in the E6N2H4 → E7N2H5 transition when S2B has dissociated.
In conclusion, our results show that later part of the reaction mechanism of Mo-nitrogenase give redox reactions that are more exothermic than that of the E
0/E
1H redox couple. Thus, we see no evidence that a stronger driving force is needed for the reaction, as has been suggested by Siegbahn [
22]. Moreover, there is no large difference between the mechanisms with S2B bound or dissociated.




{kind=link}