
Lewis Acid-Catalyzed Formal (4+2)-Cycloaddition between Cross-Conjugated Azatrienes and Styrylmalonates: The Way to Functionalized Quinolizidine Precursors

Abstract
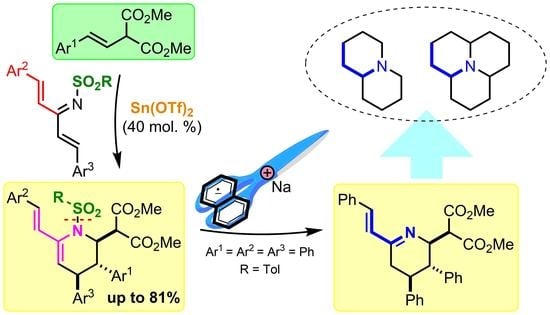
:
1. Introduction
2. Results and Discussion
3. Materials and Methods
3.1. General Methods
3.2. Synthetic Procedures
3.2.1. General Synthetic Procedure and Spectroscopic Data for Azatrienes 2
3.2.2. General Synthetic Procedure and Spectroscopic Data for Vinyltetrahydropyridines 3
3.2.3. General Synthetic Procedure and Spectroscopic Data for Heterocycles 5
3.2.4. General Synthetic Procedure and Spectroscopic Data for Heterocycle 6
3.2.5. General Synthetic Procedure and Spectroscopic Data for Azadiene 4
3.2.6. Gram-Scale Synthetic Procedure for Vinyltetrahydropyridine 3b
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Karunakaran, T.; Ngew, K.Z.; Zailan, A.A.D.; Mian, J.V.Y.; Abu Bakar, M.H. The chemical and pharmacological properties of mitragynine and its diastereomers: An insight review. Front. Pharmacol. 2022, 13, 805986–805997. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.-H.; Yu, Z.-P.; Capon, R.J.; Zhang, H. Natural enantiomers: Occurrence, biogenesis and biological properties. Molecules 2022, 27, 1279–1352. [Google Scholar] [CrossRef] [PubMed]
- Althagbi, H.I.; Alarif, W.M.; Al-Footy, K.O.; Abdel-Lateff, A. Marine-derived macrocyclic alkaloids (MDMAs): Chemical and biological diversity. Mar. Drugs 2020, 18, 368–402. [Google Scholar] [CrossRef] [PubMed]
- Michael, J.P. The Alkaloids: Chemistry and Biology, 1st ed.; Knölker, H.-J., Ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2016; Volume 75, pp. 1–498. [Google Scholar]
- Li, Y.; Wang, G.; Liu, J.; Ouyang, L. Quinolizidine alkaloids derivatives from Sophora alopecuroides Linn: Bioactivities, structure-activity relationships and preliminary molecular mechanisms. Eur. J. Med. Chem. 2020, 188, 111972–111997. [Google Scholar] [CrossRef] [PubMed]
- Toya, H.; Satoh, T.; Okano, K.; Takasu, K.; Ihara, M.; Takahashi, A.; Tanaka, H.; Tokuyama, H. Stereocontrolled total synthesis and biological evaluation of (−)- and (+)-petrosin and its derivatives. Tetrahedron 2014, 70, 8129–8141. [Google Scholar] [CrossRef]
- Guerola, M.; Sánchez-Roselló, M.; Mulet, C.; del Pozo, C.; Fustero, S. Asymmetric intramolecular aza-Michael reaction in desymmetrization processes. Total synthesis of hippodamine and epi-hippodamine. Org. Lett. 2015, 17, 960–963. [Google Scholar] [CrossRef] [PubMed]
- Alujas-Burgos, S.; Oliveras-González, C.; Álvarez-Larena, Á.; Bayón, P.; Figueredo, M. Iterative synthetic strategy for azaphenalene alkaloids. Total synthesis of (−)-9a epi-hippocasine. J. Org. Chem. 2018, 83, 5052–5057. [Google Scholar] [CrossRef] [Green Version]
- Alujas-Burgos, S.; Bayón, P.; Figueredo, M. Recent advances in the synthesis of azaphenalene alkaloids: First enantioselective approaches. Org. Biomol. Chem. 2018, 16, 8218–8229. [Google Scholar] [CrossRef] [Green Version]
- Borisov, D.D.; Novikov, R.A.; Tomilov, Y.V. GaCl3-Mediated reactions of donor–acceptor cyclopropanes with aromatic aldehydes. Angew. Chem. Int. Ed. 2016, 55, 12233–12237. [Google Scholar] [CrossRef]
- Borisov, D.D.; Novikov, R.A.; Eltysheva, A.S.; Tkachev, Y.V.; Tomilov, Y.V. Styrylmalonates as an alternative to donor–acceptor cyclopropanes in the reactions with aldehydes: A route to 5,6-dihydropyran-2-ones. Org. Lett. 2017, 19, 3731–3734. [Google Scholar] [CrossRef]
- Novikov, R.A.; Levina, A.A.; Borisov, D.D.; Volodin, A.D.; Korlyukov, A.A.; Tkachev, Y.V.; Platonova, Y.B.; Tomilova, L.G.; Tomilov, Y.V. Synthesis of the cationic gallium phthalocyanines and their catalytic application in gallium (III)-activated processes for donor–acceptor substrates. Organometallics 2020, 39, 2580–2593. [Google Scholar] [CrossRef]
- Borisov, D.D.; Novikov, R.A.; Tomilov, Y.V. Reactions of styrylmalonates with aromatic aldehydes: Detailed synthetic and mechanistic studies. J. Org. Chem. 2021, 86, 4457–4471. [Google Scholar] [CrossRef] [PubMed]
- Borisov, D.D.; Chermashentsev, G.R.; Novikov, R.A.; Tomilov, Y.V. Coupling of styrylmalonates with furan and benzofuran carbaldehydes: Synthesis and chemistry of substituted (4-oxocyclopent-2-enyl) malonates. J. Org. Chem. 2021, 86, 8489–8499. [Google Scholar] [CrossRef] [PubMed]
- Sergeev, P.G.; Novikov, R.A.; Tomilov, Y.V. Lewis acid-catalyzed formal (4 + 2)- and (2 + 2 + 2)-cycloaddition between 1-azadienes and styrylmalonates as analogues of donor-acceptor cyclopropanes. Adv. Synth. Catal. 2021, 363, 5292–5299. [Google Scholar] [CrossRef]
- Snyder, H.R.; Heckert, R.E. A method for the rapid cleavage of sulfonamides. J. Am. Chem. Soc. 1952, 74, 2006–2009. [Google Scholar] [CrossRef]
- Javorskis, T.; Orentas, E. Chemoselective deprotection of sulfonamides under acidic conditions: Scope, sulfonyl group migration, and synthetic applications. J. Org. Chem. 2017, 82, 13423–13439. [Google Scholar] [CrossRef]
- Birkinshaw, T.N.; Holmes, A.B. Synthesis of (±)-isoprosopinines A and B. Tetrahedron Lett. 1987, 28, 813–816. [Google Scholar] [CrossRef]
- Tanner, D.; Ming, H.H.; Bergdahl, M. Stereocontrolled synthesis of the spirocyclic alkaloid (±)-nitramine. Tetrahedron Lett. 1988, 29, 6493–6495. [Google Scholar] [CrossRef]
- Roemmele, R.C.; Rapoport, H. Removal of N-arylsulfonyl groups from hydroxy α-amino acids. J. Org. Chem. 1988, 53, 2367–2371. [Google Scholar] [CrossRef]
- Ji, S.; Gortler, L.B.; Waring, A.; Battisti, A.J.; Bank, S.; Closson, W.D.; Wriede, P.A. Cleavage of sulfonamides with sodium naphthalene. J. Am. Chem. Soc. 1967, 89, 5311–5312. [Google Scholar] [CrossRef]
- Alonso, E.; Ramon, D.J.; Yus, M. Reductive deprotection of allyl, benzyl and sulfonyl substituted alcohols, amines and amides using a naphthalene-catalysed lithiation. Tetrahedron 1997, 53, 14355–14368. [Google Scholar] [CrossRef]
- Teng, M.; Zi, W.; Ma, D. Total synthesis of the monoterpenoid indole alkaloid (±)-aspidophylline A. Angew. Chem. Int. Ed. 2014, 53, 1814–1817. [Google Scholar] [CrossRef] [PubMed]
- Blair, L.M.; Sperry, J. Total syntheses of (±)-spiroindimicins B and C enabled by a late-stage Schöllkopf–Magnus–Barton–Zard (SMBZ) reaction. Chem. Commun. 2016, 52, 800–802. [Google Scholar] [CrossRef] [PubMed]
- Wua, P.; Zhoub, Q.; Liua, X.-Y.; Xuea, F.; Qin, Y. Synthetic studies towards (–)-deserpidine: Total synthesis of the stereoisomer and derivative of (–)-deserpidine. Chin. Chem. Lett. 2021, 32, 401–404. [Google Scholar] [CrossRef]
- Vedejs, E.; Lin, S. Deprotection of arenesulfonamides with samarium iodide. J. Org. Chem. 1994, 59, 1602–1603. [Google Scholar] [CrossRef]
- Alonso, D.A.; Andersson, P.G. Deprotection of sulfonyl aziridines. J. Org. Chem. 1998, 63, 9455–9461. [Google Scholar] [CrossRef]
- Hayashi, T.; Kawai, M.; Tokunaga, N. Asymmetric synthesis of diarylmethyl amines by rhodium-catalyzed asymmetric addition of aryl titanium reagents to imines. Angew. Chem. Int. Ed. 2004, 43, 6125–6128. [Google Scholar] [CrossRef]
- Kuriyama, M.; Soeta, T.; Hao, X.; Chen, Q.; Tomioka, K. N-Boc-L-valine-connected amidomonophosphane rhodium (I) catalyst for asymmetric arylation of N-tosylarylimines with arylboroxines. J. Am. Chem. Soc. 2004, 126, 8128–8129. [Google Scholar] [CrossRef]
- Grach, G.; Santos, J.S.-d.O.; Lohier, J.; Mojovic, L.; Plé, N.; Turck, A.; Reboul, V.; Metzner, P. Diastereoselective addition of enantiopure lithium tert-butylsulfinylferrocene to imines. J. Org. Chem. 2006, 71, 9572–9579. [Google Scholar] [CrossRef]
- Duan, H.; Jia, Y.; Wang, L.; Zhou, Q. Enantioselective Rh-catalyzed arylation of N-tosylarylimines with arylboronic acids. Org. Lett. 2006, 8, 2567–2569. [Google Scholar] [CrossRef]
- Ankner, T.; Hilmersson, G. Instantaneous deprotection of tosylamides and esters with SmI2/amine/water. Org. Lett. 2009, 11, 503–506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berhal, F.; Wu, Z.; Zhang, Z.; Ayad, T.; Ratovelomanana-Vidal, V. Enantioselective synthesis of 1-aryl-tetrahydroisoquinolines through iridium catalyzed asymmetric hydrogenation. Org. Lett. 2012, 14, 3308–3311. [Google Scholar] [CrossRef] [PubMed]
- Ritzen, B.; Hoekman, S.; Verdasco, E.D.; van Delft, F.L.; Rutjes, F.P.J.T. Enantioselective chemoenzymatic synthesis of cis- and trans-2,5-disubstituted morpholines. J. Org. Chem. 2010, 75, 3461–3464. [Google Scholar] [CrossRef] [PubMed]
- Goulaouic-Dubois, C.; Guggisberg, A.; Hesse, M. Protection of amines by the pyridine-2-sulfonyl group and its cleavage under mild conditions (SmI2 or electrolysis). J. Org. Chem. 1995, 60, 5969–5972. [Google Scholar] [CrossRef]
- Aaseng, J.E.; Gautun, O.R. Synthesis of substituted (S)-2-aminotetralins via ring-opening of aziridines prepared from l-aspartic acid β-tert-butyl ester. Tetrahedron 2010, 66, 8982–8991. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, Y.; Tanaka, S.; Serizawa, R.; Morohashi, N.; Hattori, T. Synthesis of mono-and 1,3-diaminocalix[4]arenes via Ullmann-type amination and amidation of 1,3-bistriflate esters of calix [4] arenes J. Org. Chem. 2011, 76, 2168–2179. [Google Scholar] [CrossRef]
- Kumar, V.; Ramesh, N.G. Iodine catalyzed one-pot diamination of glycals with chloramine-T: A new approach to 2-amino-β-glycosylamines for applications in N-glycopeptide synthesis. Chem. Commun. 2006, 4952–4954. [Google Scholar] [CrossRef]
- Kumar, V.; Ramesh, N.G. A versatile strategy for the synthesis of N-linked glycoamino acids from glycals. Org. Biomol. Chem. 2007, 5, 3847–3858. [Google Scholar] [CrossRef]
- Jensen, K.L.; Franke, P.T.; Nielsen, L.T.; Daasbjerg, K.; Jørgensen, K.A. Anodic oxidation and organocatalysis: Direct regio- and stereoselective access to meta-substituted anilines by α-arylation of aldehydes. Angew. Chem. Int. Ed. 2010, 49, 129–133. [Google Scholar] [CrossRef]
- Yamagishi, T.; Ichikawa, H.; Haruki, T.; Yokomatsu, T. Diastereoselective synthesis of α,β’-disubstituted aminomethyl (2-carboxyethyl) phosphinates as phosphinyl dipeptide isosteres. Org. Lett. 2008, 10, 4347–4350. [Google Scholar] [CrossRef]
- Blay, G.; Cardona, L.; Climent, E.; Pedro, J.R. Highly enantioselective zinc/binol-catalyzed alkynylation of N-sulfonyl aldimines. Angew. Chem. Int. Ed. 2008, 47, 5593–5596. [Google Scholar] [CrossRef] [PubMed]
- Hill, D.C.; Flugge, L.A.; Petillo, P.A. SmI2-promoted deprotection of N-(arylsulfonyl) glucosamines. J. Org. Chem. 1997, 62, 4864–4866. [Google Scholar] [CrossRef]
- Gaston, J.J.; Tague, A.J.; Smyth, J.E.; Butler, N.M.; Willis, A.C.; van Eikema Hommes, N.; Yu, H.; Clark, T.; Keller, P.A. The detosylation of chiral 1,2-bis (tosylamides). J. Org. Chem. 2021, 86, 9163–9180. [Google Scholar] [CrossRef]
- Okabe, K.; Natsume, M. The second generation synthesis of a tumor promoter pendolmycin. Tetrahedron 1991, 47, 7615–7624. [Google Scholar] [CrossRef]
- Pak, C.S.; Lim, D.S. Deprotection of 2-pyridyl sulfonyl group from pyridine-2-sulfonamides by magnesium in methanol. Synth. Commun. 2001, 31, 2209–2214. [Google Scholar] [CrossRef]
- Kan, T.; Fukuyama, T. Ns strategies: A highly versatile synthetic method for amines. Chem. Commun. 2004, 353–359. [Google Scholar] [CrossRef] [PubMed]
- Fukuyama, T.; Jow, C.-K.; Cheung, M. 2- and 4-Nitrobenzenesulfonamides: Exceptionally versatile means for preparation of secondary amines and protection of amines. Tetrahedron Lett. 1995, 36, 6373–6374. [Google Scholar] [CrossRef]
- Kobayashi, S.; Furuya, T.; Otani, T.; Saito, T. A novel and facile stereocontrolled synthetic method for polyhydro-quinolines and pyridopyridazines via a diene-transmissive Diels–Alder reaction involving inverse electron-demand hetero Diels–Alder cycloaddition of cross-conjugated azatrienes. Tetrahedron 2008, 64, 9705–9716. [Google Scholar] [CrossRef]
- Borisov, D.D.; Chermashentsev, G.R.; Novikov, R.A.; Tomilov, Y.V. Synthesis of substituted β-styrylmalonates by sequential isomerization of 2-arylcyclopropane-1,1-dicarboxylates and (2-arylethylidene) malonates. Synthesis 2021, 53, 2253–2259. [Google Scholar] [CrossRef]
- Chagarovskiy, A.O.; Ivanova, O.A.; Rakhmankulov, E.R.; Budynina, E.M.; Trushkov, I.V.; Melnikov, M.Y. Lewis acid-catalyzed isomerization of 2-arylcyclopropane-1,1-dicarboxylates: A new efficient route to 2-styrylmalonates. Adv. Synth. Catal. 2010, 352, 3179–3184. [Google Scholar] [CrossRef]
- Tsedilin, A.M.; Fakhrutdinov, A.N.; Eremin, D.B.; Zalesskiy, S.S.; Chizhov, A.O.; Kolotyrkina, N.G.; Ananikov, V.P. How sensitive and accurate are routine NMR and MS measurements? Mendeleev Commun. 2015, 25, 454–456. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| Entry * | X | Y | Z | T, °C | t, Days | Products, | Total Yields, % ** | Ratio 3/3′ ** |
| 1 | 4-Cl | 4-MeO | 4-NO2 | 20 | 0.5 | 3k, 3k′ | 72 | 1/1 |
| 2 *** | 4-Cl | 4-MeO | 4-NO2 | –20 | 3 | 3k, 3k′ | 61 | 1/1 |
| 3 | 4-Cl | 2,6-Cl2 | H | 20 | 0.5 | 3l, 3l′ | 52 | 1/1 |
| 4 | 4-Cl | 2,6-Cl2 | H | –20 | 5 | 3l, 3l′ | 56 | 1.2/1 |
| 5 | 2-Cl | 2,6-Cl2 | H | 20 | 0.5 | 3m, 3m′ | 74 | 2/1 |
| 6 | 2-Cl | 2,6-Cl2 | H | –20 | 5 | 3m, 3m′ | 54 | 4/1 |
| Entry | Oxide | T, °C | t, h | Yield 6, % |
|---|---|---|---|---|
| 1 | SiO2 | 20 | 12 | 16 |
| 2 | SiO2 | –20 | 12 | 26 |
| 3 * | Al2O3 | 20 | 72 | 11 ** |
| 4 | Al2O3 | 40 | 6 | c.m. *** |
 | |||||
|---|---|---|---|---|---|
| No | Reagent | Solvent | T, °C | t, h | Yield 4, % |
| 1 | SmI2 (6 eq) | THF | 20 → 65 | 6 | n.r. |
| 2 | SmI2 (6 eq), HMPA (18 eq) | THF | 20 | 0.25 | traces |
| 3 * | SmI2 (6 eq), HMPA (18 eq) | THF | –78 | 1 | traces |
| 4 | SmI2 (10 eq), Et3N (20 eq), H2O (30 eq) | THF | 20 | 0.25 | n.r. |
| 5 | Mg (40 eq), ))) | MeOH | 20 | 1 | further reduction (traces) |
| 6 | Mg (10 eq), ))) | MeOH | 20 | 0.25 | |
| 7 | Mg (3 eq), HgCl2 (cat.) | MeOH | –20 | 1 | n.r. |
| 8 | TfOH (1 eq) | DCE ** | 60 | 1 | c.m. |
| 9 * | TfOH (1 eq) | CH2Cl2 | –20 → 20 | 1 | c.m. |
| 10 | HBr/AcOH | 100 | 1 | c.m. | |
| 11 | Na-naphthalenide (10 eq) | THF | –78 | 0.75 | 30 |
| 12 | Na-naphthalenide (6 eq) | THF | –78 | 0.25 | 40 |
| 13 | Na-naphthalenide(6 eq) | DME | –60 | 0.25 | 29 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sergeev, P.G.; Novikov, R.A.; Tomilov, Y.V. Lewis Acid-Catalyzed Formal (4+2)-Cycloaddition between Cross-Conjugated Azatrienes and Styrylmalonates: The Way to Functionalized Quinolizidine Precursors. Molecules 2023, 28, 88. https://doi.org/10.3390/molecules28010088
Sergeev PG, Novikov RA, Tomilov YV. Lewis Acid-Catalyzed Formal (4+2)-Cycloaddition between Cross-Conjugated Azatrienes and Styrylmalonates: The Way to Functionalized Quinolizidine Precursors. Molecules. 2023; 28(1):88. https://doi.org/10.3390/molecules28010088
Chicago/Turabian StyleSergeev, Pavel G., Roman A. Novikov, and Yury V. Tomilov. 2023. "Lewis Acid-Catalyzed Formal (4+2)-Cycloaddition between Cross-Conjugated Azatrienes and Styrylmalonates: The Way to Functionalized Quinolizidine Precursors" Molecules 28, no. 1: 88. https://doi.org/10.3390/molecules28010088