
Aryl Hydrocarbon Receptor as an Anticancer Target: An Overview of Ten Years Odyssey

 , and
, and
Abstract
:1. Introduction
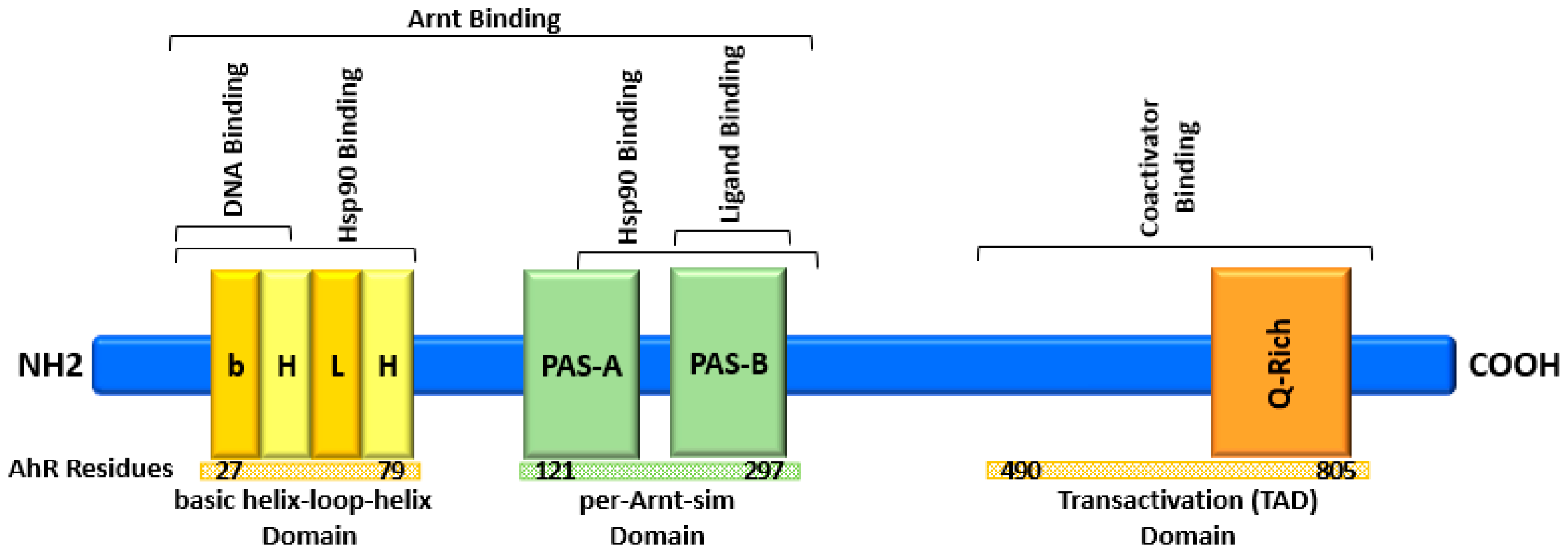
2. AhR Structure and Activation
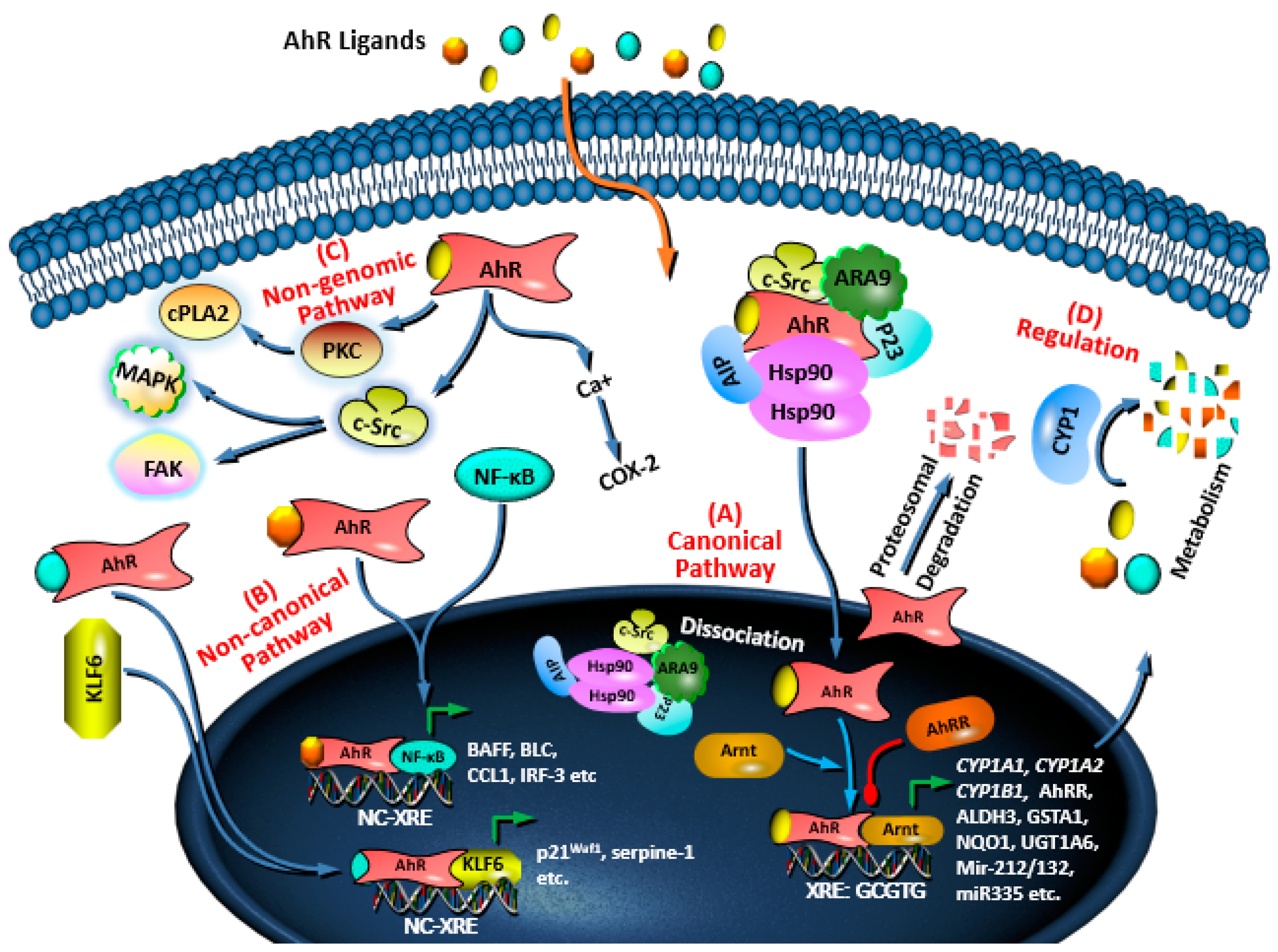
3. AhR Signaling Pathways and Regulation
4. Role of AhR in Cancer at Glance
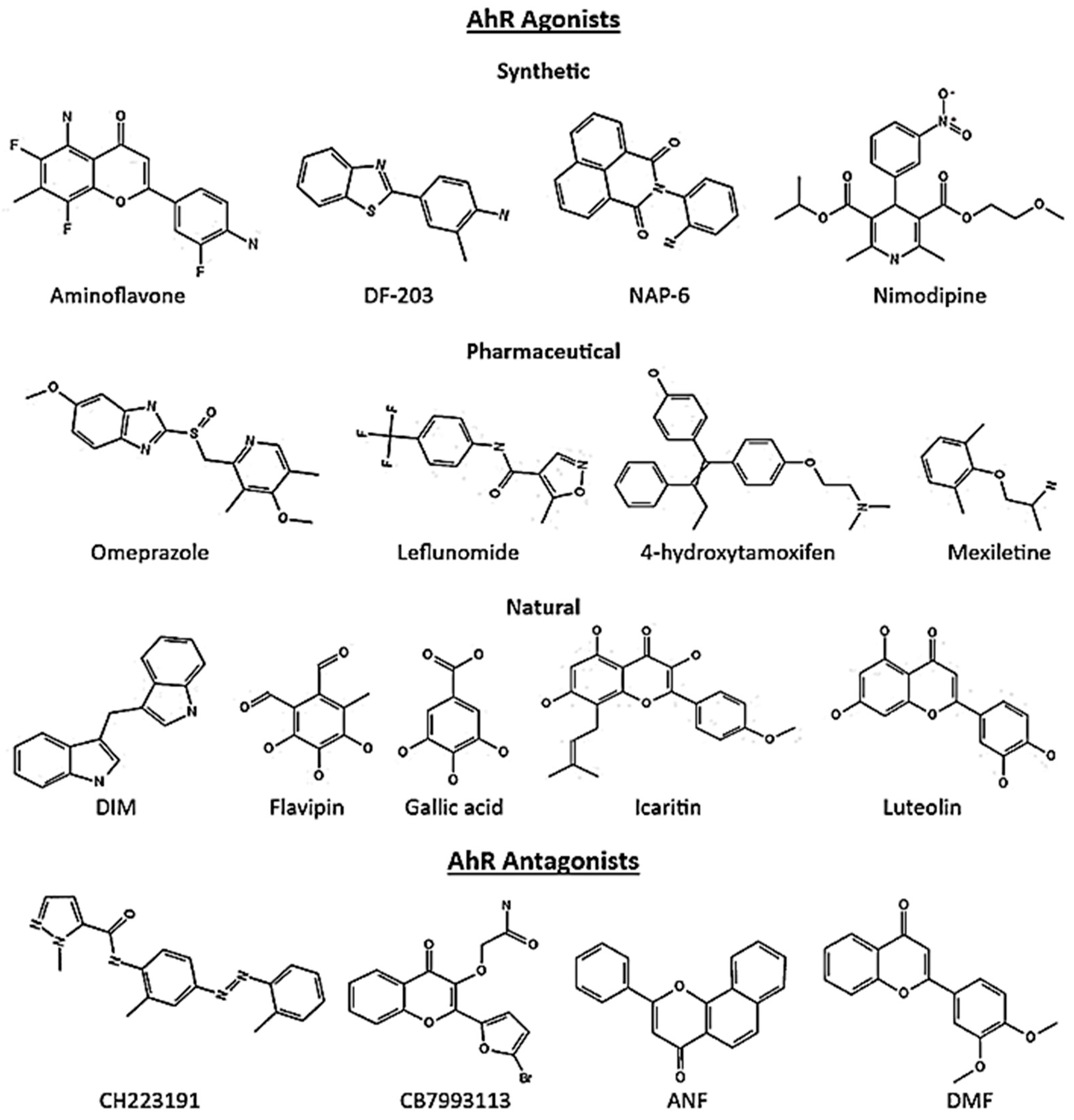
5. AhR Ligands in Cancer
5.1. Breast Cancer

{kind=link}
{kind=link}
{kind=link}
| Compound | Ligand | Response | Cells | Refs. |
|---|---|---|---|---|
| TCDD | Agonist | Proliferation, Migration, Invasion Metastasis | MDA-MB-231, T47D | [58] |
| MCDF | Agonist | Invasion | MDA-MB-231, BT474 | [59] |
| NAP-6 | Agonist | Proliferation, Cell cycle, Checkpoint, DNA damage | MDA-MB-468, MDA-MB-231, ZR-75-1, SKBR3, T47D, MCF-7, BT474, BT20 | [100] |
| 10-Cl-BBQ | Agonist | Proliferation | MDA-MB-468, T47D, ZR-75-1, SKBR3 | [101] |
| CGS-15943 | Antagonist | Apoptosis | MDA-MB-486 | [103] |
| ANI-7 | Agonist | Proliferation, DNA damage, Cell cycle, Checkpoint | MDA-MB-468, MDA-MB-231, ZR-75-1, SKBR3, T47D, MCF-7, BT474 and BT20 | [28,101] |
| 13f (acrylonitrile) | Agonist | Proliferation | MCF-7 | [29] |
| Compound 12 (quinazoline) | Antagonist | Apoptosis, Cell cycle arrest, Growth | MCF-7 | [31] |
| 2-phenylacrylonitriles (analogues) | Agonist | Proliferation (Predicted) | MCF-7 | [102] |
| FDI-6 | Agonist | Tumorsphere formation | MCF-7 | [110] |
| CB7993113 | Antagonist | Migration, Invasion | BP1, Hs578T, and SUM149 | [32] |
| CH223191 | Antagonist | Growth, Migration | MDA-MB-231 | [111] |
| DMBA | Agonist | Migration, Invasion | BP1, Hs5787 | [32] |
| Bap, 3MC, | Agonist | Mammosphere | MCF-7 | [104] |
| 5F-203 | Agonist | DNA damage, Single strand breaks (SSBs) | MD-AMB-468 | [112] |
| Raloxifene | Agonist | Apoptosis | MDA-MB-231 | [103] |
| Omeprazole | Agonist | Invasion, Metastasis | MDA-MB-231 | [92] |
| Leflunomide | Agonist | Migration | MDA-MB-468 | [107] |
| Sulindac | Agonist | Migration | MDA-MB-468 | [107] |
| Nimodipine | Agonist | Migration | MDA-MB-468 | [107] |
| Flutamide | Agonist | Migration, Proliferation | MDA-MB-468, MCF-7 | [107,113] |
| Tranilast | Agonist | Migration | MDA-MB-468 | [107] |
| Flavipin | Agonist | Proliferation, Migration, Invasion | MDA-MB-231, T47D | [108] |
| Gallic acid | Agonist | Proliferation, Migration, Invasion, Growth | MDA-MB-231, T47D | [109] |
| Luteolin | Agonist | Migration, Growth, Metastasis | MDA-MB-231 | [114] |
| Icaritin | Agonist | Growth | MCF-7 | [115] |
| DIM | Agonist | Proliferation, Migration, Invasion, Growth | MDA-MB-231, T47D | [58,116] |
| Galangin | Antagonist | Proliferation, Apoptosis | MCF-7 | [117] |
5.2. Colon Cancer
5.3. Lung Cancer
5.4. Other Cancers
6. AhR: A Potential Target in Cancer Immunotherapy
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Markham, A. Mobocertinib: First approval. Drugs 2021, 81, 2069–2074. [Google Scholar] [CrossRef]
- Passi, I.; Kumar, B. US-FDA Approved Drugs in 2020 and 2021: A Review. Mini Rev. Med. Chem. 2022, 23, 1–25. [Google Scholar] [CrossRef]
- Mak, G.; Soria, J.-C.; Blagden, S.P.; Plummer, R.; Fleming, R.A.; Nebot, N.; Zhang, J.; Mazumdar, J.; Rogan, D.; Gazzah, A. A phase Ib dose-finding, pharmacokinetic study of the focal adhesion kinase inhibitor GSK2256098 and trametinib in patients with advanced solid tumours. Br. J. Cancer 2019, 120, 975–981. [Google Scholar] [CrossRef] [PubMed]
- Stein, E.M.; DeAngelo, D.J.; Chromik, J.; Chatterjee, M.; Bauer, S.; Lin, C.-C.; Suarez, C.; De Vos, F.; Steeghs, N.; Cassier, P.A. Results from a first-in-human phase I study of siremadlin (HDM201) in patients with advanced wild-type TP53 solid tumors and acute leukemia. Clin. Cancer Res. 2022, 28, 870–881. [Google Scholar] [CrossRef]
- Tanaka, T.; Nakatani, T.; Kamitani, T. Negative regulation of NEDD8 conjugation pathway by novel molecules and agents for anticancer therapy. Curr. Pharm. Des. 2013, 19, 4131–4139. [Google Scholar] [CrossRef]
- Puri, S.; Ahmad, I.; Patel, H.; Kumar, K.; Juvale, K. Evaluation of oxindole derivatives as a potential anticancer agent against breast carcinoma cells: In vitro, in silico, and molecular docking study. Toxicol. In Vitro 2023, 86, 105517. [Google Scholar] [CrossRef]
- Salem, M.G.; El-Maaty, D.M.A.; El-Deen, Y.I.M.; Elesawy, B.H.; Askary, A.E.; Saleh, A.; Saied, E.M.; Behery, M.E. Novel 1, 3-thiazole analogues with potent activity against breast cancer: A design, synthesis, in vitro, and in silico study. Molecules 2022, 27, 4898. [Google Scholar] [CrossRef] [PubMed]
- Yousef, R.G.; Elkady, H.; Elkaeed, E.B.; Gobaara, I.M.; Al-Ghulikah, H.A.; Husein, D.Z.; Ibrahim, I.M.; Metwaly, A.M.; Eissa, I.H. (E)-N-(3-(1-(2-(4-(2, 2, 2-Trifluoroacetamido) benzoyl) hydrazono) ethyl) phenyl) nicotinamide: A Novel Pyridine Derivative for Inhibiting Vascular Endothelial Growth Factor Receptor-2: Synthesis, Computational, and Anticancer Studies. Molecules 2022, 27, 7719. [Google Scholar] [CrossRef]
- Elmaaty, A.A.; Darwish, K.M.; Chrouda, A.; Boseila, A.A.; Tantawy, M.A.; Elhady, S.S.; Shaik, A.B.; Mustafa, M.; Al-Karmalawy, A.A. In silico and in vitro studies for benzimidazole anthelmintics repurposing as VEGFR-2 antagonists: Novel mebendazole-loaded mixed micelles with enhanced dissolution and anticancer activity. ACS Omega 2021, 7, 875–899. [Google Scholar] [CrossRef] [PubMed]
- Gunder, L.C.; Moyer, T.H.; Johnson, H.R.; Auyeung, A.S.; Leverson, G.E.; Zhang, W.; Matkowskyj, K.A.; Carchman, E.H. Anal Cancer Prevention Through the Topical Use of Single or Dual PI3K/mTOR Inhibitors. J. Surg. Res. 2023, 282, 137–146. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Shukla, A.; Kalani, K.; Dubey, V.; Luqman, S.; Srivastava, S.K.; Khan, F. In-silico & in-vitro identification of structure-activity relationship pattern of serpentine & gallic acid targeting PI3Kγ as potential anticancer target. Curr. Cancer Drug Targets 2017, 17, 722–734. [Google Scholar]
- Wu, X.; Xu, Y.; Liang, Q.; Yang, X.; Huang, J.; Wang, J.; Zhang, H.; Shi, J. Recent advances in dual PI3K/mTOR inhibitors for tumour treatment. Front. Pharmacol. 2022, 13, 875372. [Google Scholar] [CrossRef]
- Lee, J.B.; Jung, M.; Beom, S.H.; Kim, G.M.; Kim, H.R.; Choi, H.J.; Sohn, J.H.; Ahn, J.B.; Rha, S.Y.; Chung, H.C. Phase 2 study of TAS-117, an allosteric akt inhibitor in advanced solid tumors harboring phosphatidylinositol 3-kinase/v-akt murine thymoma viral oncogene homolog gene mutations. Investig. New Drugs 2021, 39, 1366–1374. [Google Scholar] [CrossRef] [PubMed]
- Stockinger, B.; Meglio, P.D.; Gialitakis, M.; Duarte, J.H. The aryl hydrocarbon receptor: Multitasking in the immune system. Annu. Rev. Immunol. 2014, 32, 403–432. [Google Scholar] [CrossRef]
- Rejano-Gordillo, C.M.; Marín-Díaz, B.; Ordiales-Talavero, A.; Merino, J.M.; González-Rico, F.J.; Fernández-Salguero, P.M. From Nucleus to Organs: Insights of Aryl Hydrocarbon Receptor Molecular Mechanisms. Int. J. Mol. Sci. 2022, 23, 14919. [Google Scholar] [CrossRef]
- Nakahama, T.; Hanieh, H.; Nguyen, N.T.; Chinen, I.; Ripley, B.; Millrine, D.; Lee, S.; Nyati, K.K.; Dubey, P.K.; Chowdhury, K. Aryl hydrocarbon receptor-mediated induction of the microRNA-132/212 cluster promotes interleukin-17–producing T-helper cell differentiation. Proc. Natl. Acad. Sci. USA 2013, 110, 11964–11969. [Google Scholar] [CrossRef] [PubMed]
- Chinen, I.; Nakahama, T.; Kimura, A.; Nguyen, N.T.; Takemori, H.; Kumagai, A.; Kayama, H.; Takeda, K.; Lee, S.; Hanieh, H. The aryl hydrocarbon receptor/microRNA-212/132 axis in T cells regulates IL-10 production to maintain intestinal homeostasis. Int. Immunol. 2015, 27, 405–415. [Google Scholar] [CrossRef] [PubMed]
- Safe, S.; Zhang, L. The Role of the Aryl Hydrocarbon Receptor (AhR) and Its Ligands in Breast Cancer. Cancers 2022, 14, 5574. [Google Scholar] [CrossRef]
- Fujiwara, Y.; Kato, S.; Nesline, M.K.; Conroy, J.M.; DePietro, P.; Pabla, S.; Kurzrock, R. Indoleamine 2, 3-dioxygenase (IDO) inhibitors and cancer immunotherapy. Cancer Treat. Rev. 2022, 110, 102461. [Google Scholar] [CrossRef]
- Nakahama, T.; Kimura, A.; Nguyen, N.T.; Chinen, I.; Hanieh, H.; Nohara, K.; Fujii-Kuriyama, Y.; Kishimoto, T. Aryl hydrocarbon receptor deficiency in T cells suppresses the development of collagen-induced arthritis. Proc. Natl. Acad. Sci. USA 2011, 108, 14222–14227. [Google Scholar] [CrossRef]
- Abdulla, O.A.; Neamah, W.; Sultan, M.; Chatterjee, S.; Singh, N.; Nagarkatti, M.; Nagarkatti, P. Ahr ligands differentially regulate Mirna-132 which targets Hmgb1 and to control the differentiation of Tregs and Th-17 cells during delayed-type hypersensitivity response. Front. Immunol. 2021, 12, 635903. [Google Scholar] [CrossRef]
- Masuda, K.; Kimura, A.; Hanieh, H.; Nguyen, N.T.; Nakahama, T.; Chinen, I.; Otoyo, Y.; Murotani, T.; Yamatodani, A.; Kishimoto, T. Aryl hydrocarbon receptor negatively regulates LPS-induced IL-6 production through suppression of histamine production in macrophages. Int. Immunol. 2011, 23, 637–645. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Bao, L.; Qiu, M.; Feng, L.; Chen, L.; Liu, Z.; Duan, S.; Zhao, Y.; Wu, K.; Zhang, N. Dietary tryptophan-mediated aryl hydrocarbon receptor activation by the gut microbiota alleviates Escherichia coli-induced endometritis in mice. Microbiol. Spectr. 2022, 10, e00811–e00822. [Google Scholar] [CrossRef] [PubMed]
- Abdullah, A.; Maged, M.; Hairul-Islam, M.I.; Osama, I.A.; Manal, A.; Hamza, H. Activation of aryl hydrocarbon receptor signaling by a novel agonist ameliorates autoimmune encephalomyelitis. PLoS ONE 2019, 14, e0215981. [Google Scholar] [CrossRef]
- Alzahrani, A.; Hanieh, H. Differential modulation of Ahr and Arid5a: A promising therapeutic strategy for autoimmune encephalomyelitis. Saudi Pharm. J. 2020, 28, 1605–1615. [Google Scholar] [CrossRef] [PubMed]
- Díaz-Díaz, C.J.; Ronnekleiv-Kelly, S.M.; Nukaya, M.; Geiger, P.G.; Balbo, S.; Dator, R.; Megna, B.W.; Carney, P.R.; Bradfield, C.A.; Kennedy, G.D. The aryl hydrocarbon receptor is a repressor of inflammation-associated colorectal tumorigenesis in mouse. Ann. Surg. 2016, 264, 429–436. [Google Scholar] [CrossRef]
- Alzahrani, A.M.; Hanieh, H.; Ibrahim, H.-I.M.; Mohafez, O.; Shehata, T.; Ismail, M.B.; Alfwuaires, M. Enhancing miR-132 expression by aryl hydrocarbon receptor attenuates tumorigenesis associated with chronic colitis. Int. Immunopharmacol. 2017, 52, 342–351. [Google Scholar] [CrossRef]
- Gilbert, J.; De Iuliis, G.N.; Tarleton, M.; McCluskey, A.; Sakoff, J.A. (Z)-2-(3, 4-Dichlorophenyl)-3-(1H-pyrrol-2-yl) acrylonitrile exhibits selective antitumor activity in breast cancer cell lines via the aryl hydrocarbon receptor pathway. Mol. Pharmacol. 2018, 93, 168–177. [Google Scholar] [CrossRef]
- Baker, J.R.; Russell, C.C.; Gilbert, J.; McCluskey, A.; Sakoff, J.A. Amino alcohol acrylonitriles as broad spectrum and tumour selective cytotoxic agents. RSC Med. Chem. 2021, 12, 929–942. [Google Scholar] [CrossRef]
- Metidji, A.; Omenetti, S.; Crotta, S.; Li, Y.; Nye, E.; Ross, E.; Li, V.; Maradana, M.R.; Schiering, C.; Stockinger, B. The environmental sensor AHR protects from inflammatory damage by maintaining intestinal stem cell homeostasis and barrier integrity. Immunity 2019, 50, 1542. [Google Scholar] [CrossRef]
- Wang, K.; Zhong, H.; Li, N.; Yu, N.; Wang, Y.; Chen, L.; Sun, J. Discovery of novel anti-breast-cancer inhibitors by synergistically antagonizing microtubule polymerization and aryl hydrocarbon receptor expression. J. Med. Chem. 2021, 64, 12964–12977. [Google Scholar] [CrossRef] [PubMed]
- Parks, A.J.; Pollastri, M.P.; Hahn, M.E.; Stanford, E.A.; Novikov, O.; Franks, D.G.; Haigh, S.E.; Narasimhan, S.; Ashton, T.D.; Hopper, T.G. In silico identification of an aryl hydrocarbon receptor antagonist with biological activity in vitro and in vivo. Mol. Pharmacol. 2014, 86, 593–608. [Google Scholar] [CrossRef]
- Wang, C.; Zhang, X.; Teng, Z.; Zhang, T.; Li, Y. Downregulation of PI3K/Akt/mTOR signaling pathway in curcumin-induced autophagy in APP/PS1 double transgenic mice. Eur. J. Pharmacol. 2014, 740, 312–320. [Google Scholar] [CrossRef]
- Vázquez-Gómez, G.; Rocha-Zavaleta, L.; Rodríguez-Sosa, M.; Petrosyan, P.; Rubio-Lightbourn, J. Benzo [a] pyrene activates an AhR/Src/ERK axis that contributes to CYP1A1 induction and stable DNA adducts formation in lung cells. Toxicol. Lett. 2018, 289, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Cheong, J.E.; Sun, L. Targeting the IDO1/TDO2–KYN–AhR pathway for cancer immunotherapy–challenges and opportunities. Trends Pharmacol. Sci. 2018, 39, 307–325. [Google Scholar] [CrossRef]
- Kubli, S.P.; Bassi, C.; Roux, C.; Wakeham, A.; Göbl, C.; Zhou, W.; Jafari, S.M.; Snow, B.; Jones, L.; Palomero, L. AhR controls redox homeostasis and shapes the tumor microenvironment in BRCA1-associated breast cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 3604–3613. [Google Scholar] [CrossRef] [PubMed]
- Sun, L. Recent advances in the development of AHR antagonists in immuno-oncology. RSC Med. Chem. 2021, 12, 902–914. [Google Scholar] [CrossRef]
- Hahn, M.E.; Karchner, S.I.; Shapiro, M.A.; Perera, S.A. Molecular evolution of two vertebrate aryl hydrocarbon (dioxin) receptors (AHR1 and AHR2) and the PAS family. Proc. Natl. Acad. Sci. USA 1997, 94, 13743–13748. [Google Scholar] [CrossRef]
- Hanieh, H. Toward understanding the role of aryl hydrocarbon receptor in the immune system: Current progress and future trends. BioMed Res. Int. 2014, 2014, 520763. [Google Scholar] [CrossRef]
- Zhu, K.; Meng, Q.; Zhang, Z.; Yi, T.; He, Y.; Zheng, J.; Lei, W. Aryl hydrocarbon receptor pathway: Role, regulation and intervention in atherosclerosis therapy. Mol. Med. Rep. 2019, 20, 4763–4773. [Google Scholar] [CrossRef]
- Nguyen, N.T.; Hanieh, H.; Nakahama, T.; Kishimoto, T. The roles of aryl hydrocarbon receptor in immune responses. Int. Immunol. 2013, 25, 335–343. [Google Scholar] [CrossRef]
- Lin, L.; Dai, Y.; Xia, Y. An overview of aryl hydrocarbon receptor ligands in the Last two decades (2002–2022): A medicinal chemistry perspective. Eur. J. Med. Chem. 2022, 244, 114845. [Google Scholar] [CrossRef] [PubMed]
- Soshilov, A.; Denison, M.S. Ligand displaces heat shock protein 90 from overlapping binding sites within the aryl hydrocarbon receptor ligand-binding domain. J. Biol. Chem. 2011, 286, 35275–35282. [Google Scholar] [CrossRef]
- Kumar, M.B.; Ramadoss, P.; Reen, R.K.; Heuvel, J.P.V.; Perdew, G.H. The Q-rich subdomain of the human AhReceptor transactivation domain is required for dioxin-mediated transcriptional activity. J. Biol. Chem. 2001, 276, 42302–42310. [Google Scholar] [CrossRef] [PubMed]
- Dai, S.; Qu, L.; Li, J.; Zhang, Y.; Jiang, L.; Wei, H.; Guo, M.; Chen, X.; Chen, Y. Structural insight into the ligand binding mechanism of aryl hydrocarbon receptor. Nat. Commun. 2022, 13, 6234. [Google Scholar] [CrossRef]
- Gruszczyk, J.; Grandvuillemin, L.; Lai-Kee-Him, J.; Paloni, M.; Savva, C.G.; Germain, P.; Grimaldi, M.; Boulahtouf, A.; Kwong, H.-S.; Bous, J. Cryo-EM structure of the agonist-bound Hsp90-XAP2-AHR cytosolic complex. Nat. Commun. 2022, 13, 7010. [Google Scholar] [CrossRef]
- Ikuta, T.; Eguchi, H.; Tachibana, T.; Yoneda, Y.; Kawajiri, K. Nuclear localization and export signals of the human aryl hydrocarbon receptor. J. Biol. Chem. 1998, 273, 2895–2904. [Google Scholar] [CrossRef]
- Petrulis, J.R.; Kusnadi, A.; Ramadoss, P.; Hollingshead, B.; Perdew, G.H. The hsp90 co-chaperone XAP2 alters importin β recognition of the bipartite nuclear localization signal of the Ah receptor and represses transcriptional activity. J. Biol. Chem. 2003, 278, 2677–2685. [Google Scholar] [CrossRef] [PubMed]
- Kudo, I.; Hosaka, M.; Haga, A.; Tsuji, N.; Nagata, Y.; Okada, H.; Fukuda, K.; Kakizaki, Y.; Okamoto, T.; Grave, E. The regulation mechanisms of AhR by molecular chaperone complex. J. Biochem. 2018, 163, 223–232. [Google Scholar] [CrossRef]
- Dolciami, D.; Ballarotto, M.; Gargaro, M.; López-Cara, L.C.; Fallarino, F.; Macchiarulo, A. Targeting Aryl hydrocarbon receptor for next-generation immunotherapies: Selective modulators (SAhRMs) versus rapidly metabolized ligands (RMAhRLs). Eur. J. Med. Chem. 2020, 185, 111842. [Google Scholar] [CrossRef]
- Swanson, H.I.; Chan, W.K.; Bradfield, C.A. DNA Binding Specificities and Pairing Rules of the Ah Receptor, ARNT, and SIM Proteins. J. Biol. Chem. 1995, 270, 26292–26302. [Google Scholar] [CrossRef] [PubMed]
- Denison, M.; Phelan, D.; Winter, G.; Ziccardi, M. Carbaryl, a carbamate insecticide, is a ligand for the hepatic Ah (dioxin) receptor. Toxicol. Appl. Pharmacol. 1998, 152, 406–414. [Google Scholar] [CrossRef]
- Lamas, B.; Natividad, J.M.; Sokol, H. Aryl hydrocarbon receptor and intestinal immunity. Mucosal Immunol. 2018, 11, 1024–1038. [Google Scholar] [CrossRef]
- Friling, R.S.; Bensimon, A.; Tichauer, Y.; Daniel, V. Xenobiotic-inducible expression of murine glutathione S-transferase Ya subunit gene is controlled by an electrophile-responsive element. Proc. Natl. Acad. Sci. USA 1990, 87, 6258–6262. [Google Scholar] [CrossRef] [PubMed]
- Shen, G.; Kong, A.N. Nrf2 plays an important role in coordinated regulation of Phase II drug metabolism enzymes and Phase III drug transporters. Biopharm. Drug Dispos. 2009, 30, 345–355. [Google Scholar] [CrossRef]
- Hughes, T.; Becknell, B.; Freud, A.G.; McClory, S.; Briercheck, E.; Yu, J.; Mao, C.; Giovenzana, C.; Nuovo, G.; Wei, L. Interleukin-1β selectively expands and sustains interleukin-22+ immature human natural killer cells in secondary lymphoid tissue. Immunity 2010, 32, 803–814. [Google Scholar] [CrossRef] [PubMed]
- Hamza, H.; Abdullah, A. MicroRNA-132 suppresses autoimmune encephalomyelitis by inducing cholinergic anti-inflammation: A new Ahr-based exploration. Eur. J. Immunol. 2013, 43, 2771–2782. [Google Scholar] [CrossRef] [PubMed]
- Hanieh, H. Aryl hydrocarbon receptor-microRNA-212/132 axis in human breast cancer suppresses metastasis by targeting SOX4. Mol. Cancer 2015, 14, 1–13. [Google Scholar] [CrossRef]
- Zhang, S.; Kim, K.; Jin, U.H.; Pfent, C.; Cao, H.; Amendt, B.; Liu, X.; Wilson-Robles, H.; Safe, S. Aryl Hydrocarbon Receptor Agonists Induce MicroRNA-335 Expression and Inhibit Lung Metastasis of Estrogen Receptor Negative Breast Cancer CellsAHR-Dependent Inhibition of Breast Cancer Metastasis. Mol. Cancer Ther. 2012, 11, 108–118. [Google Scholar] [CrossRef]
- Neamah, W.H.; Singh, N.P.; Alghetaa, H.; Abdulla, O.A.; Chatterjee, S.; Busbee, P.B.; Nagarkatti, M.; Nagarkatti, P. AhR activation leads to massive mobilization of myeloid-derived suppressor cells with immunosuppressive activity through regulation of CXCR2 and microRNA miR-150-5p and miR-543-3p that target anti-inflammatory genes. J. Immunol. 2019, 203, 1830–1844. [Google Scholar] [CrossRef]
- Yu, J.; Feng, Y.; Wang, Y.; An, R. Aryl hydrocarbon receptor enhances the expression of miR-150-5p to suppress in prostate cancer progression by regulating MAP3K12. Arch. Biochem. Biophys. 2018, 654, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Vogel, C.F.; Sciullo, E.; Li, W.; Wong, P.; Lazennec, G.; Matsumura, F. RelB, a new partner of aryl hydrocarbon receptor-mediated transcription. Mol. Endocrinol. 2007, 21, 2941–2955. [Google Scholar] [CrossRef]
- Jackson, D.P.; Li, H.; Mitchell, K.A.; Joshi, A.D.; Elferink, C.J. Ah Receptor–Mediated Suppression of Liver Regeneration through NC-XRE–Driven p21Cip1 Expression. Mol. Pharmacol. 2014, 85, 533–541. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.; Elferink, C.J. A novel nonconsensus xenobiotic response element capable of mediating aryl hydrocarbon receptor-dependent gene expression. Mol. Pharmacol. 2012, 81, 338–347. [Google Scholar] [CrossRef] [PubMed]
- Tomkiewicz, C.; Herry, L.; Bui, L.; Metayer, C.; Bourdeloux, M.; Barouki, R.; Coumoul, X. The aryl hydrocarbon receptor regulates focal adhesion sites through a non-genomic FAK/Src pathway. Oncogene 2013, 32, 1811–1820. [Google Scholar] [CrossRef]
- Larigot, L.; Juricek, L.; Dairou, J.; Coumoul, X. AhR signaling pathways and regulatory functions. Biochim. Open 2018, 7, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, T.; Suzuki, T.; Aizawa, K.; Sawaki, D.; Munemasa, Y.; Ishida, J.; Nagai, R. Regulation of transforming growth factor-β-dependent cyclooxygenase-2 expression in fibroblasts. J. Biol. Chem. 2009, 284, 35861–35871. [Google Scholar] [CrossRef] [PubMed]
- Larigot, L.; Benoit, L.; Koual, M.; Tomkiewicz, C.; Barouki, R.; Coumoul, X. Aryl hydrocarbon receptor and its diverse ligands and functions: An exposome receptor. Annu. Rev. Pharmacol. Toxicol. 2022, 62, 383–404. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Lei, P.; Liu, X.; Li, X.; Walker, K.; Kotha, L.; Rowlands, C.; Safe, S. The aryl hydrocarbon receptor as a target for estrogen receptor-negative breast cancer chemotherapy. Endocr. Relat. Cancer 2009, 16, 835. [Google Scholar] [CrossRef]
- Li, B.B.; Scott, E.Y.; Olafsen, N.E.; Matthews, J.; Wheeler, A.R. Analysis of the effects of aryl hydrocarbon receptor expression on cancer cell invasion via three-dimensional microfluidic invasion assays. Lab Chip 2022, 22, 313–325. [Google Scholar] [CrossRef]
- Karasová, M.; Procházková, J.; Tylichová, Z.; Fedr, R.; Ciganek, M.; Machala, M.; Dvořák, Z.; Vyhlídalová, B.; Zůvalová, I.; Ehrmann, J. Inhibition of Aryl Hydrocarbon Receptor (AhR) Expression Disrupts Cell Proliferation and Alters Energy Metabolism and Fatty Acid Synthesis in Colon Cancer Cells. Cancers 2022, 14, 4245. [Google Scholar] [CrossRef] [PubMed]
- Bunaciu, R.P.; Yen, A. Activation of the Aryl hydrocarbon receptor ahr promotes retinoic acid–induced differentiation of myeloblastic leukemia cells by restricting expression of the stem cell transcription factor Oct4. Cancer Res. 2011, 71, 2371–2380. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Yang, Y.; Hong, W.; Huang, M.; Wu, M.; Zhao, X. Applications of genome editing technology in the targeted therapy of human diseases: Mechanisms, advances and prospects. Signal Transduct. Target. Ther. 2020, 5, 1. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Kanno, Y.; Nakayama, M.; Makimura, M.; Ohara, S.; Inouye, Y. Activation of the aryl hydrocarbon receptor represses mammosphere formation in MCF-7 cells. Cancer Lett. 2012, 317, 192–198. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Wu, X.A.; Zhang, F.; Han, L.; Bao, G.; He, X.; Xu, Z. AhR expression is increased in hepatocellular carcinoma. J. Mol. Histol. 2013, 44, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Lin, P.; Chang, H.; Tsai, W.-T.; Wu, M.-H.; Liao, Y.-S.; Chen, J.-T.; Su, J.-M. Overexpression of aryl hydrocarbon receptor in human lung carcinomas. Toxicol. Pathol. 2003, 31, 22–30. [Google Scholar] [CrossRef]
- Sarić, N.; Selby, M.; Ramaswamy, V.; Kool, M.; Stockinger, B.; Hogstrand, C.; Williamson, D.; Marino, S.; Taylor, M.D.; Clifford, S.C. The AHR pathway represses TGFβ-SMAD3 signalling and has a potent tumour suppressive role in SHH medulloblastoma. Sci. Rep. 2020, 10, 148. [Google Scholar] [CrossRef]
- Mohamed, H.T.; Gadalla, R.; El-Husseiny, N.; Hassan, H.; Wang, Z.; Ibrahim, S.A.; El-Shinawi, M.; Sherr, D.H.; Mohamed, M.M. Inflammatory breast cancer: Activation of the aryl hydrocarbon receptor and its target CYP1B1 correlates closely with Wnt5a/b-β-catenin signalling, the stem cell phenotype and disease progression. J. Adv. Res. 2019, 16, 75–86. [Google Scholar] [CrossRef]
- Wang, C.; Xu, C.-X.; Bu, Y.; Bottum, K.M.; Tischkau, S.A. Beta-naphthoflavone (DB06732) mediates estrogen receptor-positive breast cancer cell cycle arrest through AhR-dependent regulation of PI3K/AKT and MAPK/ERK signaling. Carcinogenesis 2014, 35, 703–713. [Google Scholar] [CrossRef]
- Chuang, C.-Y.; Chang, H.; Lin, P.; Sun, S.-J.; Chen, P.-H.; Lin, Y.-Y.; Sheu, G.-T.; Ko, J.-L.; Hsu, S.-L.; Chang, J.T. Up-regulation of osteopontin expression by aryl hydrocarbon receptor via both ligand-dependent and ligand-independent pathways in lung cancer. Gene 2012, 492, 262–269. [Google Scholar] [CrossRef]
- Reyes-Reyes, E.; Ramos, K. Aryl hydrocarbon receptor regulates LINE-1 expression through epigenetic mechanisms that are linked to the canonical TGF-β1 signaling pathway. Toxicol. Lett. 2016, 259, S54. [Google Scholar] [CrossRef]
- Zhu, P.; Yu, H.; Zhou, K.; Bai, Y.; Qi, R.; Zhang, S. 3, 3′-Diindolylmethane modulates aryl hydrocarbon receptor of esophageal squamous cell carcinoma to reverse epithelial-mesenchymal transition through repressing RhoA/ROCK1-mediated COX2/PGE2 pathway. J. Exp. Clin. Cancer Res. 2020, 39, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Matthews, J.; Gustafsson, J.-Å. Estrogen receptor and aryl hydrocarbon receptor signaling pathways. Nucl. Recept. Signal. 2006, 4, nrs-04016. [Google Scholar] [CrossRef] [PubMed]
- Xie, G.; Peng, Z.; Raufman, J.-P. Src-mediated aryl hydrocarbon and epidermal growth factor receptor cross talk stimulates colon cancer cell proliferation. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 302, G1006–G1015. [Google Scholar] [CrossRef]
- Yang, T.; Feng, Y.-L.; Chen, L.; Vaziri, N.D.; Zhao, Y.-Y. Dietary natural flavonoids treating cancer by targeting aryl hydrocarbon receptor. Crit. Rev. Toxicol. 2019, 49, 445–460. [Google Scholar] [CrossRef]
- Kronenberg, S.; Esser, C.; Carlberg, C. An aryl hydrocarbon receptor conformation acts as the functional core of nuclear dioxin signaling. Nucleic Acids Res. 2000, 28, 2286–2291. [Google Scholar] [CrossRef]
- Murray, I.A.; Morales, J.L.; Flaveny, C.A.; DiNatale, B.C.; Chiaro, C.; Gowdahalli, K.; Amin, S.; Perdew, G.H. Evidence for ligand-mediated selective modulation of aryl hydrocarbon receptor activity. Mol. Pharmacol. 2010, 77, 247–254. [Google Scholar] [CrossRef]
- Waller, C.L.; McKinney, J.D. Three-dimensional quantitative structure-activity relationships of dioxins and dioxin-like compounds: Model validation and Ah receptor characterization. Chem. Res. Toxicol. 1995, 8, 847–858. [Google Scholar] [CrossRef]
- Henry, E.C.; Kende, A.S.; Rucci, G.; Totleben, M.J.; Willey, J.J.; Dertinger, S.D.; Pollenz, R.S.; Jones, J.P.; Gasiewicz, T.A. Flavone antagonists bind competitively with 2, 3, 7, 8-tetrachlorodibenzo-p-dioxin (TCDD) to the aryl hydrocarbon receptor but inhibit nuclear uptake and transformation. Mol. Pharmacol. 1999, 55, 716–725. [Google Scholar]
- Turyanska, L.; Itkin, B.; Breen, A.; Loaiza-Perez, A.I.; Sandes, E.O.; Bradshaw, T.D. New Treatments in Renal Cancer: The AhR Ligands. Int. J. Mol. Sci. 2020, 21, 3551. [Google Scholar]
- Hu, W.; Sorrentino, C.; Denison, M.S.; Kolaja, K.; Fielden, M.R. Induction of cyp1a1 is a nonspecific biomarker of aryl hydrocarbon receptor activation: Results of large scale screening of pharmaceuticals and toxicants in vivo and in vitro. Mol. Pharmacol. 2007, 71, 1475–1486. [Google Scholar] [CrossRef] [PubMed]
- Jin, U.-H.; Lee, S.-O.; Pfent, C.; Safe, S. The aryl hydrocarbon receptor ligand omeprazole inhibits breast cancer cell invasion and metastasis. BMC Cancer 2014, 14, 498. [Google Scholar] [CrossRef] [PubMed]
- Goya-Jorge, E.; Jorge Rodríguez, M.E.; Veitía, M.S.-I.; Giner, R.M. Plant occurring flavonoids as modulators of the aryl hydrocarbon receptor. Molecules 2021, 26, 2315. [Google Scholar] [CrossRef]
- Burgoon, L.D.; Ding, Q.; N’jai, A.; Dere, E.; Burg, A.R.; Rowlands, J.C.; Budinsky, R.A.; Stebbins, K.E.; Zacharewski, T.R. Automated dose-response analysis of the relative hepatic gene expression potency of TCDF in C57BL/6 mice. Toxicol. Sci. 2009, 112, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Kopec, A.K.; Burgoon, L.D.; Ibrahim-Aibo, D.; Burg, A.R.; Lee, A.W.; Tashiro, C.; Potter, D.; Sharratt, B.; Harkema, J.R.; Rowlands, J.C. Automated dose-response analysis and comparative toxicogenomic evaluation of the hepatic effects elicited by TCDD, TCDF, and PCB126 in C57BL/6 mice. Toxicol. Sci. 2010, 118, 286–297. [Google Scholar] [CrossRef]
- Gouédard, C.; Barouki, R.; Morel, Y. Dietary polyphenols increase paraoxonase 1 gene expression by an aryl hydrocarbon receptor-dependent mechanism. Mol. Cell. Biol. 2004, 24, 5209–5222. [Google Scholar] [CrossRef]
- Giani Tagliabue, S.; Faber, S.C.; Motta, S.; Denison, M.S.; Bonati, L. Modeling the binding of diverse ligands within the Ah receptor ligand binding domain. Sci. Rep. 2019, 9, 10693. [Google Scholar] [CrossRef]
- Whelan, F.; Hao, N.; Furness, S.G.; Whitelaw, M.L.; Chapman-Smith, A. Amino acid substitutions in the aryl hydrocarbon receptor ligand binding domain reveal YH439 as an atypical AhR activator. Mol. Pharmacol. 2010, 77, 1037–1046. [Google Scholar] [CrossRef]
- Perkins, A.; Phillips, J.L.; Kerkvliet, N.I.; Tanguay, R.L.; Perdew, G.H.; Kolluri, S.K.; Bisson, W.H. A structural switch between agonist and antagonist bound conformations for a ligand-optimized model of the human aryl hydrocarbon receptor ligand binding domain. Biology 2014, 3, 645–669. [Google Scholar] [CrossRef]
- Gilbert, J.; De Iuliis, G.; McCluskey, A.; Sakoff, J. A novel naphthalimide that selectively targets breast cancer via the arylhydrocarbon receptor pathway. Sci. Rep. 2020, 10, 13978. [Google Scholar] [CrossRef]
- Baker, J.R.; Pollard, B.L.; Lin, A.J.; Gilbert, J.; Paula, S.; Zhu, X.; Sakoff, J.A.; McCluskey, A. Modelling and Phenotypic Screening of NAP-6 and 10-Cl-BBQ, AhR Ligands Displaying Selective Breast Cancer Cytotoxicity In Vitro. ChemMedChem 2021, 16, 1499–1512. [Google Scholar] [CrossRef] [PubMed]
- Stanton, D.T.; Baker, J.R.; McCluskey, A.; Paula, S. Development and interpretation of a QSAR model for in vitro breast cancer (MCF-7) cytotoxicity of 2-phenylacrylonitriles. J. Comput. Aided Mol. Des. 2021, 35, 613–628. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, E.F., III.; Jang, H.S.; Liefwalker, D.F.; Kerkvliet, N.I.; Kolluri, S.K. Discovery and mechanistic characterization of a select modulator of AhR-regulated transcription (SMAhRT) with anti-cancer effects. Apoptosis 2021, 26, 307–322. [Google Scholar] [CrossRef] [PubMed]
- Saito, N.; Kanno, Y.; Yamashita, N.; Degawa, M.; Yoshinari, K.; Nemoto, K. The differential selectivity of aryl hydrocarbon receptor (AHR) agonists towards AHR-dependent suppression of mammosphere formation and gene transcription in human breast cancer cells. Biol. Pharm. Bull. 2021, 44, 571–578. [Google Scholar] [CrossRef]
- Zhao, J.; Zou, H.; Han, C.; Ma, J.; Zhao, J.; Tang, J. Circlular RNA BARD1 (Hsa_circ_0001098) overexpression in breast cancer cells with TCDD treatment could promote cell apoptosis via miR-3942/BARD1 axis. Cell Cycle 2018, 17, 2731–2744. [Google Scholar] [CrossRef] [PubMed]
- O’donnell, E.; Koch, D.; Bisson, W.; Jang, H.; Kolluri, S. The aryl hydrocarbon receptor mediates raloxifene-induced apoptosis in estrogen receptor-negative hepatoma and breast cancer cells. Cell Death Dis. 2014, 5, e1038. [Google Scholar] [CrossRef]
- Jin, U.-H.; Lee, S.-O.; Safe, S. Aryl hydrocarbon receptor (AHR)-active pharmaceuticals are selective AHR modulators in MDA-MB-468 and BT474 breast cancer cells. J. Pharmacol. Exp. Ther. 2012, 343, 333–341. [Google Scholar] [CrossRef] [PubMed]
- Hanieh, H.; Mohafez, O.; Hairul-Islam, V.I.; Alzahrani, A.; Bani Ismail, M.; Thirugnanasambantham, K. Novel aryl hydrocarbon receptor agonist suppresses migration and invasion of breast cancer cells. PLoS ONE 2016, 11, e0167650. [Google Scholar] [CrossRef]
- Hanieh, H.; Ibrahim, H.-I.M.; Mohammed, M.; Alwassil, O.I.; Abukhalil, M.H.; Farhan, M. Activation of aryl hydrocarbon receptor signaling by gallic acid suppresses progression of human breast cancer in vitro and in vivo. Phytomedicine 2022, 96, 153817. [Google Scholar] [CrossRef]
- Yamashita, N.; Kawai, K.; Yoshikawa, M.; Watabe, M.; Kanno, Y.; Sanada, N.; Kizu, R. FDI-6, a FOXM1 inhibitor, activates the aryl hydrocarbon receptor and suppresses tumorsphere formation. Biochem. Biophys. Res. Commun. 2023, 639, 29–35. [Google Scholar] [CrossRef]
- Dwyer, A.R.; Kerkvliet, C.P.; Krutilina, R.I.; Playa, H.C.; Parke, D.N.; Thomas, W.A.; Smeester, B.A.; Moriarity, B.S.; Seagroves, T.N.; Lange, C.A. Breast Tumor Kinase (Brk/PTK6) Mediates Advanced Cancer Phenotypes via SH2-Domain Dependent Activation of RhoA and Aryl Hydrocarbon Receptor (AhR) SignalingPTK6 Oncogenic Activity Is SH2 Domain-Dependent. Mol. Cancer Res. 2021, 19, 329–345. [Google Scholar] [CrossRef] [PubMed]
- McLean, L.S.; Watkins, C.N.; Campbell, P.; Zylstra, D.; Rowland, L.; Amis, L.H.; Scott, L.; Babb, C.E.; Livingston, W.J.; Darwanto, A. Aryl hydrocarbon receptor ligand 5F 203 induces oxidative stress that triggers DNA damage in human breast cancer cells. Chem. Res. Toxicol. 2015, 28, 855–871. [Google Scholar] [CrossRef] [PubMed]
- Koch, D.; Jang, H.; O’donnell, E.; Punj, S.; Kopparapu, P.; Bisson, W.; Kerkvliet, N.; Kolluri, S. Anti-androgen flutamide suppresses hepatocellular carcinoma cell proliferation via the aryl hydrocarbon receptor mediated induction of transforming growth factor-β1. Oncogene 2015, 34, 6092–6104. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Zheng, T.; Hou, Z.; Lv, C.; Xue, A.; Han, T.; Han, B.; Sun, X.; Wei, Y. Luteolin, an aryl hydrocarbon receptor ligand, suppresses tumor metastasis in vitro and in vivo. Oncol. Rep. 2020, 44, 2231–2240. [Google Scholar] [CrossRef] [PubMed]
- Tiong, C.T.; Chen, C.; Zhang, S.J.; Li, J.; Soshilov, A.; Denison, M.S.; Lee, L.S.-U.; Tam, V.H.; Wong, S.P.; Xu, H.E. A novel prenylflavone restricts breast cancer cell growth through AhR-mediated destabilization of ERα protein. Carcinogenesis 2012, 33, 1089–1097. [Google Scholar] [CrossRef]
- Nicastro, H.L.; Firestone, G.L.; Bjeldanes, L.F. 3, 3′-diindolylmethane rapidly and selectively inhibits hepatocyte growth factor/c-Met signaling in breast cancer cells. J. Nutr. Biochem. 2013, 24, 1882–1888. [Google Scholar] [CrossRef]
- Liu, Y.; Liang, X.; Zhang, G.; Kong, L.; Peng, W.; Zhang, H. Galangin and pinocembrin from propolis ameliorate insulin resistance in HepG2 cells via regulating Akt/mTOR signaling. Evid. Based Complement. Altern. Med. 2018, 2018, 7971842. [Google Scholar] [CrossRef]
- Pandurangan, A.; Ismail, S.; Saadatdoust, Z.; Esa, N. Allicin alleviates dextran sodium sulfate-(DSS-) induced ulcerative colitis in BALB/c mice. Oxid. Med. Cell. Longev. 2015, 2015, 605208. [Google Scholar] [CrossRef]
- Liu, D.; You, P.; Luo, Y.; Yang, M.; Liu, Y. Galangin induces apoptosis in MCF-7 human breast cancer cells through mitochondrial pathway and phosphatidylinositol 3-kinase/Akt inhibition. Pharmacology 2018, 102, 58–66. [Google Scholar] [CrossRef]
- Stone, E.L.; Citossi, F.; Singh, R.; Kaur, B.; Gaskell, M.; Farmer, P.B.; Monks, A.; Hose, C.; Stevens, M.F.; Leong, C.-O. Antitumour benzothiazoles. Part 32: DNA adducts and double strand breaks correlate with activity; synthesis of 5F203 hydrogels for local delivery. Bioorg. Med. Chem. 2015, 23, 6891–6899. [Google Scholar] [CrossRef]
- Bradshaw, T.D.; Stone, E.L.; Trapani, V.; Leong, C.-O.; Matthews, C.S.; Te Poele, R.; Stevens, M.F. Mechanisms of acquired resistance to 2-(4-Amino-3-methylphenyl) benzothiazole in breast cancer cell lines. Breast Cancer Res. Treat. 2008, 110, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Terzuoli, E.; Puppo, M.; Rapisarda, A.; Uranchimeg, B.; Cao, L.; Burger, A.M.; Ziche, M.; Melillo, G. Aminoflavone, a ligand of the aryl hydrocarbon receptor, inhibits HIF-1α expression in an AhR-independent fashion. Cancer Res. 2010, 70, 6837–6848. [Google Scholar] [CrossRef] [PubMed]
- Campbell, P.S.; Mavingire, N.; Khan, S.; Rowland, L.K.; Wooten, J.V.; Opoku-Agyeman, A.; Guevara, A.; Soto, U.; Cavalli, F.; Loaiza-Pérez, A.I. AhR ligand aminoflavone suppresses α6-integrin–Src–Akt signaling to attenuate tamoxifen resistance in breast cancer cells. J. Cell. Physiol. 2019, 234, 108–121. [Google Scholar] [CrossRef] [PubMed]
- Baker, J.R.; Russell, C.C.; Gilbert, J.; Sakoff, J.A.; McCluskey, A. Amino alcohol acrylonitriles as activators of the aryl hydrocarbon receptor pathway: An unexpected MTT phenotypic screening outcome. ChemMedChem 2020, 15, 490–505. [Google Scholar] [CrossRef]
- Megna, B.W.; Carney, P.R.; Depke, M.G.; Nukaya, M.; McNally, J.; Larsen, L.; Rosengren, R.J.; Kennedy, G.D. The aryl hydrocarbon receptor as an antitumor target of synthetic curcuminoids in colorectal cancer. J. Surg. Res. 2017, 213, 16–24. [Google Scholar] [CrossRef]
- Kabátková, M.; Zapletal, O.; Tylichová, Z.; Neča, J.; Machala, M.; Milcová, A.; Topinka, J.; Kozubík, A.; Vondráček, J. Inhibition of β-catenin signalling promotes DNA damage elicited by benzo [a] pyrene in a model of human colon cancer cells via CYP1 deregulation. Mutagenesis 2015, 30, 565–576. [Google Scholar] [CrossRef]
- Ronnekleiv-Kelly, S.M.; Nukaya, M.; Díaz-Díaz, C.J.; Megna, B.W.; Carney, P.R.; Geiger, P.G.; Kennedy, G.D. Aryl hydrocarbon receptor-dependent apoptotic cell death induced by the flavonoid chrysin in human colorectal cancer cells. Cancer Lett. 2016, 370, 91–99. [Google Scholar] [CrossRef]
- Megna, B.W.; Carney, P.R.; Nukaya, M.; Geiger, P.; Kennedy, G.D. Indole-3-carbinol induces tumor cell death: Function follows form. J. Surg. Res. 2016, 204, 47–54. [Google Scholar] [CrossRef]
- Nguyen, B.D.; Stevens, B.L.; Elson, D.J.; Finlay, D.; Gamble, J.T.; Kopparapu, P.R.; Tanguay, R.L.; Buermeyer, A.B.; Kerkvliet, N.I.; Kolluri, S.K. 11-Cl-BBQ, a select modulator of AhR-regulated transcription, suppresses lung cancer cell growth via activation of p53 and p27Kip1. FEBS J. 2022, 290, 2064–2084. [Google Scholar] [CrossRef]
- Nothdurft, S.; Thumser-Henner, C.; Breitenbücher, F.; Okimoto, R.A.; Dorsch, M.; Opitz, C.A.; Sadik, A.; Esser, C.; Hölzel, M.; Asthana, S. Functional screening identifies aryl hydrocarbon receptor as suppressor of lung cancer metastasis. Oncogenesis 2020, 9, 102. [Google Scholar] [CrossRef]
- Holbrook, M.B.; Schindler, R.M. Nostalgic bonding: Exploring the role of nostalgia in the consumption experience. J. Consum. Behav. Int. Res. Rev. 2003, 3, 107–127. [Google Scholar] [CrossRef]
- Cai, D.-J.; Zhang, Z.-Y.; Bu, Y.; Li, L.; Deng, Y.-Z.; Sun, L.-Q.; Hu, C.-P.; Li, M. Asparagine synthetase regulates lung-cancer metastasis by stabilizing the β-catenin complex and modulating mitochondrial response. Cell Death Dis. 2022, 13, 566. [Google Scholar] [CrossRef] [PubMed]
- Hýžďalová, M.; Procházková, J.; Strapáčová, S.; Svržková, L.; Vacek, O.; Fedr, R.; Andrysík, Z.; Hrubá, E.; Líbalová, H.; Kléma, J. A prolonged exposure of human lung carcinoma epithelial cells to benzo [a] pyrene induces p21-dependent epithelial-to-mesenchymal transition (EMT)-like phenotype. Chemosphere 2021, 263, 128126. [Google Scholar] [CrossRef] [PubMed]
- Zgarbová, E.; Vrzal, R. The Impact of Indoles Activating the Aryl Hydrocarbon Receptor on Androgen Receptor Activity in the 22Rv1 Prostate Cancer Cell Line. Int. J. Mol. Sci. 2023, 24, 502. [Google Scholar] [CrossRef] [PubMed]
- Arabnezhad, M.-R.; Montazeri-Najafabady, N.; Chatrabnous, N.; Bahreman, A.G.; Mohammadi-Bardbori, A. Anti-androgenic effect of 6-formylindolo [3,2-b] carbazole (FICZ) in LNCaP cells is mediated by the aryl hydrocarbon-androgen receptors cross-talk. Steroids 2020, 153, 108508. [Google Scholar] [CrossRef]
- Sun, F.; Indran, I.R.; Zhang, Z.W.; Tan, M.E.; Li, Y.; Lim, Z.R.; Hua, R.; Yang, C.; Soon, F.-F.; Li, J. A novel prostate cancer therapeutic strategy using icaritin-activated arylhydrocarbon-receptor to co-target androgen receptor and its splice variants. Carcinogenesis 2015, 36, 757–768. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Cai, A.; Zheng, H.; Huang, H.; Sun, R.; Cui, X.; Ye, W.; Yao, Q.; Chen, R.; Kou, L. Carbidopa suppresses prostate cancer via aryl hydrocarbon receptor-mediated ubiquitination and degradation of androgen receptor. Oncogenesis 2020, 9, 49. [Google Scholar] [CrossRef] [PubMed]
- Jin, U.-H.; Kim, S.-B.; Safe, S. Omeprazole inhibits pancreatic cancer cell invasion through a nongenomic aryl hydrocarbon receptor pathway. Chem. Res. Toxicol. 2015, 28, 907–918. [Google Scholar] [CrossRef]
- Jin, U.-H.; Karki, K.; Kim, S.-B.; Safe, S. Inhibition of pancreatic cancer Panc1 cell migration by omeprazole is dependent on aryl hydrocarbon receptor activation of JNK. Biochem. Biophys. Res. Commun. 2018, 501, 751–757. [Google Scholar] [CrossRef]
- Takenaka, M.C.; Gabriely, G.; Rothhammer, V.; Mascanfroni, I.D.; Wheeler, M.A.; Chao, C.-C.; Gutiérrez-Vázquez, C.; Kenison, J.; Tjon, E.C.; Barroso, A. Control of tumor-associated macrophages and T cells in glioblastoma via AHR and CD39. Nat. Neurosci. 2019, 22, 729–740. [Google Scholar] [CrossRef]
- Korac, K.; Rajasekaran, D.; Sniegowski, T.; Schniers, B.K.; Ibrahim, A.F.; Bhutia, Y.D. Carbidopa, an activator of aryl hydrocarbon receptor, suppresses IDO1 expression in pancreatic cancer and decreases tumor growth. Biochem. J. 2022, 479, 1807–1824. [Google Scholar] [CrossRef] [PubMed]
- Platten, M.; von Knebel Doeberitz, N.; Oezen, I.; Wick, W.; Ochs, K. Cancer immunotherapy by targeting IDO1/TDO and their downstream effectors. Front. Immunol. 2015, 5, 673. [Google Scholar] [CrossRef] [PubMed]
- D’Amato, N.C.; Rogers, T.J.; Gordon, M.A.; Greene, L.I.; Cochrane, D.R.; Spoelstra, N.S.; Nemkov, T.G.; D’Alessandro, A.; Hansen, K.C.; Richer, J.K. A TDO2-AhR signaling axis facilitates anoikis resistance and metastasis in triple-negative breast cancer. Cancer Res. 2015, 75, 4651–4664. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Stone, E.; Triplett, T.A.; Triplett, K.; Lamb, C.; Karamitros, C.S.; Blazek, J.; Georgiou, G.; Manfredi, M.G. A novel approach to targeting the IDO/TDO pathway through degradation of the immunosuppressive metabolite kynurenine. Cancer Res. 2017, 77, 5570. [Google Scholar] [CrossRef]
- Triplett, T.A.; Garrison, K.C.; Marshall, N.; Donkor, M.; Blazeck, J.; Lamb, C.; Qerqez, A.; Dekker, J.D.; Tanno, Y.; Lu, W.-C. Reversal of indoleamine 2, 3-dioxygenase–mediated cancer immune suppression by systemic kynurenine depletion with a therapeutic enzyme. Nat. Biotechnol. 2018, 36, 758–764. [Google Scholar] [CrossRef]
- Beatty, G.L.; O’Dwyer, P.J.; Clark, J.; Shi, J.G.; Bowman, K.J.; Scherle, P.A.; Newton, R.C.; Schaub, R.; Maleski, J.; Leopold, L. First-in-Human Phase I Study of the Oral Inhibitor of Indoleamine 2, 3-Dioxygenase-1 Epacadostat (INCB024360) in Patients with Advanced Solid MalignanciesIDO1 Inhibitor in Advanced Solid Cancers. Clin. Cancer Res. 2017, 23, 3269–3276. [Google Scholar] [CrossRef]
- Vogel, C.F.A.; Li, W.; Wu, D.; Miller, J.K.; Sweeney, C.; Lazennec, G.; Fujisawa, Y.; Matsumura, F. Interaction of aryl hydrocarbon receptor and NF-κB subunit RelB in breast cancer is associated with interleukin-8 overexpression. Arch. Biochem. Biophys. 2011, 512, 78–86. [Google Scholar] [CrossRef]
- Sato, Y.; Fujimura, T.; Hidaka, T.; Lyu, C.; Tanita, K.; Matsushita, S.; Yamamoto, M.; Aiba, S. Possible roles of proinflammatory signaling in keratinocytes through aryl hydrocarbon receptor ligands for the development of squamous cell carcinoma. Front. Immunol. 2020, 11, 534323. [Google Scholar] [CrossRef]
- Liu, Y.; Zhou, N.; Zhou, L.; Wang, J.; Zhou, Y.; Zhang, T.; Fang, Y.; Deng, J.; Gao, Y.; Liang, X. IL-2 regulates tumor-reactive CD8+ T cell exhaustion by activating the aryl hydrocarbon receptor. Nat. Immunol. 2021, 22, 358–369. [Google Scholar] [CrossRef]
- Mo, Z.; Li, P.; Cao, Z.; Zhang, S. A comprehensive pan-cancer analysis of 33 human cancers reveals the immunotherapeutic value of aryl hydrocarbon receptor. Front. Immunol. 2021, 12, 564948. [Google Scholar] [CrossRef]
- Wang, G.-Z.; Zhang, L.; Zhao, X.-C.; Gao, S.-H.; Qu, L.-W.; Yu, H.; Fang, W.-F.; Zhou, Y.-C.; Liang, F.; Zhang, C. The Aryl hydrocarbon receptor mediates tobacco-induced PD-L1 expression and is associated with response to immunotherapy. Nat. Commun. 2019, 10, 1125. [Google Scholar] [CrossRef] [PubMed]
| Compound | Ligand | Response | Cells | Refs. |
|---|---|---|---|---|
| Colon Cancer | ||||
| Compound 12g (acetamide) | Agonist | Proliferation | HT29 | [124] |
| RL66 | Agonist | Apoptosis, Proliferation | DLD1, HCT116, LS513, RKO | [125] |
| RL118 | Agonist | Apoptosis, Proliferation | DLD1, HCT116, LS513, RKO | [125] |
| Bap | Agonist | DNA damage | HCT116, FHC, HT29 | [126] |
| Chrysin | Agonist | Apoptosis, Proliferation | HCT116, DLD-1, SW837 | [127] |
| I3C | Agonist | Apoptosis, Proliferation | DLD1, HCT116, HT-29, LS513, and RKO | [128] |
| Lung Cancer | ||||
| 11-Cl-BBQ | Agonist | Cell Cycle Arrest, Proliferation | H460 | [129] |
| Bap | Agonist | Proliferation, Migration, Invasion, OPN | H1355, A549 | [80,113] |
| Omeprazole | Agonist | ASNS, ATF4 | H1975, A549, H1299 | [130] |
| Compound | Ligand | Response | Cells | Refs. |
|---|---|---|---|---|
| Prostate | ||||
| 3MI | Agonist | Proliferation | 22Rv1 | [134] |
| 4MI | Agonist | Proliferation | 22Rv1 | [134] |
| 2,3,7TMI | Agonist | Proliferation | 22Rv1 | [134] |
| 7MeO4MI | Agonist | Proliferation | 22Rv1 | [134] |
| Flutamide | Agonist | Proliferation | LNCaP, PC3 | [113,135] |
| Carbidopa | Agonist | Proliferation, Migration, Growth | LNCaP | [137] |
| Icaritin | Agonist | Apoptosis, Proliferation, Growth | LNCaP, C4-2, 22Rv1 | [136] |
| Hepatocellular Carcinoma | ||||
| FDI-6 | Agonist | Tumorsphere formation | HepG2 | [110,140] |
| Flutamide | Agonist | Proliferation | HepG2, HuH-7, 5L | [113] |
| Pancreatic Cancer | ||||
| Omeprazole | Agonist | Invasion | Panc1, MiaPaCa2 | [138] |
| Tranilast | Agonist | Invasion | Panc1, MiaPaCa2 | [138] |
| Carbidopa | Agonist | Growth | PDAC | [141] |
| Ovarian | ||||
| Compound 12a (acrylonitrile) | Proliferation | A2780 | [29] | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hanieh, H.; Bani Ismail, M.; Alfwuaires, M.A.; Ibrahim, H.-I.M.; Farhan, M. Aryl Hydrocarbon Receptor as an Anticancer Target: An Overview of Ten Years Odyssey. Molecules 2023, 28, 3978. https://doi.org/10.3390/molecules28103978
Hanieh H, Bani Ismail M, Alfwuaires MA, Ibrahim H-IM, Farhan M. Aryl Hydrocarbon Receptor as an Anticancer Target: An Overview of Ten Years Odyssey. Molecules. 2023; 28(10):3978. https://doi.org/10.3390/molecules28103978
Chicago/Turabian StyleHanieh, Hamza, Mohammad Bani Ismail, Manal A. Alfwuaires, Hairul-Islam M. Ibrahim, and Mahdi Farhan. 2023. "Aryl Hydrocarbon Receptor as an Anticancer Target: An Overview of Ten Years Odyssey" Molecules 28, no. 10: 3978. https://doi.org/10.3390/molecules28103978
APA StyleHanieh, H., Bani Ismail, M., Alfwuaires, M. A., Ibrahim, H.-I. M., & Farhan, M. (2023). Aryl Hydrocarbon Receptor as an Anticancer Target: An Overview of Ten Years Odyssey. Molecules, 28(10), 3978. https://doi.org/10.3390/molecules28103978