
Lipidomic Characterization of Oocytes at Single-Cell Level Using Nanoflow Chromatography-Trapped Ion Mobility Spectrometry-Mass Spectrometry

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
2.1. Development of the Single-Cell Lipidomic Workflow Based on nanoLC-TIMS-MS
2.2. Single-Cell Lipidomic Analysis of Porcine Oocytes at Different Maturation Stages
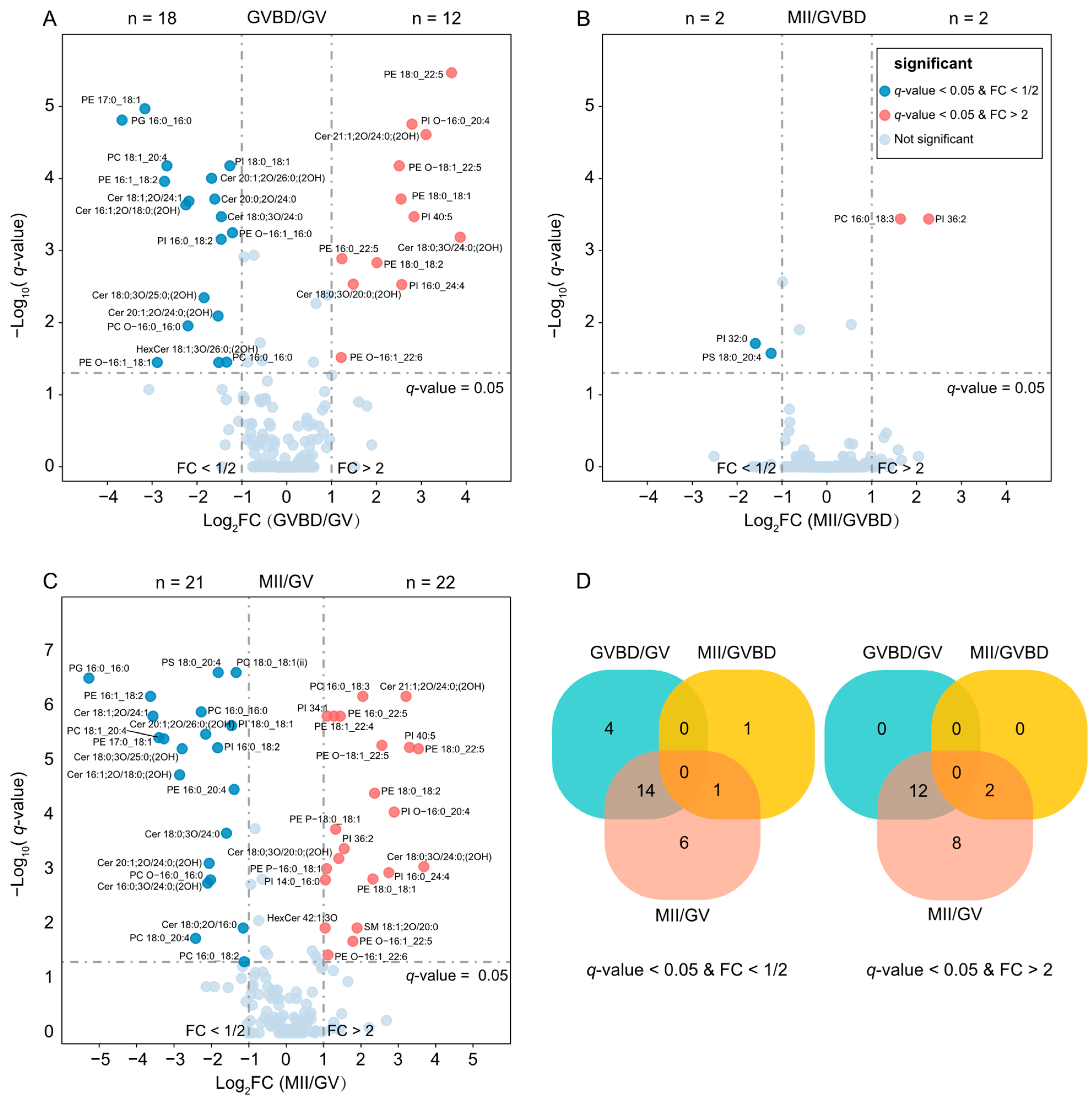
2.3. Lipid Changes during Oocyte Maturation
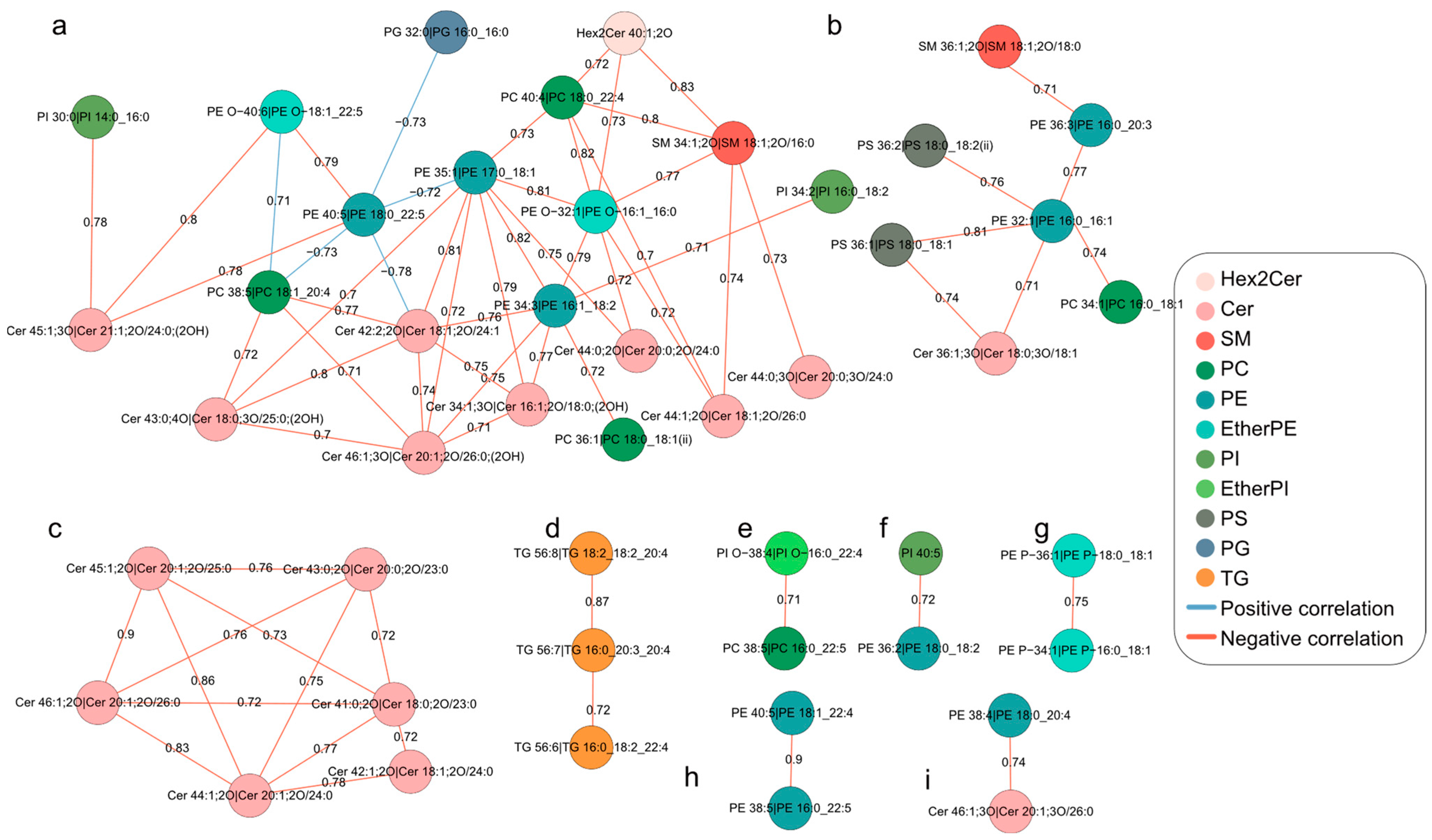
2.4. Differential Correlation Network Analyses of Lipids during Oocyte Maturation
3. Experimental Section
3.1. Chemicals and Materials
3.2. Oocyte Recovery
3.3. Lipid Extraction
3.4. Liquid Chromatography
3.5. Trapped Ion Mobility Spectrometry-Mass Spectrometric Analysis
3.6. Data Analysis and Bioinformatics
3.7. Lipid Identification and Report
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Feng, G.; Gao, M.; Wang, L.; Chen, J.; Hou, M.; Wan, Q.; Lin, Y.; Xu, G.; Qi, X.; Chen, S. Dual-resolving of positional and geometric isomers of C=C bonds via bifunctional photocycloaddition-photoisomerization reaction system. Nat. Commun. 2022, 13, 2652. [Google Scholar] [CrossRef] [PubMed]
- Han, X. Lipidomics for studying metabolism. Nat. Rev. Endocrinol. 2016, 12, 668–679. [Google Scholar] [CrossRef] [PubMed]
- Xie, P.; Zhang, J.; Wu, P.; Wu, Y.; Hong, Y.; Wang, J.; Cai, Z. Multicellular tumor spheroids bridge the gap between two-dimensional cancer cells and solid tumors: The role of lipid metabolism and distribution. Chin. Chem. Lett. 2023, 34, 107349. [Google Scholar] [CrossRef]
- Hao, Y.; Zhang, Z.; Feng, G.; Chen, M.; Wan, Q.; Lin, J.; Wu, L.; Nie, W.; Chen, S. Distinct lipid metabolic dysregulation in asymptomatic COVID-19. iScience 2021, 24, 102974. [Google Scholar] [CrossRef]
- Murphy, R.C. Tandem Mass Spectrometry of Lipids: Molecular Analysis of Complex Lipids, 1st ed.; Royal Society of Chemistry: Cambridge, UK, 2014; Volume 4. [Google Scholar]
- Chen, M.; Hao, Y.; Chen, S. A protocol for investigating lipidomic dysregulation and discovering lipid biomarkers from human serums. STAR Protoc. 2022, 3, 101125. [Google Scholar] [CrossRef]
- Sun, C.; Ma, C.; Li, L.; Han, Y.; Wang, D.; Wan, X. A novel on-tissue cycloaddition reagent for mass spectrometry imaging of lipid C=C position isomers in biological tissues. Chin. Chem. Lett. 2022, 33, 2073–2076. [Google Scholar] [CrossRef]
- Nguyen, A.; Rudge, S.A.; Zhang, Q.; Wakelam, M.J. Using lipidomics analysis to determine signalling and metabolic changes in cells. Curr. Opin. Biotechnol. 2017, 43, 96–103. [Google Scholar] [CrossRef]
- Li, Z.; Cheng, S.; Lin, Q.; Cao, W.; Yang, J.; Zhang, M.; Shen, A.; Zhang, W.; Xia, Y.; Ma, X. Single-cell lipidomics with high structural specificity by mass spectrometry. Nat. Commun. 2021, 12, 2869. [Google Scholar] [CrossRef]
- Wang, C.; Hu, W.; Guan, L.; Yang, X.; Liang, Q. Single-cell metabolite analysis on a microfluidic chip. Chin. Chem. Lett. 2022, 33, 2883–2892. [Google Scholar] [CrossRef]
- Pirro, V.; Oliveri, P.; Ferreira, C.R.; González-Serrano, A.F.; Machaty, Z.; Cooks, R.G. Lipid characterization of individual porcine oocytes by dual mode DESI-MS and data fusion. Anal. Chim. Acta 2014, 848, 51–60. [Google Scholar] [CrossRef]
- Bergman, H.-M.; Lanekoff, I. Profiling and quantifying endogenous molecules in single cells using nano-DESI MS. Analyst 2017, 142, 3639–3647. [Google Scholar] [CrossRef]
- Ferreira, C.R.; Pirro, V.; Jarmusch, A.K.; Alfaro, C.M.; Cooks, R.G. Single Cell Metabolism. In Methods in Molecular Biology; Shrestha, B., Ed.; Humana: New York, NY, USA, 2020; Volume 2064, pp. 159–179. [Google Scholar]
- Li, Y.Z.; Jia, K.; Pan, Y.X.; Han, J.; Chen, J.Y.; Wang, Y.R.; Ma, X.B.; Chen, H.W.; Wang, S.J.; Xie, D.C.; et al. Pocket-Size Wireless Nanoelectrospray Ionization Mass Spectrometry for Metabolic Analysis of Salty Biofluids and Single Cells. Anal. Chem. 2023, 95, 4612–4618. [Google Scholar] [CrossRef]
- Bodzon-Kulakowska, A.; Arena, R.; Mielczarek, P.; Hartman, K.; Kozoł, P.; Gibuła-Tarlowska, E.; Wrobel, T.P.; Gąsior, Ł.; Polański, Z.; Ptak, G.E. Mouse single oocyte imaging by MALDI-TOF MS for lipidomics. Cytotechnology 2020, 72, 455–468. [Google Scholar] [CrossRef]
- Ferreira, C.R.; Saraiva, S.A.; Catharino, R.R.; Garcia, J.S.; Gozzo, F.C.; Sanvido, G.B.; Santos, L.F.A.; Turco, E.G.L.; Pontes, J.H.F.; Basso, A.C. Single embryo and oocyte lipid fingerprinting by mass spectrometry. J. lipid Res. 2010, 51, 1218–1227. [Google Scholar] [CrossRef]
- Dai, Y.; Li, C.; Yi, J.; Qin, Q.; Liu, B.; Qiao, L. Plasmonic Colloidosome-Coupled MALDI-TOF MS for Bacterial Heteroresistance Study at Single-Cell Level. Anal. Chem. 2020, 92, 8051–8057. [Google Scholar] [CrossRef]
- Passarelli, M.K.; Ewing, A.G.; Winograd, N. Single-cell lipidomics: Characterizing and imaging lipids on the surface of individual Aplysia californica neurons with cluster secondary ion mass spectrometry. Anal. Chem. 2013, 85, 2231–2238. [Google Scholar] [CrossRef]
- Kawai, T.; Matsumori, N.; Otsuka, K. Recent advances in microscale separation techniques for lipidome analysis. Analyst 2021, 146, 7418–7430. [Google Scholar] [CrossRef]
- Wang, Z.; Cao, M.; Lam, S.M.; Shui, G. Embracing lipidomics at single-cell resolution: Promises and pitfalls. TrAC Trends Anal. Chem. 2023, 160, 116973. [Google Scholar] [CrossRef]
- He, Y.; Brademan, D.R.; Hutchins, P.D.; Overmyer, K.A.; Coon, J.J. Maximizing MS/MS Acquisition for Lipidomics Using Capillary Separation and Orbitrap Tribrid Mass Spectrometer. Anal. Chem. 2022, 94, 3394–3399. [Google Scholar] [CrossRef]
- Vasilopoulou, C.G.; Sulek, K.; Brunner, A.-D.; Meitei, N.S.; Schweiger-Hufnagel, U.; Meyer, S.W.; Barsch, A.; Mann, M.; Meier, F. Trapped ion mobility spectrometry and PASEF enable in-depth lipidomics from minimal sample amounts. Nat. Commun. 2020, 11, 331. [Google Scholar] [CrossRef]
- Sirait, B.; Wiweko, B.; Jusuf, A.A.; Iftitah, D.; Muharam, R. Oocyte competence biomarkers associated with oocyte maturation: A review. Front. Cell Dev. Biol. 2021, 9, 710292. [Google Scholar] [CrossRef] [PubMed]
- Somfai, T.; Kaneda, M.; Akagi, S.; Watanabe, S.; Haraguchi, S.; Mizutani, E.; Dang-Nguyen, T.Q.; Geshi, M.; Kikuchi, K.; Nagai, T. Enhancement of lipid metabolism with L-carnitine during in vitro maturation improves nuclear maturation and cleavage ability of follicular porcine oocytes. Reprod. Fertil. Dev. 2011, 23, 912–920. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.X.; Lee, S.; Taweechaipaisankul, A.; Kim, G.A.; Lee, B.C. Melatonin regulates lipid metabolism in porcine oocytes. J. Pineal Res. 2017, 62, e12388. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.H.; Kim, G.A.; Taweechaipaisankul, A.; Ridlo, M.R.; Lee, S.H.; Ra, K.; Ahn, C.; Lee, B.C. Phytanic acid-derived peroxisomal lipid metabolism in porcine oocytes. Theriogenology 2020, 157, 276–285. [Google Scholar] [CrossRef] [PubMed]
- Meier, F.; Brunner, A.-D.; Koch, S.; Koch, H.; Lubeck, M.; Krause, M.; Goedecke, N.; Decker, J.; Kosinski, T.; Park, M.A. Online parallel accumulation–serial fragmentation (PASEF) with a novel trapped ion mobility mass spectrometer. Mol. Cell. Proteom. 2018, 17, 2534–2545. [Google Scholar] [CrossRef]
- Brunner, A.D.; Thielert, M.; Vasilopoulou, C.; Ammar, C.; Coscia, F.; Mund, A.; Hoerning, O.B.; Bache, N.; Apalategui, A.; Lubeck, M.; et al. Ultra-high sensitivity mass spectrometry quantifies single-cell proteome changes upon perturbation. Mol. Syst. Biol. 2022, 18, e10798. [Google Scholar] [CrossRef]
- Chen, X.; Yin, Y.; Luo, M.; Zhou, Z.; Cai, Y.; Zhu, Z.-J. Trapped ion mobility spectrometry-mass spectrometry improves the coverage and accuracy of four-dimensional untargeted lipidomics. Anal. Chim. Acta 2022, 1210, 339886. [Google Scholar] [CrossRef]
- Folch, J.; Lees, M.; Stanley, G.H.S. A simple method for the isolation and purification of total lipids from animal tissues. J. Biol. Chem. 1957, 226, 497–509. [Google Scholar] [CrossRef]
- Kong, Q.; Yang, X.; Zhang, H.; Liu, S.; Zhao, J.; Zhang, J.; Weng, X.; Jin, J.; Liu, Z. Lineage specification and pluripotency revealed by transcriptome analysis from oocyte to blastocyst in pig. FASEB J. 2020, 34, 691–705. [Google Scholar] [CrossRef]
- Ovčačíková, M.; Lísa, M.; Cífková, E.; Holčapek, M. Retention behavior of lipids in reversed-phase ultrahigh-performance liquid chromatography–electrospray ionization mass spectrometry. J. Chromatogr. A 2016, 1450, 76–85. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, P.; Bu, G.; Hu, R.; Ruan, X.; Fu, R.; Zhang, Z.; Wan, Q.; Liu, X.; Miao, Y.; Chen, S. Lipidomic Characterization of Oocytes at Single-Cell Level Using Nanoflow Chromatography-Trapped Ion Mobility Spectrometry-Mass Spectrometry. Molecules 2023, 28, 4202. https://doi.org/10.3390/molecules28104202
Zhu P, Bu G, Hu R, Ruan X, Fu R, Zhang Z, Wan Q, Liu X, Miao Y, Chen S. Lipidomic Characterization of Oocytes at Single-Cell Level Using Nanoflow Chromatography-Trapped Ion Mobility Spectrometry-Mass Spectrometry. Molecules. 2023; 28(10):4202. https://doi.org/10.3390/molecules28104202
Chicago/Turabian StyleZhu, Pujia, Guowei Bu, Ruifeng Hu, Xianqin Ruan, Rongrong Fu, Zhourui Zhang, Qiongqiong Wan, Xin Liu, Yiliang Miao, and Suming Chen. 2023. "Lipidomic Characterization of Oocytes at Single-Cell Level Using Nanoflow Chromatography-Trapped Ion Mobility Spectrometry-Mass Spectrometry" Molecules 28, no. 10: 4202. https://doi.org/10.3390/molecules28104202