


Rational Engineering of (S)-Norcoclaurine Synthase for Efficient Benzylisoquinoline Alkaloids Biosynthesis


Abstract
:
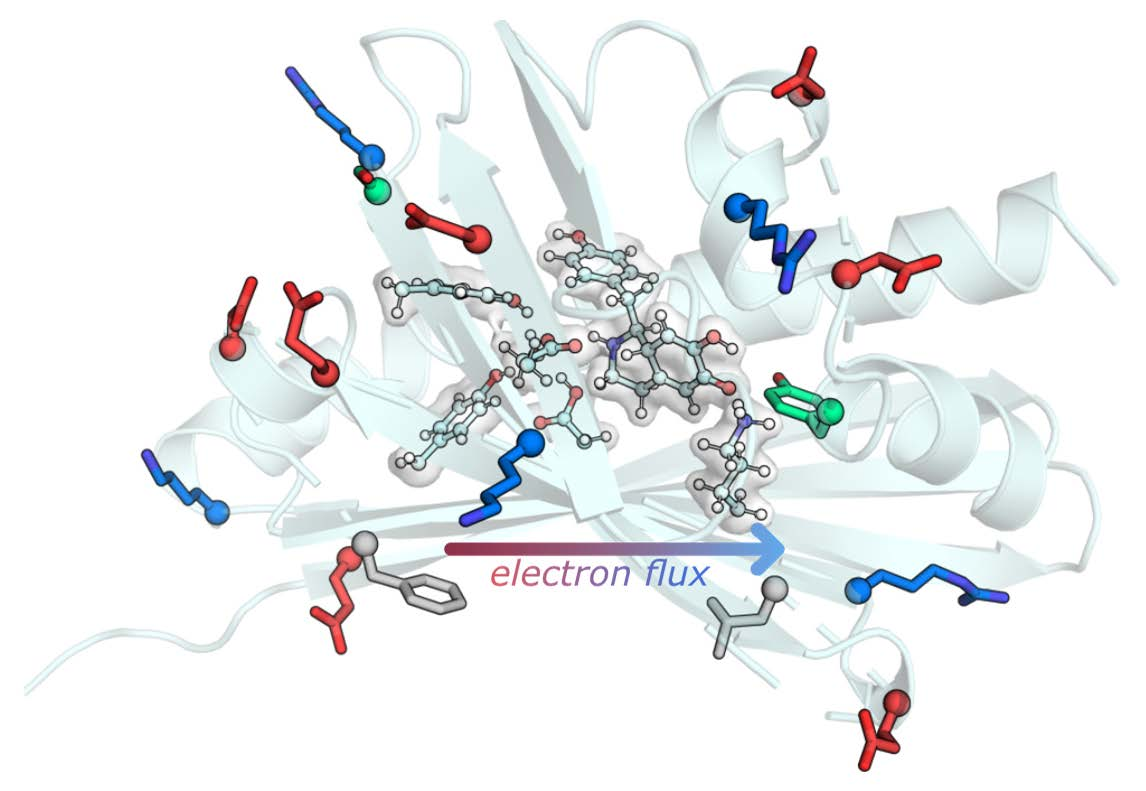{kind=link}
{kind=link}
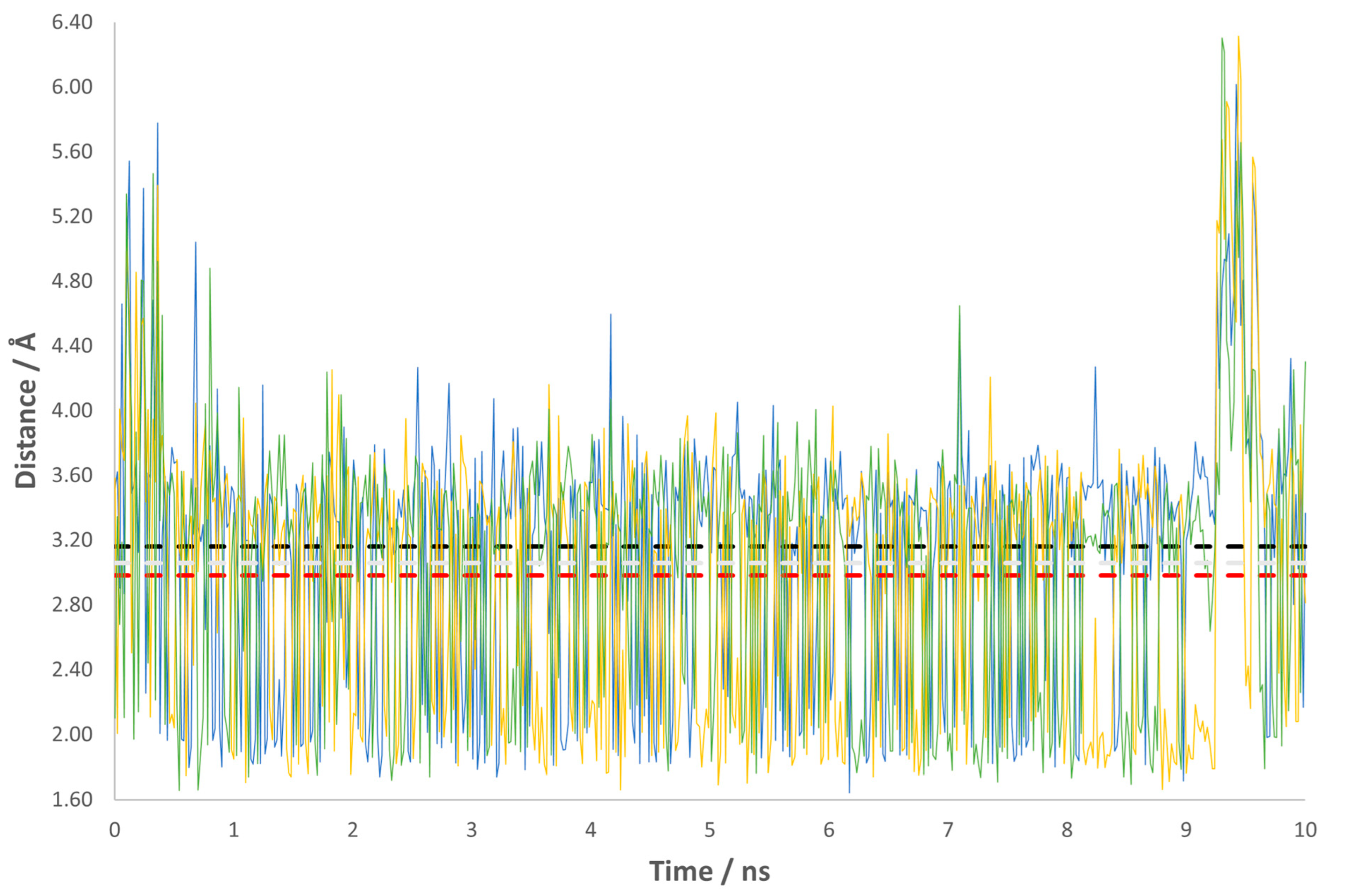{kind=link}
{kind=link}
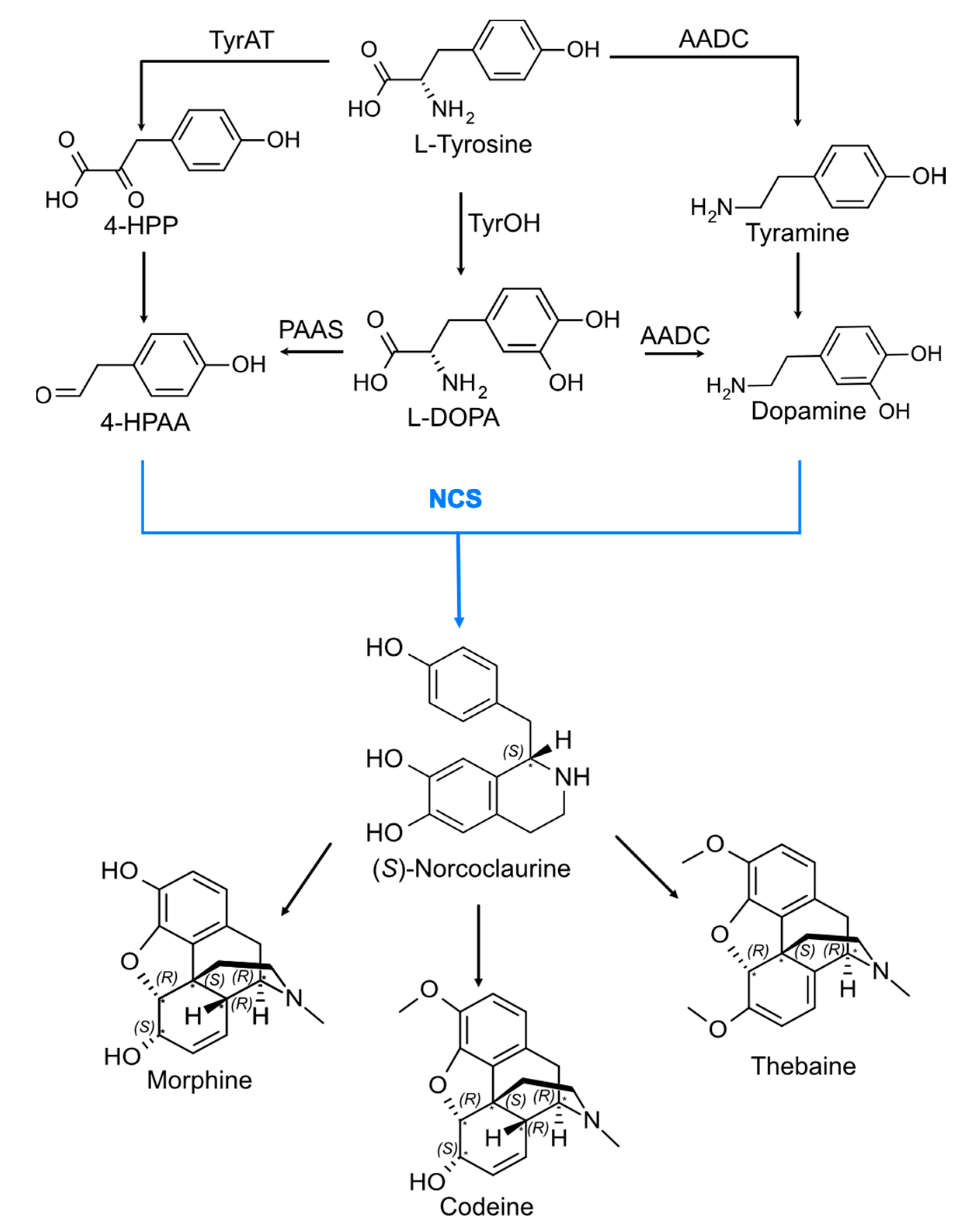
{kind=link}
{kind=link}
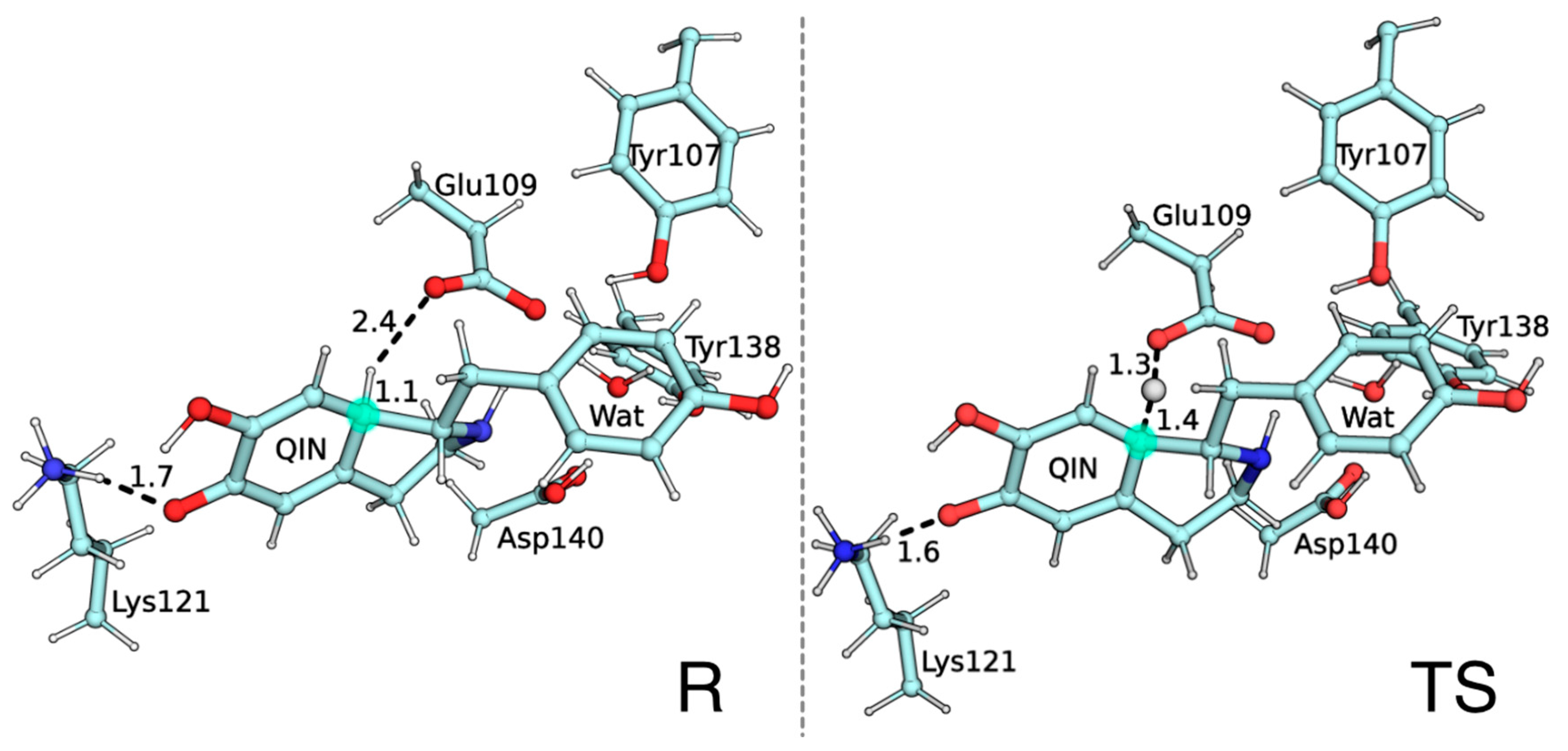
{kind=link}
{kind=link}
1. Introduction
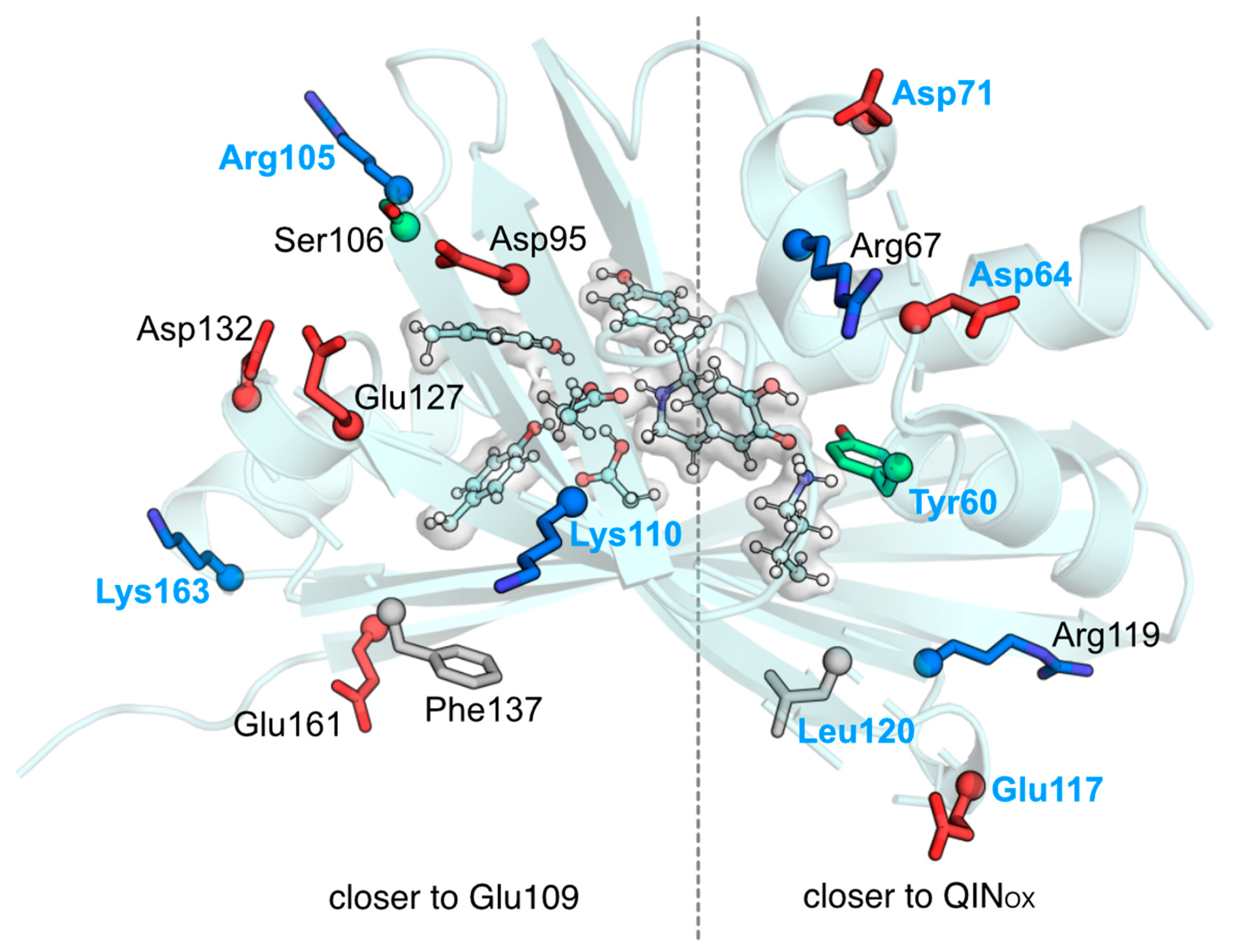
2. Results and Discussion
3. Methods
3.1. Construction of the Psncs:Qin Complex
3.2. MD Simulations
3.3. Oniom Model Details
3.4. Transition-State Macrodipole Stabilization
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Singh, A.; Menéndez-Perdomo, I.M.; Facchini, P.J. Benzylisoquinoline Alkaloid Biosynthesis in Opium Poppy: An Update. Phytochem. Rev. 2019, 18, 1457–1482. [Google Scholar] [CrossRef]
- World Health Organization. WHO Model List of Essential Medicines—22nd List, 2021; WHO: Geneva, Switzerland, 2021.
- Lohman, D.; Schleifer, R.; Amon, J.J. Access to Pain Treatment as a Human Right. BMC Med. 2010, 8, 8. [Google Scholar] [CrossRef]
- Atanasov, A.G.; Waltenberger, B.; Pferschy-Wenzig, E.-M.; Linder, T.; Wawrosch, C.; Uhrin, P.; Temml, V.; Wang, L.; Schwaiger, S.; Heiss, E.H. Discovery and Resupply of Pharmacologically Active Plant-Derived Natural Products: A Review. Biotechnol. Adv. 2015, 33, 1582–1614. [Google Scholar] [CrossRef] [PubMed]
- David, B.; Wolfender, J.-L.; Dias, D.A. The Pharmaceutical Industry and Natural Products: Historical Status and New Trends. Phytochem. Rev. 2015, 14, 299–315. [Google Scholar] [CrossRef]
- Sousa, J.P.; Ramos, M.J.; Fernandes, P.A. Modern Strategies for the Diversification of the Supply of Natural Compounds: The Case of Alkaloid Painkillers. ChemBioChem 2022, 23, e202100623. [Google Scholar] [CrossRef] [PubMed]
- Galanie, S.; Thodey, K.; Trenchard, I.J.; Filsinger Interrante, M.; Smolke, C.D. Complete Biosynthesis of Opioids in Yeast. Science 2015, 349, 1095–1100. [Google Scholar] [CrossRef] [PubMed]
- Pyne, M.E.; Kevvai, K.; Grewal, P.S.; Narcross, L.; Choi, B.; Bourgeois, L.; Dueber, J.E.; Martin, V.J. A Yeast Platform for High-Level Synthesis of Tetrahydroisoquinoline Alkaloids. Nat. Commun. 2020, 11, 3337. [Google Scholar] [CrossRef]
- Hawkins, K.M.; Smolke, C.D. Production of Benzylisoquinoline Alkaloids in Saccharomyces Cerevisiae. Nat. Chem. Biol. 2008, 4, 564–573. [Google Scholar] [CrossRef]
- DeLoache, W.C.; Russ, Z.N.; Narcross, L.; Gonzales, A.M.; Martin, V.J.; Dueber, J.E. An Enzyme-Coupled Biosensor Enables (S)-Reticuline Production in Yeast from Glucose. Nat. Chem. Biol. 2015, 11, 465–471. [Google Scholar] [CrossRef]
- Narcross, L.; Fossati, E.; Bourgeois, L.; Dueber, J.E.; Martin, V.J. Microbial Factories for the Production of Benzylisoquinoline Alkaloids. Trends Biotechnol. 2016, 34, 228–241. [Google Scholar] [CrossRef]
- Lichman, B.R.; Sula, A.; Pesnot, T.; Hailes, H.C.; Ward, J.M.; Keep, N.H. Structural Evidence for the Dopamine-First Mechanism of Norcoclaurine Synthase. Biochemistry 2017, 56, 5274–5277. [Google Scholar] [CrossRef] [PubMed]
- Ilari, A.; Franceschini, S.; Bonamore, A.; Arenghi, F.; Botta, B.; Macone, A.; Pasquo, A.; Bellucci, L.; Boffi, A. Structural Basis of Enzymatic (S)-Norcoclaurine Biosynthesis. J. Biol. Chem. 2009, 284, 897–904. [Google Scholar] [CrossRef]
- Roddan, R.; Ward, J.M.; Keep, N.H.; Hailes, H.C. Pictet–Spenglerases in Alkaloid Biosynthesis: Future Applications in Biocatalysis. Curr. Opin. Chem. Biol. 2020, 55, 69–76. [Google Scholar] [CrossRef]
- Lichman, B.R.; Zhao, J.; Hailes, H.C.; Ward, J.M. Enzyme Catalysed Pictet-Spengler Formation of Chiral 1, 1′-Disubstituted-and Spiro-Tetrahydroisoquinolines. Nat. Commun. 2017, 8, 14883. [Google Scholar] [CrossRef]
- Vaissier Welborn, V.; Head-Gordon, T. Computational Design of Synthetic Enzymes. Chem. Rev. 2018, 119, 6613–6630. [Google Scholar] [CrossRef]
- Sheldon, R.A.; Brady, D. Streamlining Design, Engineering, and Applications of Enzymes for Sustainable Biocatalysis. ACS Sustain. Chem. Eng. 2021, 9, 8032–8052. [Google Scholar] [CrossRef]
- Samanani, N.; Liscombe, D.K.; Facchini, P.J. Molecular Cloning and Characterization of Norcoclaurine Synthase, an Enzyme Catalyzing the First Committed Step in Benzylisoquinoline Alkaloid Biosynthesis. Plant J. 2004, 40, 302–313. [Google Scholar] [CrossRef]
- Minami, H.; Dubouzet, E.; Iwasa, K.; Sato, F. Functional Analysis of Norcoclaurine Synthase in Coptis Japonica. J. Biol. Chem. 2007, 282, 6274–6282. [Google Scholar] [CrossRef]
- Sheng, X.; Himo, F. Enzymatic Pictet–Spengler Reaction: Computational Study of the Mechanism and Enantioselectivity of Norcoclaurine Synthase. J. Am. Chem. Soc. 2019, 141, 11230–11238. [Google Scholar] [CrossRef]
- Svensson, M.; Humbel, S.; Froese, R.D.; Matsubara, T.; Sieber, S.; Morokuma, K. Oniom: A Multilayered Integrated MO + MM Method for Geometry Optimizations and Single Point Energy Predictions. A Test for Diels–Alder Reactions and Pt (P(t-Bu)3)2 + H2 Oxidative Addition. J. Phys. Chem. 1996, 100, 19357–19363. [Google Scholar] [CrossRef]
- Sousa, J.P.; Neves, R.P.; Sousa, S.F.; Ramos, M.J.; Fernandes, P.A. Reaction Mechanism and Determinants for Efficient Catalysis by Dszb, a Key Enzyme for Crude Oil Bio-Desulfurization. ACS Catal. 2020, 10, 9545–9554. [Google Scholar] [CrossRef]
- Viegas, M.F.; Neves, R.P.; Ramos, M.J.; Fernandes, P.A. Qm/Mm Study of the Reaction Mechanism of Thermophilic Glucuronoyl Esterase for Biomass Treatment. ChemPhysChem 2022, 23, e202200269. [Google Scholar] [CrossRef]
- Bertoni, M.; Kiefer, F.; Biasini, M.; Bordoli, L.; Schwede, T. Modeling Protein Quaternary Structure of Homo-and Hetero-Oligomers Beyond Binary Interactions by Homology. Sci. Rep. 2017, 7, 10480. [Google Scholar] [CrossRef]
- Studer, G.; Rempfer, C.; Waterhouse, A.M.; Gumienny, R.; Haas, J.; Schwede, T. Qmeandisco—Distance Constraints Applied on Model Quality Estimation. Bioinformatics 2020, 36, 1765–1771. [Google Scholar] [CrossRef]
- Studer, G.; Tauriello, G.; Bienert, S.; Biasini, M.; Johner, N.; Schwede, T. Promod3—A Versatile Homology Modelling Toolbox. PLoS Comput. Biol. 2021, 17, e1008667. [Google Scholar] [CrossRef]
- Bienert, S.; Waterhouse, A.; De Beer, T.A.; Tauriello, G.; Studer, G.; Bordoli, L.; Schwede, T. The Swiss-Model Repository—New Features and Functionality. Nucleic Acids Res. 2017, 45, D313–D319. [Google Scholar] [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L. Swiss-Model: Homology Modelling of Protein Structures and Complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. Blast+: Architecture and Applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef]
- Remmert, M.; Biegert, A.; Hauser, A.; Söding, J. Hhblits: Lightning-Fast Iterative Protein Sequence Searching by Hmm-Hmm Alignment. Nat. Methods 2012, 9, 173–175. [Google Scholar] [CrossRef]
- Steinegger, M.; Meier, M.; Mirdita, M.; Vöhringer, H.; Haunsberger, S.J.; Söding, J. Hh-Suite3 for Fast Remote Homology Detection and Deep Protein Annotation. BMC Bioinform. 2019, 20, 473. [Google Scholar] [CrossRef]
- Roddan, R.; Gygli, G.; Sula, A.; Méndez-Sánchez, D.; Pleiss, J.; Ward, J.M.; Keep, N.H.; Hailes, H.C. Acceptance and Kinetic Resolution of A-Methyl-Substituted Aldehydes by Norcoclaurine Synthases. ACS Catal. 2019, 9, 9640–9649. [Google Scholar] [CrossRef]
- Davis, I.W.; Leaver-Fay, A.; Chen, V.B.; Block, J.N.; Kapral, G.J.; Wang, X.; Murray, L.W.; Arendall, W.B., III; Snoeyink, J.; Richardson, J.S. Molprobity: All-Atom Contacts and Structure Validation for Proteins and Nucleic Acids. Nucleic Acids Res. 2007, 35, W375–W383. [Google Scholar] [CrossRef] [PubMed]
- Chen, V.B.; Arendall, W.B.; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. Molprobity: All-Atom Structure Validation for Macromolecular Crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.J.; Headd, J.J.; Moriarty, N.W.; Prisant, M.G.; Videau, L.L.; Deis, L.N.; Verma, V.; Keedy, D.A.; Hintze, B.J.; Chen, V.B. Molprobity: More and Better Reference Data for Improved All-Atom Structure Validation. Protein Sci. 2018, 27, 293–315. [Google Scholar] [CrossRef]
- Wiederstein, M.; Sippl, M.J. Prosa-Web: Interactive Web Service for the Recognition of Errors in Three-Dimensional Structures of Proteins. Nucleic Acids Res. 2007, 35, W407–W410. [Google Scholar] [CrossRef]
- Sippl, M.J. Recognition of Errors in Three-Dimensional Structures of Proteins. Proteins Struct. Funct. Bioinform. 1993, 17, 355–362. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Case, D.A.; Belfon, H.M.A.K.; Ben-Shalom, I.Y.; Berryman, J.T.; Brozell, S.R.; Cerutti, D.S.; Cheatham, G.A., III; Cisneros, V.W.D.; Cruzeiro, T.A.; Darden, R.E.; et al. Amber 2018; University of California: San Francisco, CA, USA, 2018. [Google Scholar]
- Frisch, M.J.; Schlegel, G.W.T.H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; Li, X.; et al. Gaussian, Inc.: Wallingford, CT, USA, 2016. Available online: https://www.scirp.org/(S(lz5mqp453ed%20snp55rrgjct55))/reference/referencespapers.aspx?referenceid=2418053 (accessed on 13 July 2022).
- Cerqueira, N.; Ribeiro, J.; Fernandes, P.; Ramos, M. Vslab—An Implementation for Virtual High-Throughput Screening Using Autodock and Vmd. Int. J. Quantum Chem. 2011, 111, 1208–1212. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. Vmd: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. Autodock4 and Autodocktools4: Automated Docking with Selective Receptor Flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Huey, R.; Morris, G.M.; Olson, A.J.; Goodsell, D.S. A Semiempirical Free Energy Force Field with Charge-Based Desolvation. J. Comput. Chem. 2007, 28, 1145–1152. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated Docking Using a Lamarckian Genetic Algorithm and an Empirical Binding Free Energy Function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and Testing of a General Amber Force Field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Bayly, C.I.; Cieplak, P.; Cornell, W.; Kollman, P.A. A Well-Behaved Electrostatic Potential Based Method Using Charge Restraints for Deriving Atomic Charges: The Resp Model. J. Phys. Chem. 1993, 97, 10269–10280. [Google Scholar] [CrossRef]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. Ff14sb: Improving the Accuracy of Protein Side Chain and Backbone Parameters from Ff99sb. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef]
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H.J. Numerical Integration of the Cartesian Equations of Motion of a System with Constraints: Molecular Dynamics of N-Alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef]
- Le Grand, S.; Götz, A.W.; Walker, R.C. Spfp: Speed without Compromise—A Mixed Precision Model for Gpu Accelerated Molecular Dynamics Simulations. Comput. Phys. Commun. 2013, 184, 374–380. [Google Scholar] [CrossRef]
- Gotz, A.W.; Williamson, M.J.; Xu, D.; Poole, D.; Le Grand, S.; Walker, R.C. Routine Microsecond Molecular Dynamics Simulations with Amber on Gpus. 1. Generalized Born. J. Chem. Theory Comput. 2012, 8, 1542–1555. [Google Scholar] [CrossRef]
- Salomon-Ferrer, R.; Gotz, A.W.; Poole, D.; Le Grand, S.; Walker, R.C. Routine Microsecond Molecular Dynamics Simulations with Amber on Gpus. 2. Explicit Solvent Particle Mesh Ewald. J. Chem. Theory Comput. 2013, 9, 3878–3888. [Google Scholar] [CrossRef]
- Roe, D.R.; Cheatham III, T.E. Ptraj and Cpptraj: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef]
- Cornell, W.D.; Cieplak, P.; Bayly, C.I.; Gould, I.R.; Merz, K.M.; Ferguson, D.M.; Spellmeyer, D.C.; Fox, T.; Caldwell, J.W.; Kollman, P.A. A Second Generation Force Field for the Simulation of Proteins, Nucleic Acids, and Organic Molecules. J. Am. Chem. Soc. 1995, 117, 5179–5197. [Google Scholar] [CrossRef]
- Chung, L.W.; Sameera, W.; Ramozzi, R.; Page, A.J.; Hatanaka, M.; Petrova, G.P.; Harris, T.V.; Li, X.; Ke, Z.; Liu, F. The Oniom Method and Its Applications. Chem. Rev. 2015, 115, 5678–5796. [Google Scholar] [CrossRef]
- Chung, L.W.; Hirao, H.; Li, X.; Morokuma, K. The Oniom Method: Its Foundation and Applications to Metalloenzymes and Photobiology. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 327–350. [Google Scholar] [CrossRef]
- Fernandes, H.S.; Ramos, M.J.; MFSA Cerqueira, N. Molup: A Vmd Plugin to Handle Qm and Oniom Calculations Using the Gaussian Software; Wiley Online Library: Hoboken, NJ, USA, 2018. [Google Scholar]
- Sousa, S.F.; Ribeiro, A.J.; Neves, R.P.; Brás, N.F.; Cerqueira, N.M.; Fernandes, P.A.; Ramos, M.J. Application of Quantum Mechanics/Molecular Mechanics Methods in the Study of Enzymatic Reaction Mechanisms. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2017, 7, e1281. [Google Scholar] [CrossRef]
- Fukui, K. The Path of Chemical Reactions-the Irc Approach. Acc. Chem. Res. 1981, 14, 363–368. [Google Scholar] [CrossRef]
- Ribeiro, A.J.; Santos-Martins, D.; Russo, N.; Ramos, M.J.; Fernandes, P.A. Enzymatic Flexibility and Reaction Rate: A Qm/Mm Study of Hiv-1 Protease. Acs Catal. 2015, 5, 5617–5626. [Google Scholar] [CrossRef]
- Medina, F.E.; Neves, R.P.; Ramos, M.J.; Fernandes, P.A. Qm/Mm Study of the Reaction Mechanism of the Dehydratase Domain from Mammalian Fatty Acid Synthase. ACS Catal. 2018, 8, 10267–10278. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Phys. Rev. B 1988, 37, 785. [Google Scholar] [CrossRef]
- Becke, A.D. Density-Functional Exchange-Energy Approximation with Correct Asymptotic Behavior. Phys. Rev. A 1988, 38, 3098. [Google Scholar] [CrossRef]
- Ditchfield, R.; Hehre, W.J.; Pople, J.A. Self-Consistent Molecular-Orbital Methods. Ix. An Extended Gaussian-Type Basis for Molecular-Orbital Studies of Organic Molecules. J. Chem. Phys. 1971, 54, 724–728. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (Dft-D) for the 94 Elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed]
- Benkert, P.; Tosatto, S.C.; Schomburg, D. Qmean: A Comprehensive Scoring Function for Model Quality Assessment. Proteins Struct. Funct. Bioinform. 2008, 71, 261–277. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Sousa, J.P.M.; Oliveira, N.C.S.A.; Fernandes, P.A. Rational Engineering of (S)-Norcoclaurine Synthase for Efficient Benzylisoquinoline Alkaloids Biosynthesis. Molecules 2023, 28, 4265. https://doi.org/10.3390/molecules28114265
De Sousa JPM, Oliveira NCSA, Fernandes PA. Rational Engineering of (S)-Norcoclaurine Synthase for Efficient Benzylisoquinoline Alkaloids Biosynthesis. Molecules. 2023; 28(11):4265. https://doi.org/10.3390/molecules28114265
Chicago/Turabian StyleDe Sousa, João P. M., Nuno C. S. A. Oliveira, and Pedro A. Fernandes. 2023. "Rational Engineering of (S)-Norcoclaurine Synthase for Efficient Benzylisoquinoline Alkaloids Biosynthesis" Molecules 28, no. 11: 4265. https://doi.org/10.3390/molecules28114265
APA StyleDe Sousa, J. P. M., Oliveira, N. C. S. A., & Fernandes, P. A. (2023). Rational Engineering of (S)-Norcoclaurine Synthase for Efficient Benzylisoquinoline Alkaloids Biosynthesis. Molecules, 28(11), 4265. https://doi.org/10.3390/molecules28114265