
Early Molecular Insights into Thanatin Analogues Binding to A. baumannii LptA

Abstract
:1. Introduction
2. Results
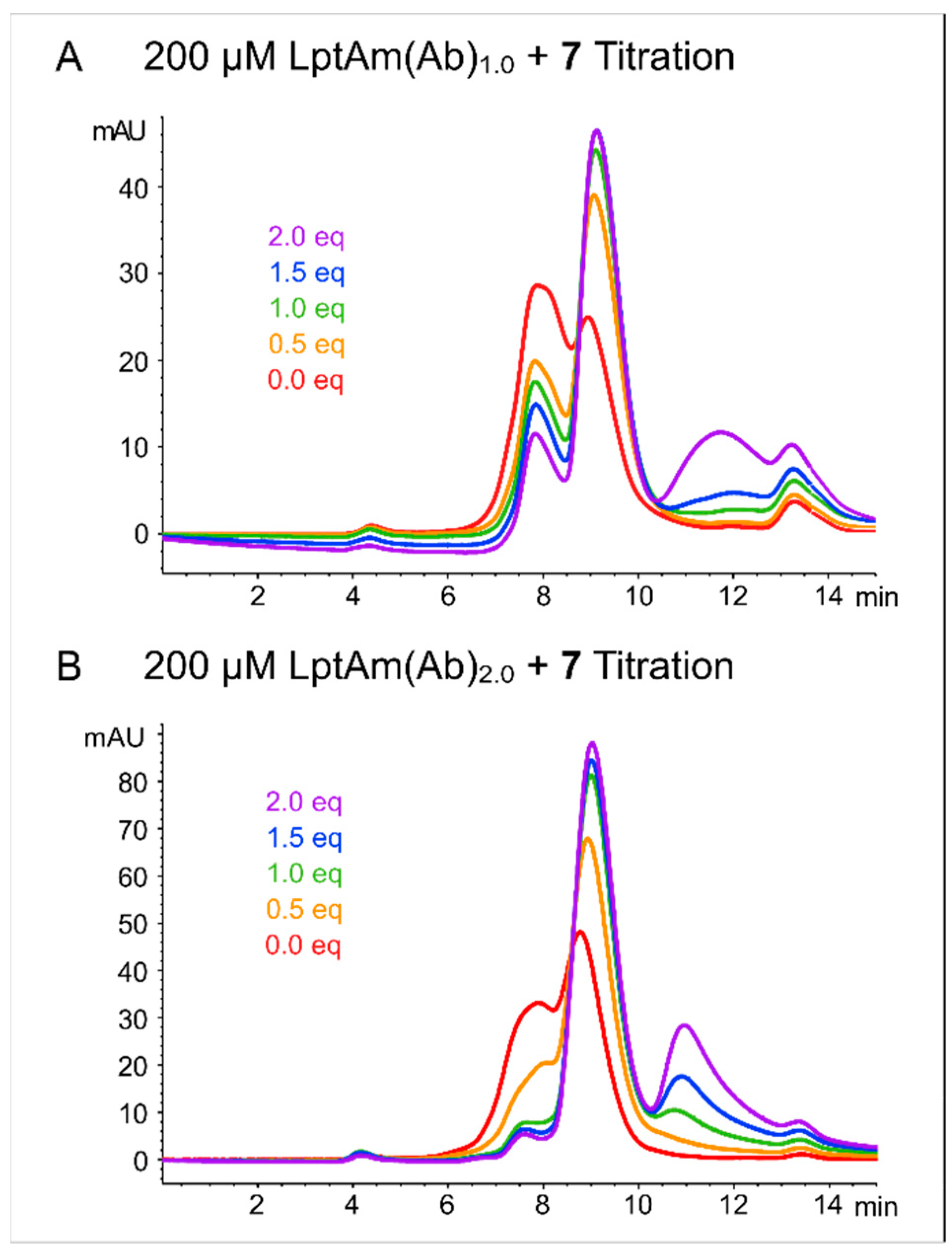
2.1. Protein Characterization and Quality Control
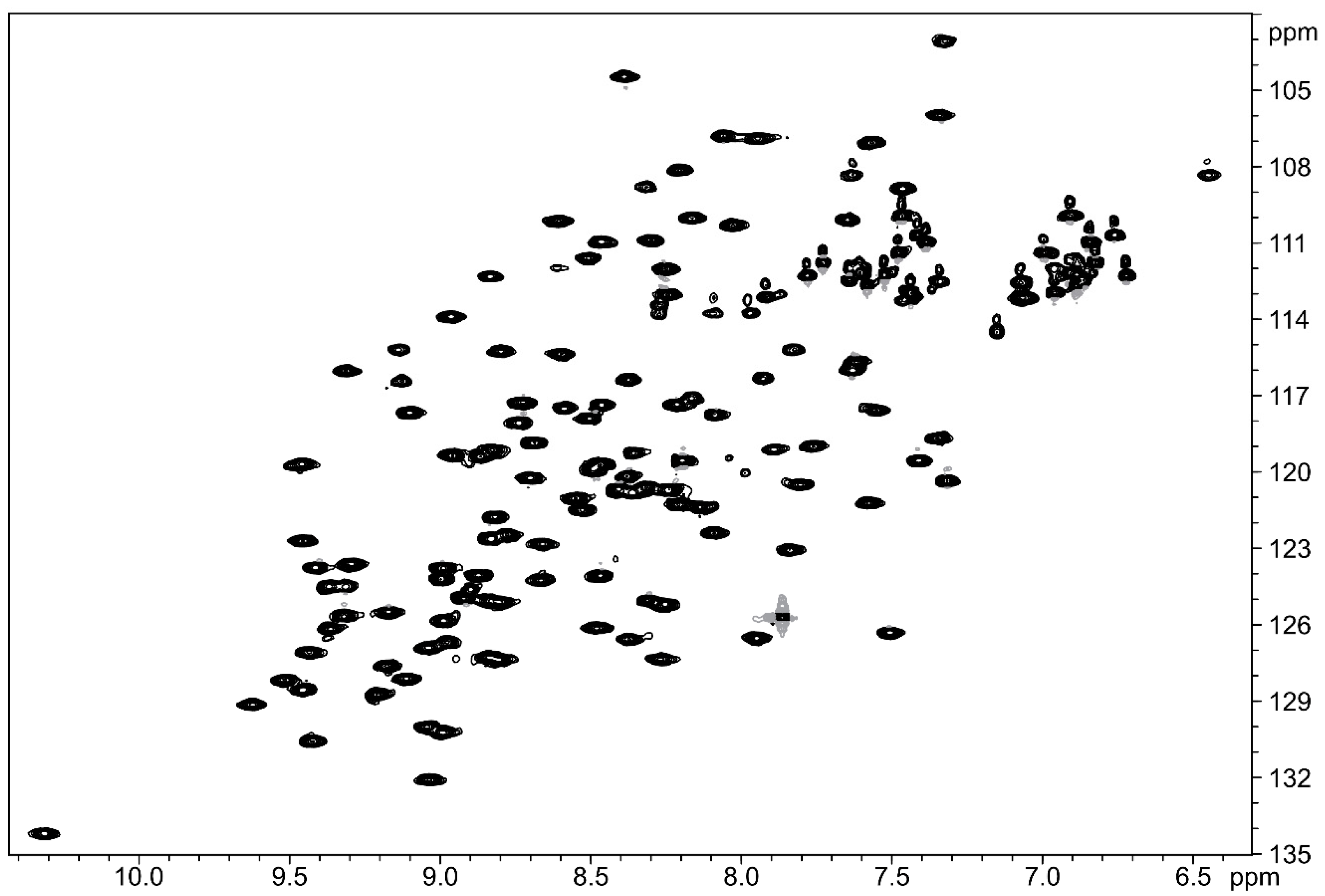
2.2. Structural Studies Using Nuclear Magnetic Resonance (NMR)
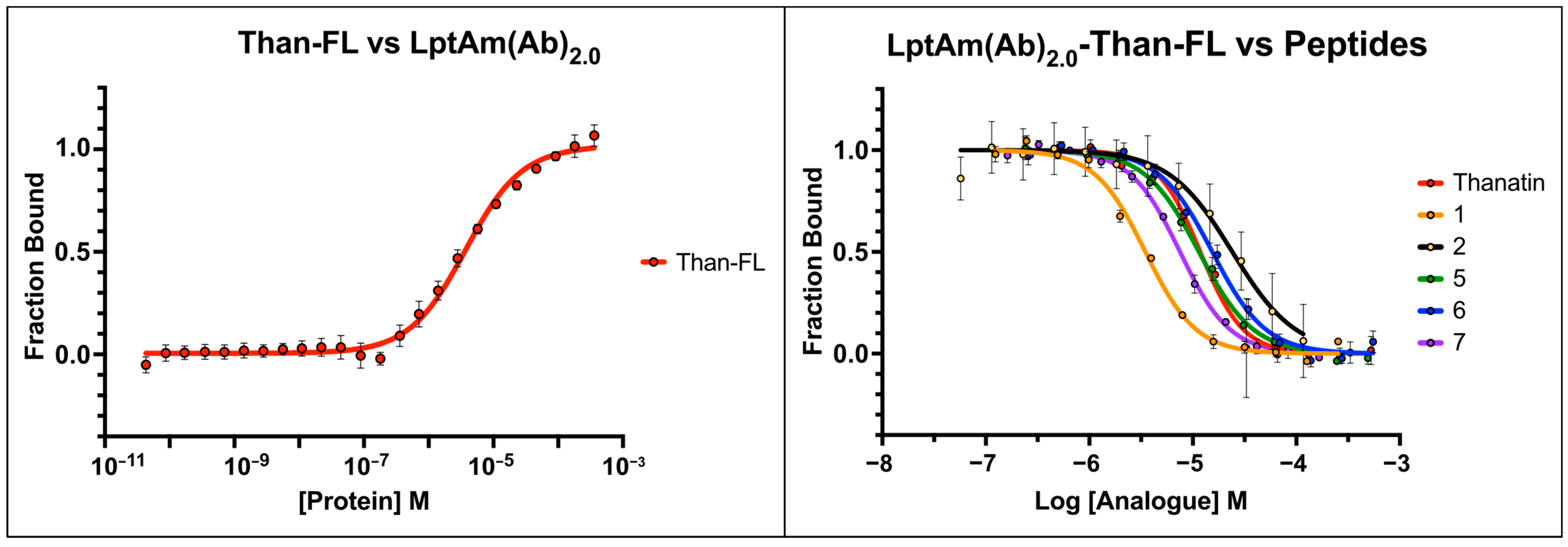
2.3. Fluorescence Polarization Binding Studies
2.4. Examining the Affinities of Thanatin Derivatives
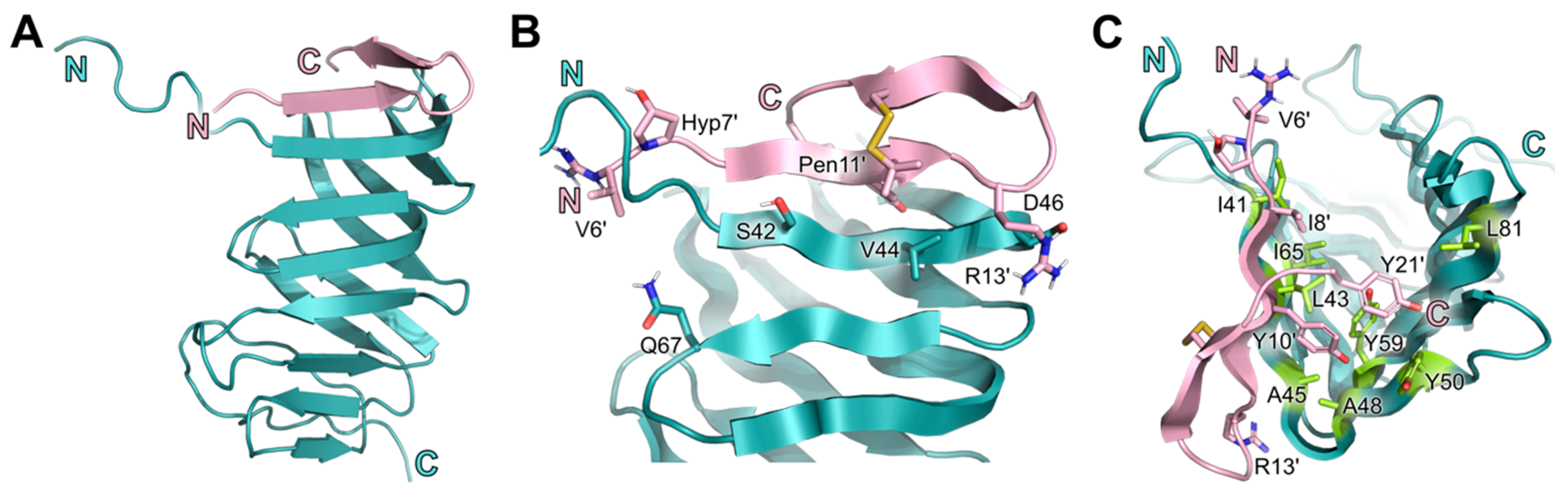
2.5. Conserved Binding Mode of 7 to LptAm across Phyla
3. Discussion and Outlook
4. Materials and Methods
4.1. Constructs, Cloning, and Protein Expression
4.2. Protein Purification
4.3. NMR Spectroscopy, Assignments, and Structure Calculations
4.3.1. Free Peptide Assignment
4.3.2. Protein–Peptide Complex Assignment and Structure Calculation
4.4. Fluorescence Polarization (FP) Assays
4.5. Protein Sequence Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Resistance, C.A. Global burden of bacterial antimicrobial resistance in 2019: A systematic analysis. Lancet 2022, 399, 629–655. [Google Scholar]
- WHO. In News: WHO Publishes List of Bacteria for Which New Antibiotics Are Urgently Needed (2017). Available online: https://www.who.int/news/item/27-02-2017-who-publishes-list-of-bacteria-for-which-new-antibiotics-are-urgently-needed (accessed on 25 January 2023).
- Richter, M.F.; Drown, B.S.; Riley, A.P.; Garcia, A.; Shirai, T.; Svec, R.L.; Hergenrother, P.J. Predictive compound accumulation rules yield a broad-spectrum antibiotic. Nature 2017, 545, 299–304. [Google Scholar] [CrossRef] [PubMed]
- Kohanski, M.A.; Dwyer, D.J.; Collins, J.J. How antibiotics kill bacteria: From targets to networks. Nat. Rev. Microbiol. 2010, 8, 423–435. [Google Scholar] [CrossRef] [PubMed]
- O’Shea, R.; Moser, H.E. Physicochemical properties of antibacterial compounds: Implications for drug discovery. J. Med. Chem. 2008, 51, 2871–2878. [Google Scholar] [CrossRef]
- Hancock, R.E. Alterations in outer membrane permeability. Annu. Rev. Microbiol. 1984, 38, 237–264. [Google Scholar] [CrossRef]
- Hancock, R.E.; Bell, A. Antibiotic uptake into gram-negative bacteria. Eur. J. Clin. Microbiol. Infect. Dis. 1988, 7, 713–720. [Google Scholar] [CrossRef]
- Zhang, L.; Dhillon, P.; Yan, H.; Farmer, S.; Hancock, R.E. Interactions of bacterial cationic peptide antibiotics with outer and cytoplasmic membranes of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2000, 44, 3317–3321. [Google Scholar] [CrossRef]
- Trimble, M.J.; Mlynarcik, P.; Kolar, M.; Hancock, R.E. Polymyxin: Alternative Mechanisms of Action and Resistance. Cold Spring Harb. Perspect. Med. 2016, 6, a025288. [Google Scholar] [CrossRef]
- Sabnis, A.; Hagart, K.L.; Klöckner, A.; Becce, M.; E Evans, L.; Furniss, R.C.D.; Mavridou, D.A.; Murphy, R.; Stevens, M.M.; Davies, J.C.; et al. Colistin kills bacteria by targeting lipopolysaccharide in the cytoplasmic membrane. Elife 2021, 10, e65836. [Google Scholar] [CrossRef]
- Guest, R.L.; Rutherford, S.T.; Silhavy, T.J. Border Control: Regulating LPS Biogenesis. Trends Microbiol. 2021, 29, 334–345. [Google Scholar] [CrossRef]
- Richter, M.F.; Hergenrother, P.J. The challenge of converting Gram-positive-only compounds into broad-spectrum antibiotics. Ann. N. Y. Acad. Sci. 2019, 1435, 18–38. [Google Scholar] [CrossRef]
- Chng, S.S.; Gronenberg, L.S.; Kahne, D. Proteins required for lipopolysaccharide assembly in Escherichia coli form a transenvelope complex. Biochemistry 2010, 49, 4565–4567. [Google Scholar] [CrossRef] [PubMed]
- Okuda, S.; Sherman, D.J.; Silhavy, T.J.; Ruiz, N.; Kahne, D. Lipopolysaccharide transport and assembly at the outer membrane: The PEZ model. Nat. Rev. Microbiol. 2016, 14, 337–345. [Google Scholar] [CrossRef] [PubMed]
- Harding, C.M.; Hennon, S.W.; Feldman, M.F. Uncovering the mechanisms of Acinetobacter baumannii virulence. Nat. Rev. Microbiol. 2018, 16, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Moffatt, J.H.; Harper, M.; Harrison, P.; Hale, J.D.; Vinogradov, E.; Seemann, T.; Henry, R.; Crane, B.; Michael, F.S.; Cox, A.D.; et al. Colistin resistance in Acinetobacter baumannii is mediated by complete loss of lipopolysaccharide production. Antimicrob. Agents Chemother. 2010, 54, 4971–4977. [Google Scholar] [CrossRef] [PubMed]
- Beceiro, A.; Llobet, E.; Aranda, J.; Bengoechea, J.A.; Doumith, M.; Hornsey, M.; Dhanji, H.; Chart, H.; Bou, G.; Livermore, D.M.; et al. Phosphoethanolamine modification of lipid A in colistin-resistant variants of Acinetobacter baumannii mediated by the pmrAB two-component regulatory system. Antimicrob. Agents Chemother. 2011, 55, 3370–3379. [Google Scholar] [CrossRef] [PubMed]
- Pelletier, M.R.; Casella, L.G.; Jones, J.W.; Adams, M.D.; Zurawski, D.V.; Hazlett, K.R.O.; Doi, Y.; Ernst, R.K. Unique structural modifications are present in the lipopolysaccharide from colistin-resistant strains of Acinetobacter baumannii. Antimicrob. Agents Chemother. 2013, 57, 4831–4840. [Google Scholar] [CrossRef]
- Boll, J.M.; Tucker, A.T.; Klein, D.R.; Beltran, A.M.; Brodbelt, J.S.; Davies, B.W.; Trent, M.S. Reinforcing Lipid A Acylation on the Cell Surface of Acinetobacter baumannii Promotes Cationic Antimicrobial Peptide Resistance and Desiccation Survival. mBio 2015, 6, e00478-15. [Google Scholar] [CrossRef]
- Boll, J.M.; Crofts, A.A.; Peters, K.; Cattoir, V.; Vollmer, W.; Davies, B.W.; Trent, M.S. A penicillin-binding protein inhibits selection of colistin-resistant, lipooligosaccharide-deficient Acinetobacter baumannii. Proc. Natl. Acad. Sci. USA 2016, 113, E6228–E6237. [Google Scholar] [CrossRef]
- Kamoshida, G.; Akaji, T.; Takemoto, N.; Suzuki, Y.; Sato, Y.; Kai, D.; Hibino, T.; Yamaguchi, D.; Ubagai, T.; Nishida, S.; et al. Lipopolysaccharide-Deficient Acinetobacter baumannii Due to Colistin Resistance Is Killed by Neutrophil-Produced Lysozyme. Front. Microbiol. 2020, 11, 573. [Google Scholar] [CrossRef]
- Zhang, G.; Meredith, T.C.; Kahne, D. On the essentiality of lipopolysaccharide to Gram-negative bacteria. Curr. Opin. Microbiol. 2013, 16, 779–785. [Google Scholar] [CrossRef] [PubMed]
- Vetterli, S.U.; Zerbe, K.; Müller, M.; Urfer, M.; Mondal, M.; Wang, S.Y.; Möhle, K.; Zerbe, O.; Vitale, A.; Pessi, G.; et al. Thanatin targets the intermembrane protein complex required for lipopolysaccharide transport in Escherichia coli. Sci. Adv. 2018, 4, eaau2634. [Google Scholar] [CrossRef] [PubMed]
- Schuster, M.; Brabet, E.; Oi, K.K.; Desjonquères, N.; Moehle, K.; Le Poupon, K.; Hell, S.; Gable, S.; Rithié, V.; Dillinger, S.; et al. Peptidomimetic Antibiotics Disrupt the Lipopolysaccharide Transport Bridge of Drug-Resistant Enterobacteriaceae. Sci. Adv. 2023; adg3683, in press. [Google Scholar]
- Laguri, C.; Sperandeo, P.; Pounot, K.; Ayala, I.; Silipo, A.; Bougault, C.M.; Molinaro, A.; Polissi, A.; Simorre, J.-P. Interaction of lipopolysaccharides at intermolecular sites of the periplasmic Lpt transport assembly. Sci. Rep. 2017, 7, 9715. [Google Scholar] [CrossRef]
- Shen, Y.; Delaglio, F.; Cornilescu, G.; Bax, A. TALOS+: A hybrid method for predicting protein backbone torsion angles from NMR chemical shifts. J. Biomol. NMR 2009, 44, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Prusoff, W.H. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 1973, 22, 3099–3108. [Google Scholar]
- Zgurskaya, H.I.; Rybenkov, V.V. Permeability barriers of Gram-negative pathogens. Ann. N. Y. Acad. Sci. 2020, 1459, 5–18. [Google Scholar] [CrossRef] [PubMed]
- Fiorentino, F.; Sauer, J.B.; Qiu, X.; Corey, R.A.; Cassidy, C.K.; Mynors-Wallis, B.; Mehmood, S.; Bolla, J.R.; Stansfeld, P.J.; Robinson, C.V. Dynamics of an LPS translocon induced by substrate and an antimicrobial peptide. Nat. Chem. Biol. 2021, 17, 187–195. [Google Scholar] [CrossRef]
- Mares, J.; Kumaran, S.; Gobbo, M.; Zerbe, O. Interactions of lipopolysaccharide and polymyxin studied by NMR spectroscopy. J. Biol. Chem. 2009, 284, 11498–11506. [Google Scholar] [CrossRef]
- Bhattacharjya, S.; Mohid, S.A.; Bhunia, A. Atomic-Resolution Structures and Mode of Action of Clinically Relevant Antimicrobial Peptides. Int. J. Mol. Sci. 2022, 23, 4558. [Google Scholar] [CrossRef]
- Ma, B.; Fang, C.; Lu, L.; Wang, M.; Xue, X.; Zhou, Y.; Li, M.; Hu, Y.; Luo, X.; Hou, Z.; et al. The antimicrobial peptide thanatin disrupts the bacterial outer membrane and inactivates the NDM-1 metallo-beta-lactamase. Nat. Commun. 2019, 10, 3517. [Google Scholar] [CrossRef] [PubMed]
- Arroyo, L.A.; Herrera, C.M.; Fernandez, L.; Hankins, J.V.; Trent, M.S.; Hancock, R.E.W. The pmrCAB operon mediates polymyxin resistance in Acinetobacter baumannii ATCC 17978 and clinical isolates through phosphoethanolamine modification of lipid A. Antimicrob. Agents Chemother. 2011, 55, 3743–3751. [Google Scholar] [CrossRef] [PubMed]
- Schuster, M.; Deluigi, M.; Pantić, M.; Vacca, S.; Baumann, C.; Scott, D.J.; Plückthun, A.; Zerbe, O. Optimizing the alpha1B-adrenergic receptor for solution NMR studies. Biochim. Biophys. Acta Biomembr. 2020, 1862, 183354. [Google Scholar] [CrossRef]
- Zhou, P.; Lugovskoy, A.A.; Wagner, G. A solubility-enhancement tag (SET) for NMR studies of poorly behaving proteins. J. Biomol. NMR 2001, 20, 11–14. [Google Scholar] [CrossRef] [PubMed]
- Sattler, M.; Schleucher, J.; Griesinger, C. Heteronuclear multidimensional NMR experiments for the structure determination of proteins in solution employing pulsed field gradients. Prog. Nucl. Magn. Reson. Spectrosc. 1999, 34, 93–158. [Google Scholar] [CrossRef]
- Otting, G.; Wüthrich, K. Heteronuclear filters in two-dimensional (1H,1H) NMR spectroscopy: Combined use with isotope labelling for studies of macromolecular conformation and intermolecular interactions. Q. Rev. Biophys. 1990, 23, 39–96. [Google Scholar] [CrossRef] [PubMed]
- Güntert, P. Automated NMR structure calculation with CYANA. Methods Mol. Biol. 2004, 278, 353–378. [Google Scholar]
- Hulme, E.C.; Trevethick, M.A. Ligand binding assays at equilibrium: Validation and interpretation. Br. J. Pharmacol. 2010, 161, 1219–1237. [Google Scholar] [CrossRef]
- Jarmoskaite, I.; AlSadhan, I.; Vaidyanathan, P.P.; Herschlag, D. How to measure and evaluate binding affinities. Elife 2020, 9, e57264. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain | E. coli ATCC 25922 | K. pneumoniae ATCC 43816 | P. aeruginosa ATCC 27853 | A. baumannii DSM 30008 |
|---|---|---|---|---|
| LptA Identity | 100% | 88% | 30% | 28% |
| Thanatin | 1 | 2 | >64 | >64 |
| 1 | 0.063 | 0.250 | >64 | >32 |
| 2 | 0.031 | 0.125 | >32 | >32 |
| 5 | 0.063 | 0.250 | >32 | >32 |
| 6 | 0.125 | 0.250 | >32 | >32 |
| 7 | 0.063 | 0.125 | >64 | >64 |
| Position | |||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Peptide | 1′ | 2′ | 3′ | 4′ | 5′ | 6′ | 7′ | 8′ | 9′ | 10′ | 11′ | 12′ | 13′ | 14′ | 15′ | 16′ | 17′ | 18′ | 19′ | 20′ | 21′ |
| Thanatin | G | S | K | K | P | V | P | I | I | Y | C | N | R | R | T | G | K | C | Q | R | M |
| 1 | K | K | P | V | P | I | I | Y | C | N | R | R | T | G | K | C | Dab | R | Nle | ||
| 2 | K | K | P | V | P | I | I | Y | Pen | N | R | Dab | T | DDab | K | C | Dab | R | Y | ||
| 5 | V | Hyp | I | I | Y | Pen | N | R | Dab | T | DDab | K | C | Dab | Dab | Y | |||||
| 6 | V | Hyp | I | T | Y | Pen | N | R | Dab | T | DDab | K | C | Dab | R | Y | |||||
| 7 | Gua | V | Hyp | I | T | Y | Pen | N | R | Dab | T | DDab | K | C | Dab | R | Y | ||||
| Protein | Ligand | Than-FL Conc. (µM) | KD ± 95CI (µM) | Experiment | |
| LptAm(Ab)2.0 | Than-FL | 5 | 3.7 ± 0.5 | Direct | |
| Protein-FL | Ligand | Than-FL conc. (µM) | IC50 ± 95CI (µM) | KI ± 95CI (µM) | Experiment |
| LptAm(Ab)2.0-than-FL | Thanatin | 10 | 11.8 ± 1.5 | 3.2 ± 0.4 | Competition |
| LptAm(Ab)2.0-than-FL | 1 | 10 | 3.5 ± 0.3 | 0.9 ± 0.1 | Competition |
| LptAm(Ab)2.0-than-FL | 2 | 10 | 24.1 ± 6.3 | 6.5 ± 1.7 | Competition |
| LptAm(Ab)2.0-than-FL | 5 | 10 | 11.5 ± 0.7 | 3.1 ± 0.2 | Competition |
| LptAm(Ab)2.0-than-FL | 6 | 10 | 15.4 ± 1.3 | 4.1 ± 0.4 | Competition |
| LptAm(Ab)2.0-than-FL | 7 | 10 | 7.5 ± 0.4 | 2.0 ± 0.1 | Competition |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oi, K.K.; Moehle, K.; Schuster, M.; Zerbe, O. Early Molecular Insights into Thanatin Analogues Binding to A. baumannii LptA. Molecules 2023, 28, 4335. https://doi.org/10.3390/molecules28114335
Oi KK, Moehle K, Schuster M, Zerbe O. Early Molecular Insights into Thanatin Analogues Binding to A. baumannii LptA. Molecules. 2023; 28(11):4335. https://doi.org/10.3390/molecules28114335
Chicago/Turabian StyleOi, Kathryn K., Kerstin Moehle, Matthias Schuster, and Oliver Zerbe. 2023. "Early Molecular Insights into Thanatin Analogues Binding to A. baumannii LptA" Molecules 28, no. 11: 4335. https://doi.org/10.3390/molecules28114335