

On the Use of Deuterated Organic Solvents without TMS to Report 1H/13C NMR Spectral Data of Organic Compounds: Current State of the Method, Its Pitfalls and Benefits, and Related Issues


Abstract
:
1. Introduction
2. Search Results and Discussion
2.1. Some History
2.2. Methods for 1H/13C Chemical Shift Reference
2.3. NMR Solvent Signals as Secondary Internal References
2.4. Reference of NMR Spectra Using Method A–Current State
2.5. Proposals Based on Current Needs and Opportunities for NMR Spectrometers

3. Summary and Conclusions
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Harris, R.K.; Becker, E.D.; de Menezes, S.M.C.; Goodfellow, R.; Granger, P. NMR nomenclature. Nuclear spin properties and conventions for chemical shifts (IUPAC Recommendations 2001). Pure Appl. Chem. 2001, 73, 1795–1818. [Google Scholar] [CrossRef]
- Harris, R.K.; Becker, E.D.; de Menezes, S.M.C.; Granger, P.; Hoffman, R.E.; Zilm, K.W. Further conventions for NMR shielding and chemical shifts (IUPAC Recommendations 2008). Pure Appl. Chem. 2008, 80, 59–84. [Google Scholar] [CrossRef]
- Chalmers, B.A.; Chen, A.P.-J.; Savage, G.P.; Williams, C.M. Cubane: A New NMR Internal Standard. Aust. J. Chem. 2010, 63, 1108–1110. [Google Scholar] [CrossRef]
- Guzman, A.L.; Hoye, T.R. TMS is Superior to Residual CHCl3 for Use as the Internal Reference for Routine 1H NMR Spectra Recorded in CDCl3. J. Org. Chem. 2022, 87, 905–909. [Google Scholar] [CrossRef]
- Hatada, K.; Kitayama, T. NMR Spectroscopy of Polymers; Springer: Berlin, Germany, 2004; pp. 1–42. [Google Scholar]
- Guizzetti, S.; Schwindeman, J.A.; Daniels, D.S.B.; Douglas, J.J.; Kosanovich, A.; Zhao, W.; Kelly, C.B.; Knight, J. Some Items of Interest to Process R&D Chemists and Engineers. Org. Process Res. Dev. 2022, 26, 239–250. [Google Scholar] [CrossRef]
- Napolitano, J.G.; Yang, C.; Conklin, B.; He, Y.; Ochoa, J.L. Toward the Development of Rapid, Automated Identification Tests for Neat Organic Liquids Using Benchtop NMR Instrumentation. Anal. Chem. 2022, 94, 16095–16102. [Google Scholar] [CrossRef]
- Tiers, G.V.D. Proton nuclear resonance spectroscopy. I. Reliable shielding values by “internal referencing” with tetramethylsilane. J. Phys. Chem. 1958, 62, 1151–1152. [Google Scholar] [CrossRef]
- Bacon, R.; Maciel, G.E. Solvent Effects on the Five Shielding Constants In Tetramethylsilane and Cyclohexane. J. Am. Chem. Soc. 1973, 95, 2413–2426. [Google Scholar] [CrossRef]
- Sugiura, M.; Takao, N.; Ueji, S. A New Method for Differentiating Between Solvent Effect Mechanisms on 13C Chemical Shifts. Org. Magn. Reson. 1982, 18, 128–133. [Google Scholar] [CrossRef]
- Residual Deuterated Solvent Peak in 1H and Deuterated Peak in 13C{1H} NMR Spectra. Available online: https://cheminfographic.wordpress.com/2020/06/01/residual-deuterated-solvent-peak-in-1h-and-deuterated-peak-in-13c-nmr-spectra (accessed on 21 May 2023).
- Rosenau, C.P.; Jelier, B.J.; Gossert, A.D.; Togni, A. Exposing the Origins of Irreproducibility in Fluorine NMR Spectroscopy. Angew. Chem. Int. Ed. 2018, 57, 9528–9533. [Google Scholar] [CrossRef]
- Jackowski, K.; Makulski, W. 13C shielding scale for MAS NMR spectroscopy. Magn. Reson. Chem. 2011, 49, 600–602. [Google Scholar] [CrossRef] [PubMed]
- Ge, H.; Spletstoser, J.T.; Yang, Y.; Kayser, M.; Georg, G.I. Synthesis of Docetaxel and Butitaxel Analogues through Kinetic Resolution of Racemic β-Lactams with 7-O-Triethylsilylbaccatin III. J. Org. Chem. 2007, 72, 756–759. [Google Scholar] [CrossRef] [PubMed]
- Totani, K.; Shinoda, Y.; Shiba, M.; Iwamoto, S.; Koizumi, A.; Matsuzaki, Y.; Hirano, M. Silyl-assisted 1,2-cis-α-glucosylation for the synthesis of a triglucoside moiety in high-mannose-type oligosaccharides. RSC Adv. 2015, 5, 75918–75922. [Google Scholar] [CrossRef]
- Cloutier, M.; Lavoie, S.; Gauthier, C. C7 Epimerization of Benzylidene-Protected β-D-Idopyranosides Brings Structural In-sights into Idose Conformational Flexibility. J. Org. Chem. 2022, 87, 12932–12953. [Google Scholar] [CrossRef] [PubMed]
- Laszlo, P.; Speert, A.; Ottinger, R.; Reisse, J. Reconsideration of the Internal Tetramethylsilane Reference for Proton Magnetic Resonance Studies. J. Chem. Phys. 1968, 48, 1732–1735. [Google Scholar] [CrossRef]
- Jutila, M. Influence of an Internal Reference on 1H NMR Solvent Shifts. Determination of Reference Independent ASIS Values. Acta Chem. Scand. B 1981, 35, 503–506. [Google Scholar] [CrossRef]
- Hoffman, R.E. Variations on the chemical shift of TMS. J. Magn. Reson. 2003, 163, 325–331. [Google Scholar] [CrossRef]
- Ziessow, D.; Carroll, M. Referencing 13C NMR Medium Shifts. Ber. Bunsenges. Phys. Chem. 1972, 76, 61–64. [Google Scholar]
- Moreau-Descoings, M.C.; Goethals, G.; Seguin, J.P.; Doucet, J.P. Measurements of actual association 13C shifts: Medium effects on the TMS carbons. Spectrochim. Acta A 1987, 43, 17–20. [Google Scholar] [CrossRef]
- de Kowalewski, D.G.; de los Santos, C.; Marceca, E. Use of an Internal Reference in 13C Chemical Shift Measurements. Magn. Reson. Chem. 1990, 48, 1–4. [Google Scholar] [CrossRef]
- Crawford, R.J.; Erickson, G.L. Thermolysis of cis- and trans-4-Deuterio-3-methyl-l-pyrazolin. J. Am. Chem. Soc. 1967, 89, 3907–3908. [Google Scholar] [CrossRef]
- Cane, D.J.; Graham, W.A.G.; Vancea, L. Synthesis and nuclear magnetic resonance study of the compounds (2-X-1,3-dithiane) (X = H, Me, SiMe3, GeMe3, SnMe3, PbMe3) and their tetracarbonyliron complexes. Can. J. Chem. 1978, 56, 1538–1544. [Google Scholar] [CrossRef]
- Huckerby, T.N. Accurate Referencing of 13C NMR Spectra from Glycosaminoglycan and other Polysaccharides in Aqueous Medium. Org. Magn. Reson. 1983, 21, 67–70. [Google Scholar] [CrossRef]
- Kalinowski, H.-O.; Berger, S.; Braun, S. 13C-NMR-Spektroskopie; Georg Thieme Verlag: Stuttgart, Germany, 1984; pp. 73–74. [Google Scholar]
- Abraham, W.; Wertz, P.W.; Downing, D.T. Linoleate-rich acylglucosylceramides of pig epidermis: Structure determination by proton magnetic resonance. J. Lipid Res. 1985, 26, 761–766. [Google Scholar] [CrossRef]
- Kalinowski, H.-O.; Berger, S.; Braun, S. Carbon-13C NMR Spectroscopy; John Wiley & Sons: Chichester, UK, 1988; pp. 85–86. [Google Scholar]
- Bercaw, J.E.; Davies, D.L.; Wolczanski, P.T. Reactions of Alkyl and Hydride Derivatives of Permethylscandocene and -zirconocene with Nitriles and Amines. Catalytic Hydrogenation of tert-Butyl Cyanide with Permethylscandocene Hydride. Organometallics 1986, 5, 443–450. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Noda, S.; Nanai, N.; Okura, I.; Inoue, Y. 1H NMR Conformational Study of Viologen-Linked Porphyrins. Bull. Chem. Soc. Jpn. 1989, 62, 2152–2158. [Google Scholar] [CrossRef]
- Silverstein, R.M.; Bassler, G.C. Spectrometric Identification of Organic Compounds, 2nd ed.; John Wiley & Sons, Inc.: New York, NY, USA, 1967; p. 135. [Google Scholar]
- Granger, D.D. 13C NMR of Crosslinked Poly(Methacrylic Anhydride). In Solid State NMR of Polymers; A collection of papers presented at The Third Chemical Congress of North America (Toronto, ON, Canada, 5–10 June 1988); Mathias, L.J., Ed.; Springer Science+Business Media: New York, NY, USA, 1991; pp. 179–200. [Google Scholar]
- Deuterated NMR Solvents–Handy Reference Data. Available online: https://scs.illinois.edu/system/files/inline-files/Deuterated_Solvents.pdf (accessed on 21 May 2023).
- Kegley, S.E.; Pinhas, A.R. Problems and Solutions in Organometallic Chemistry; University Science Books: Mill Valley, CA, USA, 1986; p. 9. [Google Scholar]
- Breitmaier, E.; Voelter, W. 13C NMR Spectroscopy. Methods and Applications, 3rd ed.; VCH Verlagsgesellschaft mbH: Weinheim, Germany, 1987; p. 109. [Google Scholar]
- Deuterated NMR Solvent Table. Available online: https://web.stanford.edu/group/chem-NMR/help_docs/nmr_solvents.htm (accessed on 21 May 2023).
- CIL’s NMR Solvent Data Chart. Available online: https://www.isotope.com (accessed on 21 May 2023).
- Zerbe, O.; Jurt, S. Applied NMR Spectroscopy for Chemists and Life Scientists; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, CA, USA, 2014; pp. 26−28, 70–71. [Google Scholar]
- Budavari, S.; O’Neil, M.J.; Smith, A.; Heckelman, P.E. The Merck Index, an Encyclopedia of Chemicals, Drugs, and Biologicals, 11th ed; Merck & Co., Inc.: Rahway, NJ, USA, 1989. [Google Scholar]
- Gordon, A.J.; Ford, R.A. The Chemist’s Companion—A Handbook of Practical Data, Techniques, and References; John Wiley & Sons: New York, NY, USA, 1972; pp. 249–251. [Google Scholar]
- Pretsch, E.; Clerc, T.; Seibl, J.; Simon, W. Tabellen zur Strukturaufklärung Organischer Verbindungen mit Spektroskopischen Methoden; Springer: Berlin, Germany, 1976; pp. C250–C260. [Google Scholar]
- Breitmaier, E.; Voelter, W. 13C NMR Spectroscopy. Methods and Applications in Organic Chemistry, 2nd ed.; Verlag Chemie: Weinheim, CA, USA, 1978; p. 69. [Google Scholar]
- Handbuch der Instrumentellen Analytik NMR-Spektroskopie; Merck: Darmstadt, Germany, 1988.
- Pouchert, C.J.; Behnke, J. (Eds.) The Aldrich Library of 13C and 1H FT NMR Spectra; Aldrich Chemical Company, Inc.: Milwaukee, WI, USA, 1993; Volume 3, pp. 620A–625A. [Google Scholar]
- Spectroscopy Catalogue; Sigma-Aldrich Co.: St. Louis, MO, USA, 1997; p. 37.
- Bruno, T.J.; Svoronos, P.D.N. CRC Handbook of Basic Tables for Chemical Analysis, 2nd ed.; CRC Press: Boca Raton, FL, USA, 2003. [Google Scholar]
- Balci, M. Basic 1H- and 13C-NMR Spectroscopy; Elsevier: London, UK, 2005; p. 35. [Google Scholar]
- Bruno, T.J.; Svoronos, P.D.N. CRC Handbook of Basic Tables for Chemical Analysis, 3rd ed.; CRC Press: Boca Raton, FL, USA, 2011; pp. 484–486. [Google Scholar]
- Gottlieb, H.E.; Kotlyar, V.; Nudelman, A. NMR Chemical Shifts of Common Laboratory Solvents as Trace Impurities. J. Org. Chem. 1997, 62, 7512–7515. [Google Scholar] [CrossRef]
- Fletton, R.A.; Page, J.E. Proton Chemical Shifts for Solvents and other Simple Substances. Analyst 1971, 96, 370–373. [Google Scholar] [CrossRef]
- Fulmer, G.R.; Miller, A.J.M.; Sherden, N.H.; Gottlieb, H.E.; Nudelman, A.; Stoltz, B.M.; Bercaw, J.E.; Goldberg, K.I. NMR Chemical Shifts of Trace Impurities: Common Laboratory Solvents, Organics, and Gases in Deuterated Solvents Relevant to the Organometallic Chemist. Organometallics 2010, 29, 2176–2179. [Google Scholar] [CrossRef]
- Gottlieb, H.E.; Graczyk-Millbrandt, G.; Inglis, G.G.A.; Nudelman, A.; Perez, D.; Qian, Y.; Shuster, L.E.; Sneddon, H.F.; Upton, R.J. Development of GSK’s NMR guides—A tool to encourage the use of more sustainable solvents. Green Chem. 2016, 18, 3867–3878. [Google Scholar] [CrossRef]
- Jones, I.C.; Sharman, G.J.; Pidgeon, J. 1H and 13C NMR data to aid the identification and quantification of residual solvents by NMR spectroscopy. Magn. Reson. Chem. 2005, 43, 497–509. [Google Scholar] [CrossRef] [PubMed]
- Babij, N.R.; McCusker, E.O.; Whiteker, G.T.; Canturk, B.; Choy, N.; Creemer, L.C.; De Amicis, C.V.; Hewlett, N.M.; Johnson, P.L.; Knobelsdorf, J.A.; et al. NMR Chemical Shifts of Trace Impurities: Industrially Preferred Solvents Used in Process and Green Chemistry. Org. Process Res. Dev. 2016, 20, 661–667. [Google Scholar] [CrossRef]
- Richards, S.A.; Hollerton, J.C. Essential Practical NMR for Organic Chemistry; John Wiley & Sons: Chichester, UK, 2011; pp. 16−18, 31–36. [Google Scholar]
- Grzonka, M.; Davies, A.N. Empirical Investigation on the Reproducibility of 13C NMR Shift Values. J. Chem. Inf. Comput. Sci. 1998, 38, 1096–1101. [Google Scholar] [CrossRef]
- Pauli, G.F.; Gödecke, T.; Jaki, B.U.; Lankin, D.C. Quantitative 1H NMR. Development and Potential of an Analytical Method: An Update. J. Nat. Prod. 2012, 75, 834–851. [Google Scholar] [PubMed]
- Alexandri, E.; Ahmed, R.; Siddiqui, H.; Choudhary, M.I.; Tsiafoulis, C.G.; Gerothanassis, I.P. High Resolution NMR Spectroscopy as a Structural and Analytical Tool for Unsaturated Lipids in Solution. Molecules 2017, 22, 1663. [Google Scholar] [CrossRef]
- NMR Guidelines for ACS Journals. According to these guidelines, residual 1H impurities of deuterated NMR solvents are allowed to be applied as internal reference standards. Available online: http://pubsapp.acs.org/paragonplus/submission/acs_nmr_guidelines.pdf (accessed on 21 May 2023).
- MestReNova Version 14.3.1. Mestrelab Research S.L.: Santiago de Compostela, Spain, 2023. Available online: https://mestrelab.com/software/mnova (accessed on 21 May 2023).
- Hoffman, R.E. Standardization of chemical shifts of TMS and solvent signals in NMR solvents. Magn. Reson. Chem. 2006, 44, 606–616. [Google Scholar] [CrossRef]
- Granger, P.; Bourdonneau, M.; Assémat, O.; Piotto, M. NMR Chemical Shift Measurements Revisited: High Precision Measure-ments. Concepts Magn. Reson. A 2007, 30, 184–193. [Google Scholar] [CrossRef]
- NMR Solvent Reference Shift. Version 4.2. Available online: http://chem.ch.huji.ac.il/nmr/software/solvent.html (accessed on 21 May 2023).
- Hoffman, R.; (The Hebrew University of Jerusalem, Jerusalem, Israel). Personal Communications, 2022–2023.
- Akitt, J.W.; Mann, B.E. NMR and Chemistry: An Introduction to Modern NMR Spectroscopy, 4th ed.; CRC Press: Boca Raton, FL, USA, 2000; pp. 37–38. [Google Scholar]
- ur-Rahman, A.; Choudhary, M.I.; tul-Wahab, A. Solving Problems with NMR Spectroscopy, 2nd ed.; Elsevier Academic Press: Amsterdam, The Netherlands, 2016; pp. 22–23. [Google Scholar]
- Chemical Shift Referencing Your NMR Spectra. Available online: http://mangia.caltech.edu/NMRshifts.html (accessed on 21 May 2023).
- Chemical Shift Referencing. Available online: https://nmr.chem.ucsb.edu/protocols/refppm.html (accessed on 21 May 2023).
- Georgiou, M.; Wöckel, S.; Konstanzer, V.; Dechert, S.; John, M.; Meyer, F. Structural Variations in Tetrasilver(I) Complexes of Pyrazolate-bridged Compartmental N-Heterocyclic Carbene Ligands. Z. Naturforsch. B 2009, 64, 1542–1552. [Google Scholar] [CrossRef]
- Mulloyarova, V.V.; Ustimchuk, D.O.; Filarowski, A.; Tolstoy, P.M. H/D Isotope Effects on 1H-NMR Chemical Shifts in Cyclic Heterodimers and Heterotrimers of Phosphinic and Phosphoric Acids. Molecules 2020, 25, 1907. [Google Scholar] [CrossRef]
- Caló, F.P.; Bistoni, G.; Auer, A.A.; Leutzsch, M.; Fürstner, A. Triple Resonance Experiments for the Rapid Detection of 103Rh NMR Shifts: A Combined Experimental and Theoretical Study into Dirhodium and Bismuth−Rhodium Paddlewheel Complexes. J. Am. Chem. Soc. 2021, 143, 12473–12479. [Google Scholar] [CrossRef]
- Bakardjiev, M.; Holub, J.; Bavol, D.; Vrána, J.; Samsonov, M.A.; Růžička, A.; Růžičková, Z.; Fanfrlík, J.; Hnyk, D. Thiaborane Icosahedral Barrier Increased by the Functionalization of all Terminal Hydrogens in closo-1-SB11H11. Inorg. Chem. 2021, 60, 8428–8431. [Google Scholar] [CrossRef]
- Khalaji, M.; Paluch, P.; Potrzebowski, M.J.; Dudek, M.K. Narrowing down the conformational space with solid-state NMR in crystal structure prediction of linezolid cocrystals. Solid State Nucl. Magn. Reson. 2022, 121, 101813. [Google Scholar] [CrossRef]
- The Unified Scale for Referencing in NMR. New IUPAC Recommendations [revised (cgf): 26 July 2010]. Available online: http://www2.chem.wisc.edu/~cic/nmr/Guides/Other/Xi_chem_shift_scale.pdf (accessed on 21 May 2023).
- Automatic Heteronuclear Referencing with setref. (Varian/Agilent instruments). Available online: https://lsa.umich.edu/content/dam/chem-assets/chem-docs/setref.pdf (accessed on 21 May 2023).
- Chemical Shift Referencing. Available online: http://chem.ch.huji.ac.il/nmr/whatisnmr/chemshift.html (accessed on 21 May 2023).
- Witanowski, M.; Stefaniak, L.; Kamieński, B.; Biernat, S.; Webb, G.A. Influence of Some Paramagnetic Relaxation Reagents on Nitrogen Nuclear Shielding. J. Magn. Reson. 1981, 43, 456–462. [Google Scholar] [CrossRef]
- Batley, M.; Redmond, J.W. 31P NMR Reference Standards for Aqueous Samples. J. Magn. Reson. 1982, 49, 172–174. [Google Scholar] [CrossRef]
- Ksenofontov, A.A.; Isaev, Y.I.; Lukanov, M.M.; Makarov, D.M.; Eventova, V.A.; Khodov, I.A.; Berezin, M.B. Accurate prediction of 11B NMR chemical shift of BODIPYs via machine learning. Phys. Chem. Chem. Phys. 2023, 25, 9472–9481. [Google Scholar] [CrossRef]
- Live, D.; Harris, R.K. Hazardous references. Chem. Eng. News 1997, 75, 7. [Google Scholar]
- Zee, D.Z.; Singer, C.P.; O’Halloran, T.V. Chemical-Shift Standards for 199Hg NMR Spectroscopy, 25 Years Later. Inorg. Chem. 2022, 61, 13657–13661. [Google Scholar] [CrossRef]
- Blayney, M.B.; Nierenberg, D.; O’Halloran, T.V.; Wilcox, D.E.; Winn, J.S. Twenty-Five Years Ago−Remembering the Life and Loss of Professor Karen, E. Wetterhahn. ACS Chem. Health Saf. 2022, 29, 325–326. [Google Scholar] [CrossRef]
- Silverstein, R.M.; Webster, F.X.; Kiemle, D.J.; Bryce, L.B. Spectrometric Identification of Organic Compounds, 8th ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2015; pp. 298, 320–324. [Google Scholar]
- Smith, J.E.; Yang, H.; Gabbaï, F.P. An Electrophilic, Intramolecularly Base-Stabilized Platinum−Antimony Complex. Organometallics 2021, 40, 3886–3892. [Google Scholar] [CrossRef]
- Sleem, H.F.; Dawe, L.N.; Rahman, S.; Georghiou, P.E. Halide ion effect on the chloroform chemical shift in supramolecular complexation studies with tetra-n-butylammonium salts: A 1H NMR and X-ray study. Supramol. Chem. 2014, 26, 579–582. [Google Scholar] [CrossRef]
- Assiri, Y.; Rahman, S.; Georghiou, P.E. Halide ion effect on the 1H NMR chemical shifts of the residual protons in commonly employed deuterated solvents with tetra-n-butylammonium chloride—Part 2. Supramol. Chem. 2016, 28, 6–9. [Google Scholar] [CrossRef]
- Sroczyński, D.; Grzejdziak, A.; Nazarski, R.B. Basic Properties and Protonation Mechanism of Some Tetraaza Macrocyclic Ligands. J. Incl. Phenom. Macrocycl. Chem. 1999, 35, 251–260. [Google Scholar] [CrossRef]
- Nazarski, R.B. Assignment of pH-dependent NMR spectra of the scorpiand macrocycle: An application of titration profiles and spectral linewidths. Magn. Reson. Chem. 2003, 41, 70–74. [Google Scholar] [CrossRef]
- Hayashi, S.; Yanagisawa, M.; Hayamizu, K. Nuclear Magnetic Resonance Chemical Shifts of Pure Organic-Solvents Determined by Magic Angle Spinning. Anal. Sci. 1991, 7, 955–957. [Google Scholar] [CrossRef]
- Hoffman, R.E.; Becker, E.D. Temperature dependence of the 1H chemical shift of tetramethylsilane in chloroform, methanol, and dimethylsulfoxide. J. Magn. Reson. 2005, 176, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, R. Magnetic susceptibility measurement by NMR: 2. The magnetic susceptibility of NMR solvents and their chemical shifts. J. Magn. Reson. 2022, 335, 107105. [Google Scholar] [CrossRef]
- Jacobsen, N.E. NMR Data Interpretation Explained: Understanding 1D and 2D NMR Spectra of Organic Compounds and Natural Products; John Wiley & Sons Inc.: Hoboken, NJ, USA, 2017; Chapter 4. [Google Scholar]
- Teipel, J.; Gottstein, V.; Hölzle, E.; Kaltenbach, K.; Lachenmeier, D.W.; Kuballa, T. An Easy and Reliable Method for the Mitigation of Deuterated Chloroform Decomposition to Stabilise Susceptible NMR Samples. Chemistry 2022, 4, 776–785. [Google Scholar] [CrossRef]
- Nazarski, R.B.; Gralak, D.K.; Kudzin, Z.H. Physical Image vs. Structure Relation, 5. Multinuclear NMR Structural Study on Some Hydrazones of O,O-Dialkyl 1-Oxoalkanephosphonates. Bull. Pol. Acad. Sci. Chem. 2000, 48, 27–33. [Google Scholar]
- Spectral Database for Organic Compounds. National Institute of Advanced Industrial Science and Technology (AIST), Japan. Available online: https://sdbs.db.aist.go.jp/sdbs/cgi-bin/direct_frame_top.cgi (accessed on 21 May 2023).
- NMR Solvents. Available online: https://www.reading.ac.uk/caf/-/media/project/functions/research/chemical-analysis-facility/documents/nmrsamplepreparationpdf (accessed on 21 May 2023).
- Wietstock, S.M.; Peterson, K.A.; Goodenough-Lashua, D.A.M.; Miller, D.A.; Johnson, J.F. NMR Spectroscopy in the Undergraduate Curriculum at the University of Notre Dame. In NMR Spectroscopy in the Undergraduate Curriculum; ACS Symposium Series; American Chemical Society: Washington, DC, USA, 2013; Volume 1128, Chapter 18; p. 279. [Google Scholar]
- Leonard, J.; Lygo, B.; Procter, G. Advanced Practical Organic Chemistry, 3rd ed.; CRC Press Taylor & Francis Group, LLC: Boca Raton, FL, USA, 2013; p. 279. [Google Scholar]
- Armarego, W.L.F.; Chai, C.L.L. Purification of Laboratory Chemicals, 7th ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2013; p. 34. [Google Scholar]
- Bogdan, A.R.; Davies, N.L.; James, K. Comparison of diffusion coefficients for matched pairs of macrocyclic and linear molecules over a drug-like molecular weight range. Org. Biomol. Chem. 2011, 9, 7727–7733. [Google Scholar] [CrossRef]
- NMR Solvents & Referencing. Available online: http://www.chm.bris.ac.uk/nmr/nmrweb/nmrsolvents.htm (accessed on 21 May 2023).
- Hoye, R.C.; Anderson, G.L.; Brown, S.G.; Schultz, E.E. Total Synthesis of Clathculins A and B. J. Org. Chem. 2010, 75, 7400–7403. [Google Scholar] [CrossRef]
- Morodo, R.; Riva, R.; van den Akker, N.M.S.; Molin, D.G.M.; Jérôme, C.; Monbaliu, J.-C.M. Accelerating the end-to-end production of cyclic phosphate monomers with modular flow chemistry. Chem. Sci. 2022, 13, 10699–10706. [Google Scholar] [CrossRef]
- Yuan, S.; Liu, X.; Huang, Z.; Gui, S.; Diao, Y.; Peng, Y.-Y.; Ding, Q. Tetrabutylammonium Chloride-Induced Cascade Radical Addition/Cyclization of O-Isocyanodiaryl Amines: A Novel Protocol for the Synthesis of 11-Trifluoromethylated Dibenzodiazepines. J. Org. Chem. 2022, 87, 16542–16549. [Google Scholar] [CrossRef]
- Tsuzuki, S.; Honda, K.; Uchimaru, T.; Mikami, M.; Tanabe, K. The Interaction of Benzene with Chloro- and Fluoromethanes: Effects of Halogenation on CH/π Interaction. J. Phys. Chem. A 2002, 106, 4423–4428. [Google Scholar] [CrossRef]
- Golubev, V.A.; Gurina, D.L.; Kumeev, R.S. Self-Diffusion and Heteroassociation in an Acetone–Chloroform Mixture at 298 K. Russ. J. Phys. Chem. A 2018, 92, 75–78. [Google Scholar] [CrossRef]
- Kuballa, T.; (Chemisches und Veterinäruntersuchungsamt (CVUA), Karlsruhe, Germany). Personal Communications, 2023.
- Zhu, G.-L.; Wan, L.-S.; Peng, X.-R.; Shi, Q.-Q.; Li, X.-N.; Chen, J.-C.; Zhou, L.; Qiu, M.-H. Cytotoxic Limonoids from the Twigs and Leaves of Toona ciliata. J. Nat. Prod. 2019, 82, 2419–2429. [Google Scholar] [CrossRef]
- Choi, B.-K.; Phan, T.H.T.; Hwang, S.; Oh, D.-C.; Kang, J.S.; Lee, H.-S.; Ngo, T.D.N.; Tran, T.T.V.; Shin, H.J. Resorcinosides A and B, Glycosylated Alkylresorcinols from a Marine-Derived Strain of the Fungus Penicillium janthinellum. J. Nat. Prod. 2019, 82, 3186–3190. [Google Scholar] [CrossRef]
- Morcombe, C.R.; Zilm, K.W. Chemical shift referencing in MAS solid state NMR. J. Magn. Reson. 2003, 162, 479–486. [Google Scholar] [CrossRef]
- Pauli, G.F.; Chen, S.-N.; Lankin, D.C.; Bisson, J.; Case, R.J.; Chadwick, L.R.; Gödecke, T.; Inui, T.; Krunic, A.; Jaki, B.U.; et al. Essential Parameters for Structural Analysis and Dereplication by 1H NMR Spectroscopy. J. Nat. Prod. 2014, 77, 1473–1487. [Google Scholar] [CrossRef]
- Nazarski, R.B. Summary of DFT calculations coupled with current statistical and/or artificial neural network (ANN) methods to assist experimental NMR data in identifying diastereomeric structures. Tetrahedron Lett. 2021, 71, 152548. [Google Scholar] [CrossRef]
- Hägele, G.; Nazarski, R.B.; Schmitz, A.; Xing, S.; Janiak, C. 1H NMR spectra, structure, and conformational exchange of S-n-alkyltetrahydrothiophenium cations of some ionic liquids. Phosphorus Sulfur Silicon Relat. Elem. 2022, 197, 788–798. [Google Scholar] [CrossRef]
- Lindon, J.C.; Ferrige, A.G. Digitisation and data processing in Fourier transform NMR. Prog. Nucl. Magn. Reson. Spectrosc. 1980, 14, 27–66. [Google Scholar] [CrossRef]
- Kupka, T.; Dzięgielewski, J.O. Improvement in the evaluation of quantitative data in FT NMR spectroscopy by the convolution difference resolution enhancement (CDRE) technique. Magn. Reson. Chem. 1988, 26, 353–357. [Google Scholar] [CrossRef]
- Anet, F.A.L.; O’Leary, D.J. H-D coupling constants and deuterium isotope effects on the proton chemical shifts in partially deuteriated methanes. Tetrahedron Lett. 1989, 30, 2755–2758. [Google Scholar] [CrossRef]
- Nazarski, R.B.; Leśniak, S. Physical Image vs. Structure Relation, 4. Configuration and Conformation Determination of Some Bicyclic Lactams by 1H NMR and Theoretical Methods. Bull. Pol. Acad. Sci. Chem. 2000, 48, 19–25. [Google Scholar]
- Nazarski, R.B.; Wałejko, P.; Witkowski, S. Multi-conformer molecules in solutions: An NMR-based DFT/MP2 conformational study of two glucopyranosides of a vitamin E model compound. Org. Biomol. Chem. 2016, 14, 3142–3158. [Google Scholar] [CrossRef] [PubMed]
- Pauli, G.F.; Niemitz, M.; Bisson, J.; Lodewyk, M.W.; Soldi, C.; Shaw, J.T.; Tantillo, D.J.; Saya, J.M.; Vos, K.; Kleinnijenhuis, R.A.; et al. Toward Structural Correctness: Aquatolide and the Importance of 1D Proton NMR FID Archiving. J. Org. Chem. 2016, 81, 878–889. [Google Scholar] [CrossRef]
- McAlpine, J.B.; Chen, S.-N.; Kutateladze, A.; MacMillan, J.B.; Appendino, G.; Barison, A.; Beniddir, M.A.; Biavatti, M.W.; Bluml, S.; Boufridi, A.; et al. The value of universally available raw NMR data for transparency, reproducibility, and integrity in natural product research. Nat. Prod. Rep. 2019, 36, 35–107. [Google Scholar] [CrossRef]
- Available online: https://pubsapp.acs.org/paragonplus/submission/fid_for_publication_joceah_orlef7.pdf (accessed on 21 May 2023).
- ACS Research Data Policy. Available online: https://publish.acs.org/publish/data_policy (accessed on 21 May 2023).
- Pupier, M.; Nuzillard, J.-M.; Wist, J.; Schlörer, N.E.; Kuhn, S.; Erdelyi, M.; Steinbeck, C.; Williams, A.J.; Butts, C.; Claridge, T.D.W.; et al. NMReDATA, a standard to report the NMR assignment and parameters of organic compounds. Magn. Reson. Chem. 2018, 56, 703–715. [Google Scholar] [CrossRef]
- Nazarski, R.B.; Justyna, K.; Leśniak, S.; Chrostowska, A. A Benefit of Using the IDSCRF- over UFF-Radii Cavities and Why Joint Correlations of NMR Chemical Shifts Can Be Advantageous: Condensed Pyridines as an IEF-PCM/GIAO/DFT Case Study. J. Phys. Chem. A 2016, 120, 9519–9528. [Google Scholar] [CrossRef]
- Fuentes-Monteverde, J.C.C.; Nath, N.; Forero, A.M.; Balboa, E.M.; Navarro-Vázquez, A.; Griesinger, C.; Jiménez, C.; Rodríguez, J. Connection of Isolated Stereoclusters by Combining 13C-RCSA, RDC, and J-Based Configurational Analyses and Structural Revision of a Tetraprenyltoluquinol Chromane Meroterpenoid from Sargassum muticum. Mar. Drugs 2022, 20, 462. [Google Scholar] [CrossRef]
- Tay, B.; van Meurs, M.; Tan, J.; Ye, S.; Borgna, A.; van Herk, A.M.; Selvaratnam, S.; Wang, C.; Taniguchi, S.; Suzuki, Y.; et al. Imidazolium-Catalyzed Formation of Bisphenol A Polycarbonate with a Reduced Level of Branching. Ind. Eng. Chem. Res. 2021, 60, 17928–17941. [Google Scholar] [CrossRef]
- Aghazada, S.; Munz, D.; Heinemann, F.W.; Scheurer, A.; Meyer, K. A Crystalline Iron Terminal Methylidene. J. Am. Chem. Soc. 2021, 143, 17219–17225. [Google Scholar] [CrossRef]
- Polukeev, A.V.; Wallenberg, R.; Uhlig, J.; Hulteberg, C.P.; Wendt, O.F. Iridium-Catalyzed Dehydrogenation in a Continuous Flow Reactor for Practical On-Board Hydrogen Generation From Liquid Organic Hydrogen Carriers. ChemSusChem 2022, e202200085. [Google Scholar] [CrossRef]
- Burns, D.C.; Reynolds, W.F. Optimizing NMR Methods for Structure Elucidation: Characterizing Natural Products and Other Organic Compounds; The Royal Society of Chemistry: Cambridge, UK, 2019; Chapter 5. [Google Scholar]
- Duchowny, A.; Adams, A. Compact NMR Spectroscopy for Low-Cost Identification and Quantification of PVC Plasticizers. Molecules 2021, 26, 1221. [Google Scholar] [CrossRef]
- Levy, G.C.; Nelson, G.L. Carbon-13 Nuclear Magnetic Resonance for Organic Chemists; Wiley-Interscience: New York, NY, USA, 1972; p. 23. [Google Scholar]
- Levy, G.C.; Cargioli, J.D. Carbon-13 Chemical Shifts on the TMS Scale. J. Magn. Reson. 1972, 6, 143–144. [Google Scholar] [CrossRef]
- Jackowski, K.; Jaszuński, M.; Wilczek, M. Alternative Approach to the Standardization of NMR Spectra. Direct Measurement of Nuclear Magnetic Shielding in Molecules. J. Phys. Chem. A 2010, 114, 2471–2475. [Google Scholar] [CrossRef]
- Garbacz, P.; Jackowski, K. Referencing of 1H and 13C NMR shielding measurements. Chem. Phys. Lett. 2019, 728, 148–152. [Google Scholar] [CrossRef]
- Reichardt, C.; Welton, T. Solvents and Solvent Effects in Organic Chemistry, 4th ed.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2011; pp. 564–567. [Google Scholar]
- Reichardt, C. Solvents and Solvent Effects in Organic Chemistry, 1st ed.; Verlag Chemie GmbH: Weinheim, Germany, 1979; pp. 277–278. [Google Scholar]
- Claridge, T.D.W. High-Resolution NMR Techniques in Organic Chemistry, 2nd ed.; Elsevier Ltd.: Amsterdam, The Netherlands, 2009; p. 60. [Google Scholar]
- Claridge, T.D.W. High-Resolution NMR Techniques in Organic Chemistry, 3rd ed.; Elsevier Ltd.: Amsterdam, The Netherlands, 2016; p. 90. [Google Scholar]
- Pretsch, E. ; Bühlmann, P; Badertscher, M. Structure Determination of Organic Compounds. Tables of Spectral Data, 4th ed.; Springer: Berlin, Germany, 2009; pp. 153–154, 239–240. [Google Scholar]
- Pretsch, E. ; Bühlmann, P; Badertscher, M. Structure Determination of Organic Compounds. Tables of Spectral Data, 5th ed.; Springer GmbH Germany: Berlin, Germany, 2020; pp. 160–162, 251–252. [Google Scholar]
- Metz, K.R. Nuclear Magnetic Resonance (NMR) Spectroscopy. In Handbook of Measurement in Science and Engineering; Kutz, M., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2016; Volume 3, p. 2547. [Google Scholar]
- NMR Deuterated Solvent Properties Reference Chart. Available online: https://www.sigmaaldrich.com/PL/pl/technical-documents/technical-article/analytical-chemistry/nuclear-magnetic-resonance/nmr-deuterated-solvent-properties-reference (accessed on 21 May 2023).
- Sleem, H.F. Synthesis of New Macrocyclic Polyamides and Polysulfonamides and a Study of their Complexation Behavior using 1H-NMR and Mass Spectrometry. Ph.D. Thesis, Memorial University of Newfoundland, St. John’s, NL, Canada, 2013; pp. 144–159. [Google Scholar]
- Assiri, Y. Complexation Properties of Upper- and Lower-rim Functionalized Calix[4]arenes. M.Sc. Thesis, Memorial University of Newfoundland, St. John’s, NL, Canada, 2014. Chapter 3. pp. 76–95. [Google Scholar]
- Georghiou, P.E.; (Memorial University of Newfoundland, St. John’s, Canada). Personal Communication, 2022.
- Desando, M.A.; Lahajnar, G.; Plavec, J. Molecular Interactions and Mechanisms in the 1H NMR Relaxation of Residual CHCl3 in Deuterochloroform Solution of a Two-Chain Ionic Surfactant. J. Solution Chem. 2018, 47, 1246–1268. [Google Scholar] [CrossRef]
- Wu, Z.; Jäger, M.; Buter, J.; Minnaard, A.J. A protecting group-free synthesis of the Colorado potato beetle pheromone. Beilstein J. Org. Chem. 2013, 9, 2374–2377. [Google Scholar] [CrossRef]
- Chan, D.; Cronin, L.; Duckett, S.B.; Hupfield, P.; Perutz, R.N. Synthesis, structure and reactivity of N,O-metallacyclic (dicarbonyldiazene) platinum complexes. New J. Chem. 1998, 22, 511–516. [Google Scholar] [CrossRef]
- Popova, T.; Dymova, M.A.; Koroleva, L.S.; Zakharova, O.D.; Lisitskiy, V.A.; Raskolupova, V.I.; Sycheva, T.; Taskaev, S.; Silnikov, V.N.; Godovikova, T.S. Homocystamide Conjugates of Human Serum Albumin as a Platform to Prepare Bimodal Multidrug Delivery Systems for Boron Neutron Capture Therapy. Molecules 2021, 26, 6537. [Google Scholar] [CrossRef] [PubMed]
- Adamson, J.; Nazarski, R.B.; Jarvet, J.; Pehk, T.; Aav, R. Shortfall of B3LYP in Reproducing NMR JCH Couplings in Some Isomeric Epoxy Structures with Strong Stereoelectronic Effects: A Benchmark Study on DFT Functionals. ChemPhysChem 2018, 19, 631–642. [Google Scholar] [CrossRef] [PubMed]
- Ciechańska, M.; Jóźwiak, A.; Nazarski, R.B.; Skorupska, E.A. Unexpected Rearrangement of Dilithiated Isoindoline-1,3-diols into 3-Aminoindan-1-ones via N-Lithioaminoarylcarbenes: A Combined Synthetic and Computational Study. J. Org. Chem. 2019, 84, 11425–11440. [Google Scholar] [CrossRef] [PubMed]
- Muhamadejev, R.; Melngaile, R.; Paegle, P.; Zibarte, I.; Petrova, M.; Jaudzems, K.; Veliks, J. Residual Solvent Signal of CDCl3 as a qNMR Internal Standard for Application in Organic Chemistry Laboratory. J. Org. Chem. 2021, 86, 3890–3896. [Google Scholar] [CrossRef]
- Deuterated Chloroform. Available online: https://www.ckgas.com/wp-content/uploads/2015/04/deuterated_chloroform.pdf (accessed on 21 May 2023).
- Kawai, S. Discussion on Decomposition of Chloroform. J. Pharm. Soc. Jpn. 1966, 86, 1125–1132. [Google Scholar] [CrossRef]
- Alapi, T.; Dombi, A. Direct VUV photolysis of chlorinated methanes and their mixtures in an oxygen stream using an ozone producing low-pressure mercury vapour lamp. Chemosphere 2007, 67, 693–701. [Google Scholar] [CrossRef]
- Hungerford, N.L.; McKinney, A.R.; Stenhouse, A.M.; McLeod, M.D. Selective manipulation of steroid hydroxyl groups with boronate esters: Efficient access to antigenic C-3 linked steroid−protein conjugates and steroid sulfate standards for drug detection. Org. Biomol. Chem. 2006, 4, 3951–3959. [Google Scholar] [CrossRef]
- Miles, W.H.; Duca, D.G.; Selfridge, B.R.; Palha De Sousa, C.A.; Hamman, K.B.; Goodzeit, E.O.; Freedman, J.T. Amine-catalyzed epimerization of γ-hydroxybutenolides. Tetrahedron Lett. 2007, 48, 7809–7812. [Google Scholar] [CrossRef]
- Dias, D.A.; Urban, S. Phytochemical Investigation of the Australian Lichens Ramalina glaucescens and Xanthoria parietina. Nat. Prod. Commun. 2009, 4, 959–964. [Google Scholar] [CrossRef]
- Dialer, L.O.; Selivanova, S.V.; Müller, C.J.; Müller, A.; Stellfeld, T.; Graham, K.; Dinkelborg, L.M.; Krämer, S.D.; Schibli, R.; Reiher, M.; et al. Studies toward the Development of New Silicon-Containing Building Blocks for the Direct 18F-Labeling of Peptides. J. Med. Chem. 2013, 56, 7552–7563. [Google Scholar] [CrossRef]
- Inayoshi, T.; Hirata, K.; Watanabe, T.; Yamazaki, M. Syntheses and properties of a new series of tetrathiafulvalene derivatives incorporating fused ethyleneoxymethylene and ethylenethiomethylene units, and their charge–transfer complexes. Synth. Met. 2015, 205, 162–177. [Google Scholar] [CrossRef]
- Sedrik, R.; Bonjour, O.; Laanesoo, S.; Liblikas, I.; Pehk, T.; Jannasch, P.; Vares, L. Chemically Recyclable Poly(β-thioether ester)s Based on Rigid Spirocyclic Ketal Diols Derived from Citric Acid. Biomacromolecules 2022, 23, 2685–2696. [Google Scholar] [CrossRef]
- Gunda, P.; Russon, L.M.; Lakshman, M.K. Pd-Catalyzed Amination of Nucleoside Arylsulfonates to yield N6-Aryl-2,6-Diaminopurine Nucleosides. Angew. Chem. Int. Ed. 2004, 43, 6372–6377. [Google Scholar] [CrossRef]
- Salih, A.M.; Ahmad, M.B.; Ibrahim, N.A.; Dahlan, K.Z.H.M.; Tajau, R.; Mahmood, M.H.; Yunus, W.M.Z.W. Synthesis of Radiation Curable Palm Oil−Based Epoxy Acrylate: NMR and FTIR Spectroscopic Investigations. Molecules 2015, 20, 14191–14211. [Google Scholar] [CrossRef]
- Nguyen, T.V.T.; Wodrich, M.D.; Waser, J. Substrate-controlled C–H or C–C alkynylation of cyclopropanes: Generation of aryl radical cations by direct light activation of hypervalent iodine reagents. Chem. Sci. 2022, 13, 12831–12839. [Google Scholar] [CrossRef]
- Zhong, T.; Wolcott, M.P.; Liu, H.; Wang, J. Propionylation-modified chitin with improved solubility in green ethanol/water binary solvents for sustainable film and coating applications. J. Clean. Prod. 2020, 250, 119458. [Google Scholar] [CrossRef]
- Mascitti, A.; Lupacchini, M.; Guerra, R.; Taydakov, I.; Tonucci, L.; d’Alessandro, N.; Lamaty, F.; Martinez, J.; Colacino, E. Poly(ethylene glycol)s as grinding additives in the mechanochemical preparation of highly functionalized 3,5-disubstituted hydantoins. Beilstein J. Org. Chem. 2017, 13, 19–25. [Google Scholar] [CrossRef]
- Nyquist, R.A.; Putzig, C.L.; Hasha, D.L. Solvent Effect Correlations for Acetone: IR versus NMR Data for the Carbonyl Group. Appl. Spectrosc. 1989, 43, 1049–1053. [Google Scholar] [CrossRef]
- Handa, M.; Kataoka, M.; Wakaumi, M.; Sasaki, Y. Physical and Donor–Acceptor Properties of 3-Propyl-4-ethylsydnone. Bull. Chem. Soc. Jpn. 1997, 70, 315–320. [Google Scholar] [CrossRef]
- He, X.; Yan, Z.; Hu, X.; Zuo, Y.; Jiang, C.; Jin, L.; Shang, Y. FeCl3-Catalyzed Cascade Reaction: An Efficient Approach to Functionalized Coumarin Derivatives. Synth. Commun. 2014, 44, 1507–1514. [Google Scholar] [CrossRef]
- Ghadimi, H.; Ghani, S.A.; Amiri, I.S. Electrochemistry of Dihydroxybenzene Compounds. Carbon Based Electrodes and Their Uses in Synthesis and Sensors; Elsevier: Amsterdam, The Netherlands, 2017; Chapter 3; pp. 47–49. [Google Scholar]
- Breitmaier, E.; Voelter, W. 13C NMR Spectroscopy. Methods and Applications, 1st ed.; Verlag Chemie GmbH: Weinheim, Germany, 1974; p. 70. [Google Scholar]
- Pretsch, E.; Clerc, T.; Seibl, J.; Simon, W. Tabellen zur Strukturaufklärung organischer Verbindungen mit spektroskopischen Methoden; Springer: Berlin, Germany, 1981. [Google Scholar]
- NMR-Solvents. Available online: https://www.science-and-fun.de/tools/solvents (accessed on 21 May 2023).
- Skorupska, E.A.; Nazarski, R.B.; Ciechańska, M.; Jóźwiak, A.; Kłys, A. Dynamic 1H NMR spectroscopic study of hindered internal rotation in selected N,N-dialkyl isonicotinamides: An experimental and DFT analysis. Tetrahedron 2013, 69, 8147–8154. [Google Scholar] [CrossRef]
- Wei, J.; Zhou, X.; Dong, M.; Yang, L.; Zhao, C.; Lu, R.; Bao, G.; Hu, F. Metabolites and novel compounds with anti-microbial or antiaging activities from Cordyceps fumosorosea. AMB Express 2022, 12, 40. [Google Scholar] [CrossRef] [PubMed]
- Manwill, P.K.; Kalsi, M.; Wu, S.; Martinez Rodriguez, E.J.; Cheng, X.; Piermarini, P.M.; Rakotondraibe, H.L. Semi-synthetic cinnamodial analogues: Structural insights into the insecticidal and antifeedant activities of drimane sesquiterpenes against the mosquito Aedes aegypti. PLoS Negl. Trop. Dis. 2020, 14, e0008073. [Google Scholar] [CrossRef] [PubMed]
- HyperChemTM-Molecular Modeling System Release 8.0.10 for Windows; Hypercube, Inc.: Gainesville, FL, USA, 2011.
- Kadam, R.U.; Garg, D.; Schwartz, J.; Visini, R.; Sattler, M.; Stocker, A.; Darbre, T.; Reymond, J.-L. CH−π “T-Shape” Interaction with Histidine Explains Binding of Aromatic Galactosides to Pseudomonas aeruginosa Lectin LecA. ACS Chem. Biol. 2013, 8, 1925–1930. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2019. [Google Scholar]
- PCMODEL for Windows. Version 8.50.0. Molecular Modeling Software for Windows Operating System, Apple Macintosh OS, Linux and Unix; Serena Software: Bloomington, IN, USA, 2003.
- Rauhut, G.; Puyear, S.; Wolinski, K.; Pulay, P. Comparison of NMR Shieldings Calculated from Hartree-Fock and Density Functional Wave Functions Using Gauge-Including Atomic Orbitals. J. Phys. Chem. 1996, 100, 6310–6316. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cancès, E. The IEF Version of the PCM Solvation Method: An Overview of a New Method Addressed to Study Molecular Solutes at the QM ab Initio Level. J. Mol. Struct. Theochem 1999, 464, 211–226. [Google Scholar] [CrossRef]
- Michalik, E.; Nazarski, R.B. Synthesis, complete NMR assignments, and NOE versus GIAO data assisted ab initio modelling the overall conformations of amide 3,4′-diquinolinyl sulfides in solution. Another approach to analysis of flexible systems. Tetrahedron 2004, 60, 9213–9222. [Google Scholar] [CrossRef]
- Chemcraft, Version 1.8 (built 523b)-A graphical visualization program for quantum chemistry computation; Chemcraft: High Point, NC, USA, 2023.
- Makulski, W.; Jackowski, K. 1H, 13C and 29Si magnetic shielding in gaseous and liquid tetramethylsilane. J. Magn. Reson. 2020, 313, 106716. [Google Scholar] [CrossRef]
- Rzepiela, K.; Kaminský, J.; Buczek, A.; Broda, M.A.; Kupka, T. Electron Correlation or Basis Set Quality: How to Obtain Converged and Accurate NMR Shieldings for the Third-Row Elements? Molecules 2022, 27, 8230. [Google Scholar] [CrossRef]
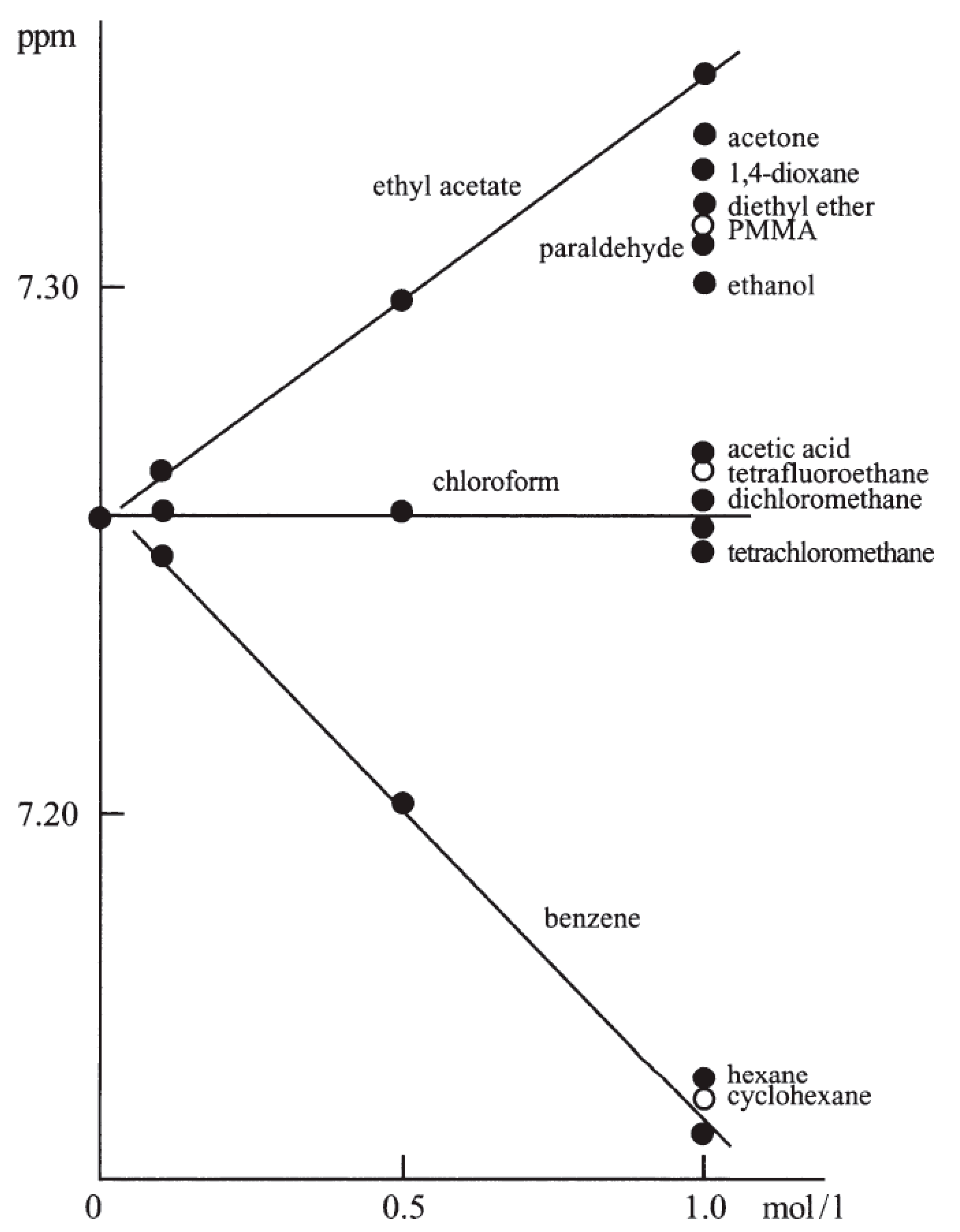



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method | Description | Notes |
|---|---|---|
| A | Residual 1H and 13C signals from NMR solvents are used as internal standards–two variants (1H and 13C) | A simplified, formally unregulated but widely applied method, the use of which is discussed in detail here |
| B | By using internal references (mainly TMS) | Standard approach–a codex of chemistry (TMS) |
| C | Use 2H known lock frequencies of the NMR solvents | Default method on all modern NMR spectrometers |
| D | An accurate IUPAC-recommended general scheme for indirect referencing based on Ξ values | Used for all NMR-observable nuclei, but only very rarely routinely for 1H and 13C |
| E | Use of external standards (typically) in coaxial tubes | Used most often for the 11B, 15N, 19F, 31P nuclei, etc. |
| CDCl3 | (CD3)2CO | (CD3)2SO | C6D6 | CD3CN | CD3OD | ||||
|---|---|---|---|---|---|---|---|---|---|
| δH | 7.260 | 2.053 | 2.502 | 7.156 | 1.939 | 3.306 | 4.848 a,b | ||
| δC | 77.01 | 29.83 | 206.15 b | 39.46 | 128.03 | 1.36 | 118.36 | 49.04 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nazarski, R.B. On the Use of Deuterated Organic Solvents without TMS to Report 1H/13C NMR Spectral Data of Organic Compounds: Current State of the Method, Its Pitfalls and Benefits, and Related Issues. Molecules 2023, 28, 4369. https://doi.org/10.3390/molecules28114369
Nazarski RB. On the Use of Deuterated Organic Solvents without TMS to Report 1H/13C NMR Spectral Data of Organic Compounds: Current State of the Method, Its Pitfalls and Benefits, and Related Issues. Molecules. 2023; 28(11):4369. https://doi.org/10.3390/molecules28114369
Chicago/Turabian StyleNazarski, Ryszard B. 2023. "On the Use of Deuterated Organic Solvents without TMS to Report 1H/13C NMR Spectral Data of Organic Compounds: Current State of the Method, Its Pitfalls and Benefits, and Related Issues" Molecules 28, no. 11: 4369. https://doi.org/10.3390/molecules28114369
APA StyleNazarski, R. B. (2023). On the Use of Deuterated Organic Solvents without TMS to Report 1H/13C NMR Spectral Data of Organic Compounds: Current State of the Method, Its Pitfalls and Benefits, and Related Issues. Molecules, 28(11), 4369. https://doi.org/10.3390/molecules28114369