Abstract
A series of nickel(II) porphyrins bearing one or two bulky nitrogen donors at the meso positions were prepared by using Ullmann methodology or more classical Buchwald–Hartwig amination reactions to create the new C-N bonds. For several new compounds, single crystals were obtained, and the X-ray structures were solved. The electrochemical data of these compounds are reported. For a few representative examples, spectroelectrochemical measurements were used to clarify the electron exchange process. In addition, a detailed electron paramagnetic resonance (EPR) study was performed to estimate the extent of delocalization of the generated radical cations. In particular, electron nuclear double resonance spectroscopy (ENDOR) was used to determine the coupling constants. DFT calculations were conducted to corroborate the EPR spectroscopic data.
1. Introduction
The selective peripheral functionalization of the porphyrin macrocycle has a long history [1,2]. The introduction of some substituents remained a synthetic challenge until recently. More specifically, introducing nitrogen nucleophiles at the periphery of porphyrin was mainly developed in the last twenty years. Several approaches were proposed, with or without metal-catalyzed reactions [3,4,5,6,7], and, for example, the classical palladium-catalyzed Buchwald–Hartwig amination reaction worked well for nucleophilic amines [8,9,10,11]. Although the introduction of many functional groups was considered a solved problem, adding bulky and/or less-nucleophilic aromatic amines with good yields at the meso positions of porphyrins remained a synthetic challenge, and this problem was really tackled over the last decade. The so-called Pd-PEPPSI complexes were chosen by the group of Osuka to prepare porphyrins bearing aromatic amines at their meso positions, which could later be fused to the aromatic core of the porphyrin [12,13,14,15]. Additionally, inexpensive copper-catalyzed Ullmann couplings were used to make these C-N connections [16]. In this particular case, porphyrins could be substituted once or twice with phenoxazine or carbazole units in good yields [17]. Such reactions are particularly useful to introduce electron donors and/or acceptors to build new molecular dyads and triads for dye-sensitized solar cells (DSSCs) or to prepare model compounds mimicking elementary steps in natural photosynthetic systems [18,19,20,21,22]. Mixed-valence compounds, with porphyrins as large π-bridges and aromatic amines as redox centers, were also studied [23,24]. The compounds described in this manuscript are shown in Figure 1 and were studied by electrochemistry, EPR spectroscopy, and DFT calculations to obtain insight into the electronic delocalization of the generated radical cation of the amines on the π-systems of the nickel(II) porphyrins.
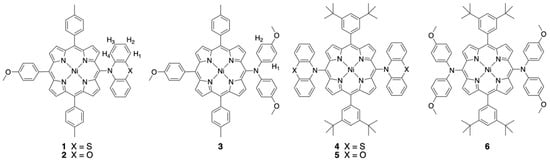
Figure 1.
Chemical structures of the nickel(II) porphyrins studied.
2. Results and Discussion
2.1. Syntheses and Characterization
Some compounds described in this study were already described earlier, but their preparation is shortly included in the synthetic procedures presented. The phenoxazine and phenothiazine-substituted porphyrins were obtained by the inexpensive Ullmann coupling between brominated porphyrin precursors and these amines (see Scheme 1) [17]. The dianisylamines were introduced by using a classical palladium-catalyzed Buchwald–Hartwig amination reaction [5].
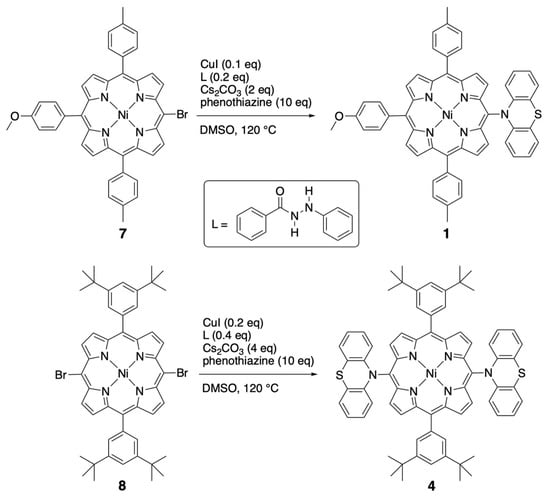
Scheme 1.
Preparation of the nickel(II) porphyrins bearing one or two meso-phenothiazine substituents.
All compounds were characterized by standard spectroscopic techniques. Single crystals were obtained for some of them, and the structures were solved. The X-ray structure of the bis-phenothiazine-substituted nickel(II) porphyrin is presented in Figure 2. As noticed before in the solid-state structures of the 5,15-bis-carbazole- or 5,15-bis-phenoxazine-substituted nickel(II) porphyrins, the porphyrin plane is almost perfectly planar, despite the fact that the metal ion inside the aromatic cavity is nickel(II). Indeed, the vast majority of the nickel(II) porphyrin structures reported in the literature are strongly ruffled because the nickel(II) ion is too small for the inner porphyrin cavity [25].
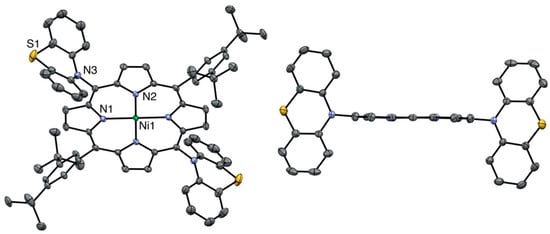
Figure 2.
Two views of the nickel(II) porphyrin 4 X-ray structure (all hydrogen atoms are omitted for clarity). Right: View showing the planarity of the porphyrin plane (two meso-aryl groups omitted for clarity).
The same planarity of the porphyrin macrocycle was observed for the 5,15-bis(dianisylamine)nickel(II) porphyrin (see Figure 3).
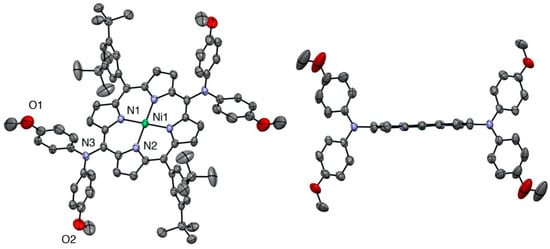
Figure 3.
Two views of the nickel(II) porphyrin 6 X-ray structure (all hydrogen atoms are omitted for clarity). Right: View showing the planarity of the porphyrin plane (two meso-aryl groups omitted for clarity).
The related nickel(II) porphyrin bearing only one phenoxazine donor was described earlier, but the X-ray structure was missing. In this case (see Figure 4), the macrocycle was slightly ruffled, and the Ni-N distances were shorter than in the two other examples (1.94–1.95 Å instead of 1.95–1.97 Å).
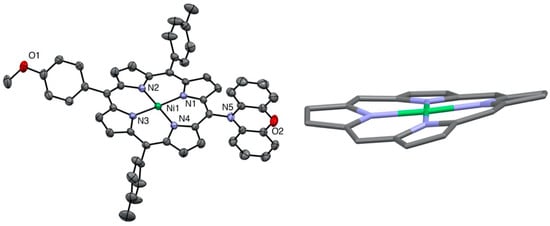
Figure 4.
X-ray structure of nickel(II) porphyrin 2 (all hydrogen atoms are omitted for clarity). Right: view showing the ruffled nickel(II) porphyrin core.
As expected for very bulky donors (phenoxazine or phenothiazine), the aromatic amines are not in the plane of the porphyrin and are almost orthogonal to this plane. The meso-carbon to nitrogen distances were all in the same range (1.43 to 1.44 Å). The main difference between dianisylamine and the two other donors resides in the flexibility of the two aromatic groups attached to the nitrogen, whereas in the case of phenoxazine and phenothiazine, the linking oxygen or sulfur atoms prevent rotation of the two phenyl groups.
2.2. Electrochemical Studies
Compounds 1, 2, 3, 4, 5, and 6 were studied by cyclic voltammetry, and the results are summarized in Table 1.

Table 1.
Electrochemical data for the six compounds a.
The reduction and oxidation steps of these molecules can be localized on the porphyrinic core and/or on the aromatic amine meso-substituents. In the case of nickel(II) porphyrins, the redox processes are localized on the aromatic moiety, and the nickel(II) ion remains formally in oxidation state 2. Generally, for nickel(II) porphyrins, two reduction and two oxidation states are present, and sometimes a third oxidation step can be observed [26]. For compounds 1–6, one or two additional oxidation states were expected due to the presence of the aromatic amines. The electrochemical data for compounds 2 and 5 were described earlier [17]. The cyclic voltammetry of compounds 1 and 4 is shown in Figure 5. For both compounds, two reduction waves corresponding to the formation of the porphyrinic radical anion and dianion were observed. In oxidation, four and five oxidation steps were, respectively, observed for compounds 1 and 4.
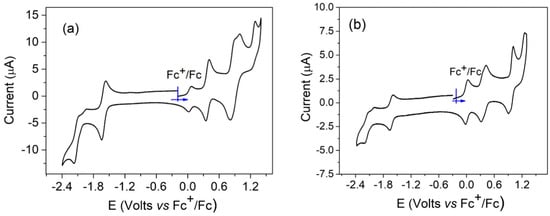
Figure 5.
Cyclic voltammetry of compounds 1 (a) and 4 (b), recorded in dichloromethane, NBu4PF6 (0.1 M), scan rate = 0.1 V/s.
To ascertain the electronic states of the oxidized species of the compounds, spectroelectrochemical studies were carried out. For the monosubstituted porphyrin 1, the first oxidation led to a new compound with almost no changes in the Soret band (from 415 to 417 nm) or the Q bands (see Figure 6). Three new bands appeared at 716, 789, and 897 nm. The very small bathochromic shift of the Soret band clearly indicated that the porphyrin ring was not involved in this redox process, and the new bands were attributed to a radical cation located on the substituent. Under the same conditions, increasing the oxidation potential to a value higher than the second oxidation modified the spectrum drastically. The intensity of the Soret band decreased drastically, showing that now the formation of the porphyrinic π-cation radical was occurring. The same behavior was observed for compound 2 (see Figure S10).
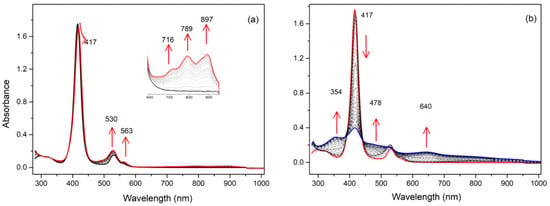
Figure 6.
UV/Vis spectra during the stepwise electrochemical oxidation of 1: (a) at the first oxidation potential; (b) at the second oxidation potential (recorded in dichloromethane, 0.1 M NBu4PF6).
Spectroelectrochemical studies of compound 3 under similar conditions led to a different result. While for compounds 1 and 2 the intensities of the Soret bands remained quasi-identical after the first oxidation step, the intensity of the Soret band of compound 3 dropped markedly after the first oxidation step (see Figure 7). In this case, the generation of the radical cation of the meso-donor nitrogen atom was partly delocalized over the aromatic porphyrinic moiety. For the three compounds bearing only one meso-donor group, the second oxidation step led to a large decrease in the Soret intensity, showing that the second oxidation steps were localized on the porphyrin aromatic ring.
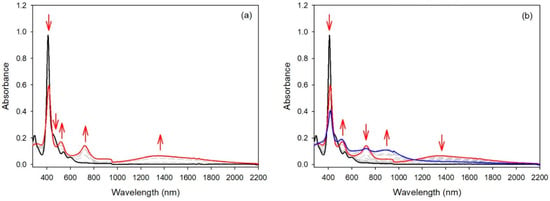
Figure 7.
UV/Vis spectra during the stepwise electrochemical oxidation of 3: (a) at the first oxidation potential; (b) at the second oxidation potential (recorded in dichloromethane, 0.1 M NBu4PF6).
The cyclic voltammetry of compound 4 (see Figure 5 right) was similar to the results described earlier for compound 5 and a bis-carbazole-substituted nickel(II) porphyrin, with small differences in potential values [17]. Again, compound 6, bearing two meso-bis-anisylamine donor groups, showed a different electrochemical behavior. The first oxidation step was reversible, but the following oxidation steps proved to be irreversible. The spectroelectrochemical study showed again that the aromatic porphyrinic core was involved in the oxidation processes because the Soret band intensity dropped markedly (see Figure 8). To confirm these observations, an EPR study and DFT calculations were carried out (vide infra).
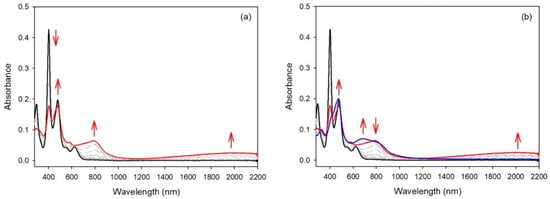
Figure 8.
UV/Vis spectra during the stepwise electrochemical oxidation of 6: (a) at the first oxidation potential; (b) at the second oxidation potential (recorded in dichloromethane, 0.1 M NBu4PF6).
2.3. EPR Studies
2.3.1. CW-EPR/ENDOR Measurements
The X-band EPR spectra of the fluid solutions of the radicals from mono-substituted nickel(II) porphyrins generated by chemical oxidation with one equivalent of AgSbF6 in CH2Cl2 at room temperature are shown in Figure 1 and in Figure S1 in SI. They exhibit a well-resolved three-line pattern centered at g = 2.003 with nitrogen hyperfine coupling constants Aiso(14N) of 19, 23, and 19 MHz for 1, 2, and 3, respectively. Additionally, each of the three lines is resolved for 1 and 2 due to further hyperfine coupling interactions with magnetically active nuclei. To gain better information about the small spin density distributions within all compounds, we used CW electron nuclear double resonance spectroscopy (ENDOR) as an alternative tool for studying radicals with many overlapping and/or incompletely resolved EPR lines. Three, four, and two additional hyperfine coupling interactions centered around the nuclear frequency of the proton are observed for 1, 2, and 3, respectively (see Figure 9, Figures S11 and S12, and Table 2). These patterns arise from the contributions of the known additional couplings described for the corresponding radical cations of phenothiazine and phenoxazine [27,28]. From these data, the EPR spectra were best simulated with the hyperfine coupling constants reported in Table 2, which were in close agreement with the DFT-computed values. Oxidations were also performed in situ electrochemically in the EPR cavity. The EPR spectra observed after a few minutes of electrolysis were identical to the spectra obtained after chemical oxidations.
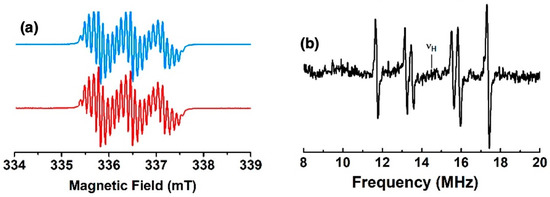
Figure 9.
(a) X-band EPR spectrum of the radical cation of 1 in CH2Cl2 fluid solution at room temperature (blue) and its simulation (red); (b) 1H ENDOR spectrum in CH2Cl2 fluid solution at 200 K.
Table 2.
Isotropic hyperfine coupling constants obtained from DfT computations compared to experimental values from CW-EPR and ENDOR spectra (values in MHz).
2.3.2. HYSCORE Results
Pulse EPR experiments were conducted using the 2D-Hyperfine sublevel correlation experiment (HYSCORE) to obtain information about the electronic structure and the delocalization of the unpaired electron all over the compounds. The use of HYSCORE spectroscopy affords the accurate assignment of various couplings with a large number of nuclei. For 1 and 2, the HYSCORE spectra revealed in the (+, +) quadrant two distinct ridges centered at 14.5 MHz (the Larmor nuclear frequency of the 1H) with no symmetric cross-peak in the (−, +) quadrant (Figure 10 and Figure S13). These results demonstrated the presence of weak hyperfine constants (|Aiso| < 2|νI|) in agreement with the CW 1H-ENDOR experiments. At low frequency, the (+,+) quadrants showed nitrogen features (νI = 1.06 MHz), which correspond also to a weak coupling situation (|Aiso| < 2|νI|) with two pairs of cross peaks at 2.1 and 3.0 MHz for 1, and are attributed to the porphyrin nitrogen atoms. In the (−, +) quadrant, a more complex pattern (i.e., the strong coupling situation where (|Aiso| > 2|νI|) is present and indicative of a hyperfine coupling with a more important anisotropy probably from a nitrogen nucleus. To interpret the low frequency part of the HYSCORE spectra, the 14N hyperfine and quadrupole data were computed, and these values were used as a starting point for the simulation. The corresponding 14N hyperfine and nuclear quadrupole couplings are given in Table 3 with the simulations shown in Figure S14 and support the presence of a second nitrogen atom with a hyperfine tensor and a quadrupole contribution very close to nitrogen from porphyrin with the phenothiazine and phenoxazine substituents. In contrast, the HYSCORE spectrum of 3 recorded at 80 K is rather different (Figure S13). A similar shape is observed at low frequency in the (+, +) quadrant, but the pattern visible in the (−, +) quadrant is different. This signal could originate from nitrogen atoms of the porphyrin with close hyperfine coupling values, as suggested by the DFT calculation. From this information, the following trends in the spin density distributions have emerged. The spin distribution over the Ni porphyrin is slightly different when species 1 and 2 are compared to 3. For the latter, the computed values (see DFT calculations in SI) predict a non-zero spin density on the four 14N porphyrin, whereas only the two nitrogen atoms close to the meso group bear spin density for 1 and 2. These results reflect the enhanced electron donating ability of the bis(p-anisyl)-amine substituent and the flexibility of this amine compared to the rigid phenoxazine and phenothiazine. Consequently, enhanced radical delocalization was observed in compound 3 when compared to compounds 1 and 2.
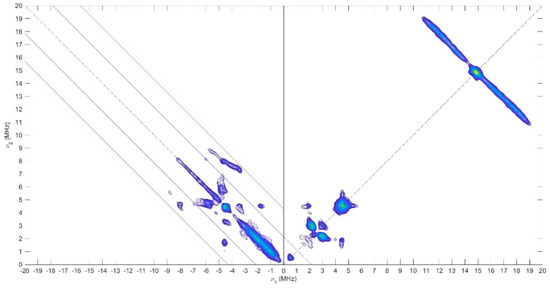
Figure 10.
X-Band HYSCORE spectrum of 1 recorded at 70 K and collected at 349.4 mT, microwave frequency 9.70 GHz, τ = 136 ns.
Table 3.
14N hyperfine and quadrupolar coupling parameters obtained from DFT computations and experimental HYSCORE spectra (values in MHz) a.
2.3.3. Magnetic Properties
The reaction of 4 and 5 with two equivalents of AgSbF6 in CH2Cl2 fluid solution yielded EPR spectra with five transitions consistent with two spin species and a 14N hyperfine coupling constant approximatively equal to the half of the hyperfine coupling constant of the monoradical species (see Figure 11 and Figure S16). This situation corresponds to the strong exchange limit and means that the exchange coupling is much higher than the hyperfine coupling constant. In contrast, one line was observed for 6 with a linewidth of 10 G and thus also suggested the formation of the biradical. The EPR spectra of the biradicals could also be obtained upon electrochemical generation if the electrolyses were carried out around the second oxidation potential. For compound 4, a spectrum corresponding to a mixture of the mono- and biradical species (see Figure S17) was obtained. The very small differences between the first and second oxidation potentials for compounds 5 and 6 led directly to the doubly oxidized species, with EPR spectra identical to those obtained by chemical oxidation.
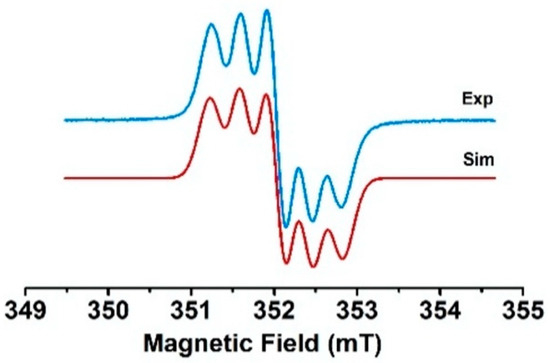
Figure 11.
Experimental EPR spectrum of the dication of compound 4 in CH2Cl2 fluid solution at room temperature and its simulation.
The frozen solution EPR spectra of three bis compounds recorded at 4 K exhibit only one intense symmetrical line (ΔBPP = 12 G) centered on g = 2.00 without any fine structure characteristic of the triplet state, but a forbidden ΔMs = ±2 transition was detected at half field. This definitely indicates a biradical nature. To compare the exchange interaction, the temperature dependence of the EPR signal intensity (I) was measured. The intensity is known to be proportional to the magnetic susceptibility. Note that the EPR susceptibility is assessed as usual with the integrated intensity for the ΔMs = ±1 line with a high S/N ratio. This is correct as long as the line shape does not vary. In all three cases, the product IT decreases when the temperature is below 20 K, indicating the presence of an antiferromagnetic coupling (see Figure 12 for 6 and Figure S18). To obtain the exchange coupling constant (J/kB where kB is the Boltzmann constant), the observed curves were adjusted with a Bleaney–Bowers law with the additional contribution of a Curie law to account for the presence of the single radical. The estimated J/kB values of −8 K for 4 and −8 K for 5 were very close, while the value of 6 was in the order of −30 K and showed much better magnetic communication between the two bis(p-anisyl)amine meso-substituents.
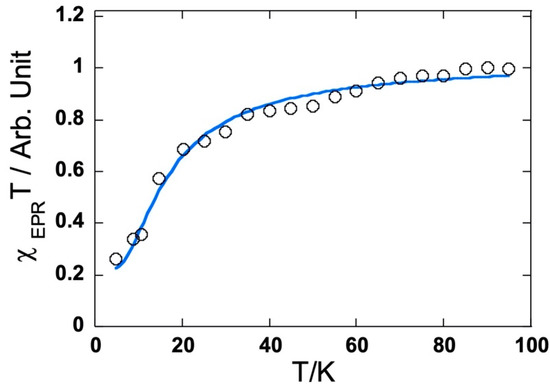
Figure 12.
Temperature dependence of the EPR susceptibility (χT product) in CH2Cl2 frozen solution for the dication of 6.
3. Materials and Methods
3.1. Materials
All commercial reagents (Aldrich, St. Louis, MI, USA; Fisher Scientific, Hampton, NH, USA; Merck, Rahway, NJ, USA) were used as received. All solvents were dried and freshly distilled before use. Column chromatography was performed using silica gel (Merck 0.40–0.63 nm).
3.2. Preparation of the Compounds
5,15-Diarylporphyrin and 5,10,15-triarylporphyrin were prepared by well-established porphyrin synthetic procedures, followed by bromination of the remaining free meso positions to obtain 7 and 8. The syntheses and characterizations of these starting materials were described previously. Similarly, the syntheses of the porphyrins 2 and 5 bearing one or two phenoxazine donors at the meso positions were described previously [17].
MonoPhenothiazine 1. A degassed solution of brominated nickel(II) porphyrin 7 (53 mg, 72 μmol), phenothiazine (136 mg, 10 eq.), CuI (2 mg, 0.1 eq.), N′-phenylbenzoylhydrazine (3 mg, 0.2 eq.), Cs2CO3 (45 mg, 2 eq.) in DMSO (1 mL) was heated under argon at 120 °C for 16 h. After cooling, the reaction mixture was diluted in dichloromethane (100 mL), washed twice with a saturated NaHCO3 aqueous solution (50 mL), and finally twice with water. The organic phase was dried over Na2SO4, and the solvents evaporated. After chromatographic purification (silica gel, CH2Cl2 then CHCl3) and crystallization (CH2Cl2, n-hexane, MeOH), the desired compound 1 was isolated as a purple solid (18 mg, 21 μmol, 29%). 1H NMR (300 MHz, CDCl3, 25 °C), δ (ppm) = 9.29 (d, J = 5.0 Hz, 2H, pyrr.), 8.77 (d, J = 5.0 Hz, 2H, pyrr.), 8.74 (m, 4H, pyrr.), 7.91 (d, J = 8.6 Hz, 2H, Ho-Anis), 7.87 (d, J = 7.9 Hz, 4H, Ho-tolyl),7.45 (d, J = 7.9 Hz, 4H, Hm-tolyl), 7.20 (d, J = 8.6 Hz, 2H, Hm-Anis), 7.12 (dd, J = 7.6 and 1.5 Hz, 2H, Ha), 6.73 (ddd, J = 7.6, 7.6, and 1.0 Hz, 2H, Hb), 6.46 (ddd, J = 8.2, 7.6, and 1.5 Hz, 2H, Hc), 5.79 (dd, J = 8.2 and 1.0 Hz, 2H, Hd), 4.03 (s, 3H, OCH3), 2.62 (s, 6H, CH3). MS (Maldi-TOF, dithranol): Calcd for [C53H37N5NiOS]+•: 849.21; found 849.19. UV-Vis. (CH2Cl2): λmax = 416 nm (ε = 24,5000 M−1.cm−1), 530 (19,000). Anal. Calcd for C53H37N5NiOS•1/2 CH3OH: C 74.14, H 4.54, N 8.08; found C 74.16, H 4.51, N 8.00.
BisPhenothiazine 4. A degassed solution of dibrominated nickel(II)porphyrin 8 (101 mg, 112 μmol), phenothiazine (220 mg, 10 eq.), CuI (4 mg, 0.2 eq.), N′-phenylbenzoylhydrazine (9 mg, 0.4 eq.), and Cs2CO3 (144 mg, 4 eq.) in DMSO (4 mL) was heated under argon at 120 °C for 16 h. After cooling, the reaction mixture was diluted in dichloromethane (100 mL), washed twice with a saturated NaHCO3 aqueous solution (50 mL), and finally twice with water. The organic phase was dried over Na2SO4, and the solvents evaporated. After chromatographic purification (silica gel, CH2Cl2 then CHCl3) and crystallization (CH2Cl2, n-hexane, MeOH), the desired compound 4 was isolated as a purple solid (32 mg, 28 μmol, 25%). 1H NMR (400 MHz, CDCl3, 25 °C), δ (ppm) = 9.38 (d, J = 5.0 Hz, 4H, pyrr.), 8.81 (d, J = 5.0 Hz, 4H, pyrr.), 7.87 (d, J = 1.9 Hz, 4H, Ho-Ar), 7.74 (t, J = 1.9 Hz, 2H, Hp-Ar), 7.15 (m, 4H, Ha), 6.75 (br, 4H, Hb), 6.50 (br, 4H, Hc), 5.80 (br, 4H, Hd), 1.47 (s, 36 H, CH3). MS (Maldi-TOF, dithranol): Calcd for [C72H66N6NiS2]+•: 1136.41; found 1136.40. UV-Vis. (CH2Cl2): λmax = 414 nm (ε = 248,000 M−1.cm−1), 533 (19,000), 566 (11,000). Anal. Calcd for C72H66N6NiS2: C 75.98, H 5.84, N 7.38; found C 75.70, H 5.90, N 7.32. Crystal data. From CH2Cl2-CH3OH, C74H70Cl4N6NiS2, M = 1307.99 g.mol−1, 0.25 × 0.20 × 0.10 red prisms, triclinic, space group P-1, a = 9.581(5) Å, b = 12.043(5) Å, c = 15.226(5) Å, α = 69.453° (5), β = 84.502° (5), γ = 83.241° (5), V = 1630.8 (12) Å3, Z = 1, T = 173 K, MoKα = 0.71073, 1.81 < θ < 30.05, 35,851 reflections measured, 9530 unique reflections, R1 = 0.0579, wR2 = 0.1505, GoF = 1.033. CCDC Nr 2261554.
Monophenoxazine 2. The preparation and characterization of this compound were described previously [17]. Crystal data. C53H37N5NiO2, M = 834.58 g.mol−1, 0.500 × 0.380 × 0.160 mm red, triclinic, space group P-1, a = 9.3928 (6) Å, b = 16.1279 (11) Å, c = 16.7580 (11) Å, α = 74.871° (2), β = 83.3960° (10), γ = 87.470° (2), V = 2434.1 (3) Å3, Z = 2, T = 173 K, MoKα = 0.71073, 1.308 < θ < 28.125, 56,323 reflections measured, 11,860 unique reflections, R1 = 0.0515, wR2 = 0.1166, GoF = 1.048. CCDC Nr 2261556.
Compound 3. A Schlenk tube filled with bis(4-methoxyphenyl)-amine (298 mg, 1.3 mmol), tBuOK (1.5 g, 13.5 mmol), Pd(OAc)2 (14 mg, 0.06 mmol), rac-BINAP (58 mg, 0.09 mmol), and 18-crown-6 (15 mg, 0.06 mmol) was dried under vacuum for two hours and backfilled with argon. A degassed solution of 5-iodo-10,20-ditolyl-15-(p-anisyl)-nickel(II) porphyrin (100 mg, 0.13 mmol) in freshly distilled THF (60 mL) was added to the previously dried solids, and the reaction mixture was stirred at 70 °C under argon. After 16 h, the mixture was cooled to room temperature and the solvent evaporated. Chromatographic purification (silica gel, CH2Cl2/C6H12 3/7) followed by crystallization (CH2Cl2, MeOH) afforded the dehalogenated porphyrin (30 mg, 35%) and the desired product 3 (35 mg, 0.04 mmol, 31%). 1H NMR (500 MHz, CDCl3, 25 °C), δ (ppm) = 9.04 (d, J = 5.0 Hz, 2H, pyrr.), 8.67 (d, J = 5.0 Hz, 2H, pyrr.), 8.66 (d, J = 5.0 Hz, 2H, pyrr.), 8.64 (d, J = 5.0 Hz, 2H, pyrr.), 7.87 (d, J = 6.7 Hz, 2H, Ho-Anis), 7.82 (d, J = 8.0 Hz, 4H, Ho-Tolyl), 7.42 (d, J = 8.0 Hz, 4H, Hm-Tolyl), 7.18 (d, J = 6.7 Hz, 2H, Hm-Anis), 7.12 (d, J = 6.7 Hz, 8H, HN-Anis), 6.70 (d, J = 6.7 Hz, 8H, HN-Anis), 4.02 (s, 4H, OCH3), 3.68 (s, 6H, OCH3), 2.60 (s, 6H, CH3). 13C (125 MHz, CDCl3): δ (ppm) = 159.3, 153.7, 164.1, 145.2, 143.2, 143.1, 142.0, 137.7, 137.4, 134.7 (CH), 133.6 (CH), 133.2, 133.1 (CH), 132.1 (CH), 132.0 (CH), 130.8 (CH), 127.6 (CH), 123.0 (CH), 122.0, 119.1, 119.0, 114.5 (CH), 112.4 (CH), 55.6 (OCH3), 55.5 (OCH3), 21.5 (CH3). UV-Vis. (CH2Cl2): λmax = 410 nm (ε = 152,000 M−1.cm−1), 470 (28,000sh), 541 (12,800), 595 (7000). Anal. Calcd for C55H43N5NiO3: C 75.01, H 4.92, N 7.95; found C 74.62, H 4.93, N 7.76.
Compound 6. A Schlenk tube filled with bis(4-methoxyphenyl)-amine (415 mg, 1.80 mmol), tBuOK (2.0 g, 18 mmol), Pd(OAc)2 (21 mg, 0.09 mmol), rac-BINAP (78.5 mg, 0.13 mmol), and 18-crown-6 (20 mg, 0.08 mmol) was dried under vacuum for one hour and backfilled with argon. A degassed solution of 5,15-diodo-10,20-di-(3,5-di-tBu-phenyl)-nickel(II) porphyrin (180 mg, 0.18 mmol) in freshly distilled THF (60 mL) was added to the previously dried solids, and the reaction mixture was stirred at 70 °C under argon. After 15 h, the mixture was cooled to room temperature and the solvent evaporated. Chromatographic purification (silica gel, CH2Cl2/C6H12 3/7) followed by crystallization (CH2Cl2, n-hexane, MeOH) afforded green crystals (49 mg, 0.041 mmol, 23%). 1H NMR (500 MHz, CDCl3, 25 °C), δ (ppm) = 9.00 (d, J = 5.0 Hz, 4H, pyrr.), 8.59 (d, J = 5.0 Hz, 4H, pyrr.), 7.75 (d, J = 1.7 Hz, 4H, Ho-Ar), 7.65 (t, J = 1.7 Hz, 2H, Hp-Ar), 7.12 (d, J = 9.1 Hz, 8H, HAnis), 6.71 (d, J = 9.1 Hz, 8H, HAnis), 3.68 (s, 12H, OCH3), 1.42 (s, 36H, CH3). 13C (125 MHz, CDCl3): δ (ppm) = 153.6, 148.9, 146.0, 145.3, 142.4, 139.5, 133.4 (CH), 130.6 (CH), 128.4 (CH), 122.9 (CH), 122.2, 121.3 (CH), 120.3, 114.5 (CH), 55.5 (OCH3), 35.0, 31.7 (CH3). MS (ESI-TOF-HRMS): Calcd for [C76H78N6NiO4]+•: 1196.5433; found 1196.5439. UV-Vis. (CH2Cl2): λmax = 401 nm (ε = 115,000 M−1.cm−1), 477 (53,000), 545 (10,500), 622 (11,300). Anal. Calcd for C76H78N6NiO4•H2O: C 75.06, H 6.63, N 6.91; found C 75.11, H 6.48, N 6.93. Crystal data. From chlorobenzene and isopropanol, C76H78Cl4N6NiO4, M = 1198.15 g.mol−1, 0.500 × 0.500 × 0.500 red, triclinic, space group P-1, a = 10.7094 (10) Å, b = 14.9585 (16) Å, c = 20.634 (2) Å, α = 97.122° (5), β = 90.811° (5), γ = 99.452° (6), V = 3233.4 (6) Å3, Z = 2, T = 173 K, CuKα = 1.54178, 3.932 < θ < 66.975, 44,205 reflections measured, 11,200 unique reflections, R1 = 0.088, wR2 = 0.2262, GoF = 1.030. CCDC Nr 2261547.
3.3. Electrochemistry
Electrochemical measurements were carried out in CH2Cl2 containing NBu4PF6 (0.1 M) in a classical three-electrode cell by cyclic voltammetry (CV) and rotating-disk voltammetry (RDV). The working electrode was a glassy carbon disk (3 mm in diameter), the auxiliary electrode was a Pt wire, and the pseudo-reference electrode was a Pt wire. The cell was connected to an Autolab PGSTAT30 potentiostat (Eco Chemie, Utrecht, Holland) driven by GPSE software running on a personal computer. All potentials are given vs. Fc+/Fc used as internal standards and are uncorrected from ohmic drop. Spectroelectrochemical investigations were carried out with a MCS600 Carl Zeiss photodiodes array spectrometer (MCS 601 UV-vis and MCS 611 NIR2.2 modules) controlled by the Aspect Plus software. A home-made Optical Transparent Thin Layer Electrode (OTTLE) spectroelectrochemical cell was placed in the light beam. The three electrodes of the cell (a platinum grid as the working electrode, platinum wire as the counter electrode, and silver/silver chloride as the reference electrode) were connected to an Autolab 302 potentiostat/galvanostat controlled by the GPES software running on a PC computer.
3.4. EPR Measurements
CW EPR experiments were performed using an X-band (9–10 GHz) Bruker ESP300 spectrometer equipped with a standard rectangular cavity (TE102). A Bruker Elexis E580 spectrometer operating at the X-band (9–10 GHz) equipped with an Oxford instrument CF935 cryostat, an ITC4 temperature controller, and a 1 kW TWT amplifier was used for pulsed EPR measurements. 2D HYSCORE (hyperfine sublevel correlation spectroscopy) spectra were measured by using a pulse sequence (π/2–τ–π/2–t1–π-t2–π/2–τ echo), with pulse lengths of 16 and 32 ns for the π/2 and π pulses, respectively. The experiments were performed with two different delay values τ (136 and 200 ns). Numerous unwanted pulse echoes were removed by four-step phase cycling. The intensity of the echo was measured after the fourth pulse with variable t1 and t2 values, keeping τ constant. The collected ESEEM and HYSCORE data were Fourier transformed by using the spectrum manipulation routines available within the Bruker program: subtraction of the relaxation decay, apodization (Hamming window), and zero filling. The HYSCORE spectra were recorded with a magnetic field corresponding to the maximum intensity of the radical signal in the two-pulse field-swept EPR spectra. The EPR and HYSCORE spectra were simulated using Easyspin V5.2.28 using the MATLAB R2020a version [29].
3.5. Computational Methods
The ORCA program package was used to perform all theoretical calculations based on density functional theory (DFT) [30]. Starting from the experimentally determined solid-state structures, all complexes were subjected to full geometry optimization using the B3LYP functional [31,32] in combination with the 6–31 g* basis sets [33,34,35]. Increased integration grids and tight SCF convergence criteria were used in the calculations. Cartesian coordinates of the DFT-optimized structures are provided in the SI. Solvent effects were accounted for according to the experimental conditions, and dichloromethane was used as a solvent (ε = 80) within the framework of the conductor-like polarizable continuum model, COSMO [36]. Single point calculations were conducted to compute electronic structures and predict EPR parameters using the B3LYP functional together with the CP(PPP) basis set for the metal center [37] and the EPR-II basis set for other atoms [38]. Optimized geometries as well as electronic structures were visualized using the program Chemdraft.
4. Conclusions
This work demonstrates that the introduction of aromatic amines on the free meso positions of diarylporphyrins via either Buchwald or Ullmann methods is very efficient. Radicals can be generated on the nitrogen atoms by electrochemistry and chemical oxidation. The degree of communication between the radicals through the π-electron density of the porphyrin ring strongly depends on the aromatic amines’ ability to contribute to their delocalization. The higher global flexibility of amine substituents able to freely rotate along both the N-Cmeso and the N-Aryl bonds allows a more efficient delocalization of the radicals.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/molecules28114405/s1, Figure S1: 1H NMR spectrum of compound 1; Figure S2: MALDI-TOF MS of 1 (top) simulation (bottom); Figure S3: 1H NMR spectrum of compound 4; Figure S4: MALDI-TOF MS of 4 (top) simulation (bottom); Figure S5: 1H NMR of compound 3 (top) and aromatic area (bottom); Figure S6: 13C NMR of compound 3 (top) and DEPT (bottom); Figure S7: 1H NMR of compound 6; Figure S8: HRMS (ESI-TOF) of compound 6 (top) and simulation (bottom); Figure S9: 13C NMR of compound 6 (top) and DEPT (bottom); Figure S10: UV/Vis spectra during the stepwise electrochemical oxidation of 2: (a) at the first oxidation potential; (b) at the second oxidation potential (recorded in dichloromethane, 0.1 M NBu4PF6); Figure S11: (a) X-band EPR spectrum of 2 in CH2Cl2 fluid solution at room temperature (blue) and its simulation (red) (b) 1H-ENDOR spectrum of 2 in CH2Cl2 fluid solution at 200K; Figure S12: (a) X-band EPR spectrum of 3 in CH2Cl2 fluid solution at room temperature (b) 1H-ENDOR spectrum of 3 in CH2Cl2 fluid solution at 200 K; Figure S13: (++) and (+ −) quadrants of the 1H and 14N of X-band HYSCORE spectrum at 80 K of (a) 2 (b) 3 showing the location of 14N cross-peaks weakly coupled nitrogen nuclei, respectively, and 1H cross-ridges. Microwave frequency of 9.71 and 9.74 GHz respectively, magnetic field 350.0 and 352.5 mT respectively and time τ of 136 ns; Figure S14: Experimental (blue) and simulated (red) X-band 14N–HYSCORE spectra of (a) 1, (b) 2 and (c) 3. The simulations are carried out using parameters given in Table 2 in the main text; Figure S15: Field-swept EPR spectra at 80 K of (a) 1 (b) 2 and (c) 3. Experimental (black) and simulated (red); Figure S16: Experimental EPR spectrum of (a) 5 and (b) 6 in CH2Cl2 fluid solution at room temperature; Figure S17: Experimental EPR spectrum of mixture 2 and 4 in CH2Cl2 fluid solution generated by electrolysis at room temperature. Simulated spectrum was obtained by an admixture of 23% of monoradical and 77% of biradical; Figure S18: Temperature dependence of the EPR susceptibility (χT product) in CH2Cl2 frozen solution for (a) 5 and (b) 4; Table S1: 14N hyperfine coupling parameters in MHz obtained from simulations and experimental Field-swept EPR spectra; and DFT calculations for compounds 1, 2, and 3.
Author Contributions
Conceptualization, J.W. and R.R.; syntheses, M.-A.C., J.H. and A.M.V.M.P.; electrochemistry, L.R.; EPR studies, K.M., N.P., V.M. and S.C.; DFT, M.O.; writing—original draft preparation, S.C. and R.R.; writing—review and editing, J.W., S.C., L.R. and R.R.; funding acquisition, S.C. and R.R. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by IR Infrananalytics FR205.
Acknowledgments
We thank L. Karmazin and C. Bailly from the X-ray facilities of the University of Strasbourg for solving the structures. We thank the CNRS and the University of Strasbourg for continuous financial support. We thank also the FRC-Labex CSC (PhD fellowship to MAC) and the Fondation Recherche Chimie (PhD fellowship to KM). Financial support from the IR Infrananalytics FR2054 for conducting the research is gratefully acknowledged.
Conflicts of Interest
The authors declare no conflict of interest.
Sample Availability
Samples of all compounds are available from the authors.
References
- Kadish, K.M.; Smith, K.M.; Guilard, R. (Eds.) The Porphyrin Handbook; Academic Press: San Diego, CA, USA, 2000; Volume 1, pp. 1–400. [Google Scholar]
- Hiroto, S.; Miyake, Y.; Shinokubo, H. Synthesis and functionalization of porphyrins through organometallic methodologies. Chem. Rev. 2017, 117, 2910–3043. [Google Scholar] [CrossRef]
- Balaban, M.C.; Chappaz-Gillot, C.; Canard, G.; Fuhr, O.; Roussel, C.; Balaban, T.S. Metal catalyst-free amination of meso-bromoporphrins: An entry to supramolecular porphyrinoid frameworks. Tetrahedron 2009, 65, 3733–3739. [Google Scholar] [CrossRef]
- Devillers, C.H.; Hebié, S.; Lucas, D.; Cattey, H.; Clément, S.; Richeter, S. Aromatic nucleophilic substitution (SNAr) of meso-nitroporphyrin with azide and amines as an alternative metal catalyst free synthetic approach to obtain meso-N-substituted porphyrins. J. Org. Chem. 2014, 79, 6424–6434. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Castillo, P.; Buchwald, S.L. Applications of palladium-catalyzed C-N cross-coupling reactions. Chem. Rev. 2016, 116, 12564–12649. [Google Scholar] [CrossRef] [PubMed]
- Imahori, H.; Matsubara, Y.; Iijima, H.; Umeyama, T.; Matano, Y.; Ito, S.; Niemi, M.; Tkachenko, N.V.; Lemmetyinen, H. Effects of meso-diarylamino group of porphyrins as sensitizers in dye-sensitized solar cells on optical, electrochemical, and photovoltaic properties. J. Phys. Chem. C 2010, 114, 10656–10665. [Google Scholar] [CrossRef]
- Nowak-Krol, A.; Gryko, D.T. Oxidative aromatic coupling of meso-arylamino-porphyrins. Org. Lett. 2013, 15, 5618–5621. [Google Scholar] [CrossRef] [PubMed]
- Fields, K.B.; Ruppel, J.V.; Snyder, N.L.; Zhang, X.P. Handbook of Porphyrin Science; Kadish, K.M., Smith, K.M., Guilard, R., Eds.; World Scientific: Singapore, 2010; Volume 3, pp. 367–427. [Google Scholar]
- Kawano, S.; Kawada, S.; Kitagawa, Y.; Teramoto, R.; Nakano, M.; Tanaka, K. Near-infrared absorption by intramolecular charge-transfer transition in 5,10,15,20-tetra-(N-carbazolyl)porphyrin through protonation. Chem. Commun. 2019, 55, 2992–2995. [Google Scholar] [CrossRef] [PubMed]
- Kawano, S.; Kawada, S.; Matsubuchi, A.; Tanaka, K. Metalloporphyrins substituted with N-carbazolyl groups quadruply at meso positions. J. Porphyr. Phthalocyanines 2022, 26, 140–146. [Google Scholar] [CrossRef]
- Pawlicki, M.; Hurej, K.; Kwiecinska, K.; Szterenberg, L.; Latos-Grazynski, L. A fused meso-aminoporphyrin: A switchable near-IR chromophore. Chem. Commun. 2015, 51, 11362–11365. [Google Scholar] [CrossRef]
- Fukui, N.; Cho, W.Y.; Lee, S.; Tokuji, S.; Kim, D.; Yorimitsu, H.; Osuka, A. Oxidative fusion reactions of meso-(diarylamino)porphyrins. Angew. Chem. Int. Ed. 2013, 52, 9728–9732. [Google Scholar] [CrossRef]
- Susuki, Y.; Fukui, N.; Murakami, K.; Yorimitsu, H.; Osuka, A. Amination of meso-bromoporphyrins and haloanthracenes with diarylamines catalyzed by a palladium-PEPPSI complex. Asian J. Org. Chem. 2013, 2, 1066–1071. [Google Scholar] [CrossRef]
- Fukui, N.; Lee, S.K.; Kato, K.; Shimizu, D.; Tanaka, T.; Lee, S.; Yorimitsu, H.; Kim, D.; Osuka, A. Regioselective phenylene-fusion reactions of Ni(II)-porphyrins controlled by an electron-withdrawing meso-substituent. Chem. Sci. 2016, 7, 4059–4066. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Osuka, A.; Song, J. Pd-catalyzed cross coupling strategy for functional porphyrin arrays. ACS Cent. Sci. 2020, 6, 2159–2178. [Google Scholar] [CrossRef] [PubMed]
- Haumesser, J.; Gisselbrecht, J.-P.; Weiss, J.; Ruppert, R. Carbene Spacers in bis-porphyrinic scaffolds. Chem. Commun. 2012, 48, 11653–11655. [Google Scholar] [CrossRef] [PubMed]
- Haumesser, J.; Pereira, A.M.V.M.; Gisselbrecht, J.-P.; Merahi, K.; Choua, S.; Weiss, J.; Cavaleiro, J.A.S.; Ruppert, R. Inexpensive and efficient Ullmann methodology to prepare donor-substituted porphyrins. Org. Lett. 2013, 15, 6282–6285. [Google Scholar] [CrossRef]
- Esdaile, L.J.; Senge, M.O.; Arnold, D.P. New palladium catalysed reactions of bromoporphyrins: Synthesis and crystal structures of nickel(II)complexes of primary 5-aminoporphyrin, 5,5′-bis(porphyrinyl) secondary amine, and 5-hydroxyporphyrin. Chem. Commun. 2006, 4192–4194. [Google Scholar] [CrossRef]
- Pereira, A.M.V.M.; Neves, M.G.P.M.S.; Cavaleiro, J.A.S.; Jeandon, C.; Gisselbrecht, J.-P.; Choua, S.; Ruppert, R. Diporphyrinylamines: Synthesis and electrochemistry. Org. Lett. 2011, 13, 4742–4745. [Google Scholar] [CrossRef] [PubMed]
- Kawamata, M.; Sugai, T.; Minoura, M.; Maruyama, Y.; Furukawa, K.; Holstrom, C.; Nemykin, V.N.; Nakano, H.; Matano, Y. Nitrogen-bridged metallodiazaporphyrin dimers: Synergistic effects of nitrogen bridges and meso-nitrogen atoms on structure and properties. Chem. Asian J. 2017, 12, 816–821. [Google Scholar] [CrossRef]
- Shimizu, D.; Fujimoto, K.; Osuka, A. Stable diporphyrinylaminyl radical and nitrenium ion. Angew. Chem. Int. Ed. 2018, 57, 9434–9438. [Google Scholar] [CrossRef]
- Yella, A.; Lee, H.W.; Tsao, H.N.; Yi, C.; Chandiran, A.K.; Nazeeruddin, M.K.; Diau, E.W.G.; Yeh, C.Y.; Zakeeruddin, S.M.; Grätzel, M. Porphyrin-sensitized solar cells with cobalt(II/III)-based redox electrolyte exceed 12 percent efficiency. Science 2011, 334, 629–634. [Google Scholar] [CrossRef]
- Sakamoto, R.; Sasaki, T.; Honda, N.; Yamamura, T. 5,15-Bis(di-p-anisylamino)-10,20-diphenylporphyrin: Distant and intense electronic communication between two amine sites. Chem. Commun. 2009, 45, 5156–5158. [Google Scholar] [CrossRef]
- Sakamoto, R.; Nishikawa, M.; Yamamura, T.; Kume, S.; Nishihira, H. A new special pair model comprising meso-di-p-anisylaminoporphyrin: Enhancement of visible-light absorptivities and quantification of electronic communication in mixed valent cation radical. Chem. Commun. 2010, 46, 2028–2030. [Google Scholar] [CrossRef] [PubMed]
- Senge, M.O.; Davis, M. 5,15-dianthracen-9-yl-10,20-dihexylporphynato)nickel(II): A planar nickel(II) porphyrin. Acta Crystallogr. Sect. E Struct. Rep. Online 2010, E66, m790. [Google Scholar] [CrossRef]
- Kadish, K.M.; Royal, G.; Van Caemelbecke, E.; Gueletti, L. The Porphyrin Handbook; Kadish, K.M., Smith, K.M., Guilard, R., Eds.; Academic Press: San Diego, CA, USA, 2000; Volume 9, pp. 1–219. [Google Scholar]
- Neugebauer, F.A.; Bamberger, S. Diarylamine radical cations. Angew. Chem. Int. Ed. Engl. 1971, 10, 71–72. [Google Scholar] [CrossRef]
- Kennedy, D.E.; Dalal, N.S.; McDowell, C.A. Endor of protons and determination of chlorine hyperfine couplings in neutral free radicals of biologically interesting compounds. Chem. Phys. Lett. 1974, 29, 521–525. [Google Scholar] [CrossRef]
- Stoll, S.; Schweiger, A. EasySpin, a comprehensive software package for spectral simulation and analysis in EPR. J. Magn. Reson. 2006, 178, 42–55. [Google Scholar] [CrossRef] [PubMed]
- Neese, F. The ORCA program system. WIREs Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar] [CrossRef]
- Becke, A.D. Density functional thermochemistry III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.T.; Yang, W.T.; Parr, R.G. Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Csonka, G.I. Proper basis set for quantum mechanical studies of potential energy surfaces of carbohydrates. J. Mol. Struct. THEOCHEM. 2002, 584, 1–4. [Google Scholar] [CrossRef]
- Boese, A.D.; Martin, J.M.L.; Handy, N.C. The role of the basis set: Assessing density functional theory. J. Chem. Phys. 2003, 119, 3005–3014. [Google Scholar] [CrossRef]
- Mackie, I.D.; DiLabio, G.A. Accurate dispersion interactions from standard density-functional theory methods with small basis sets. Phys. Chem. Chem. Phys. 2010, 12, 6092–6098. [Google Scholar] [CrossRef] [PubMed]
- Klamt, A.; Schüürmann, G. COSMO: A new approach to dielectric screening in solvents with explicit expressions for the screening energy and its gradient. J. Chem. Soc. Perkin Trans. 2 1993, 799–805. [Google Scholar] [CrossRef]
- Neese, F. Prediction and interpretation of the 57Fe isomer shift in Mössbauer spectra by density functional theory. Inorg. Chim. Acta 2002, 337, 181–192. [Google Scholar] [CrossRef]
- Barone, V. Recent Advances in Density Functional Methods, Part I; Chong, D.P., Ed.; World Scientific Publ Co.: Singapore, 1995; p. 287. [Google Scholar]













Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).