
Catalytic-CO2-Desorption Studies of BZA-AEP Mixed Absorbent by the Lewis Acid Catalyst CeO2-γ-Al2O3

Abstract
:1. Introduction
2. Results and Discussion
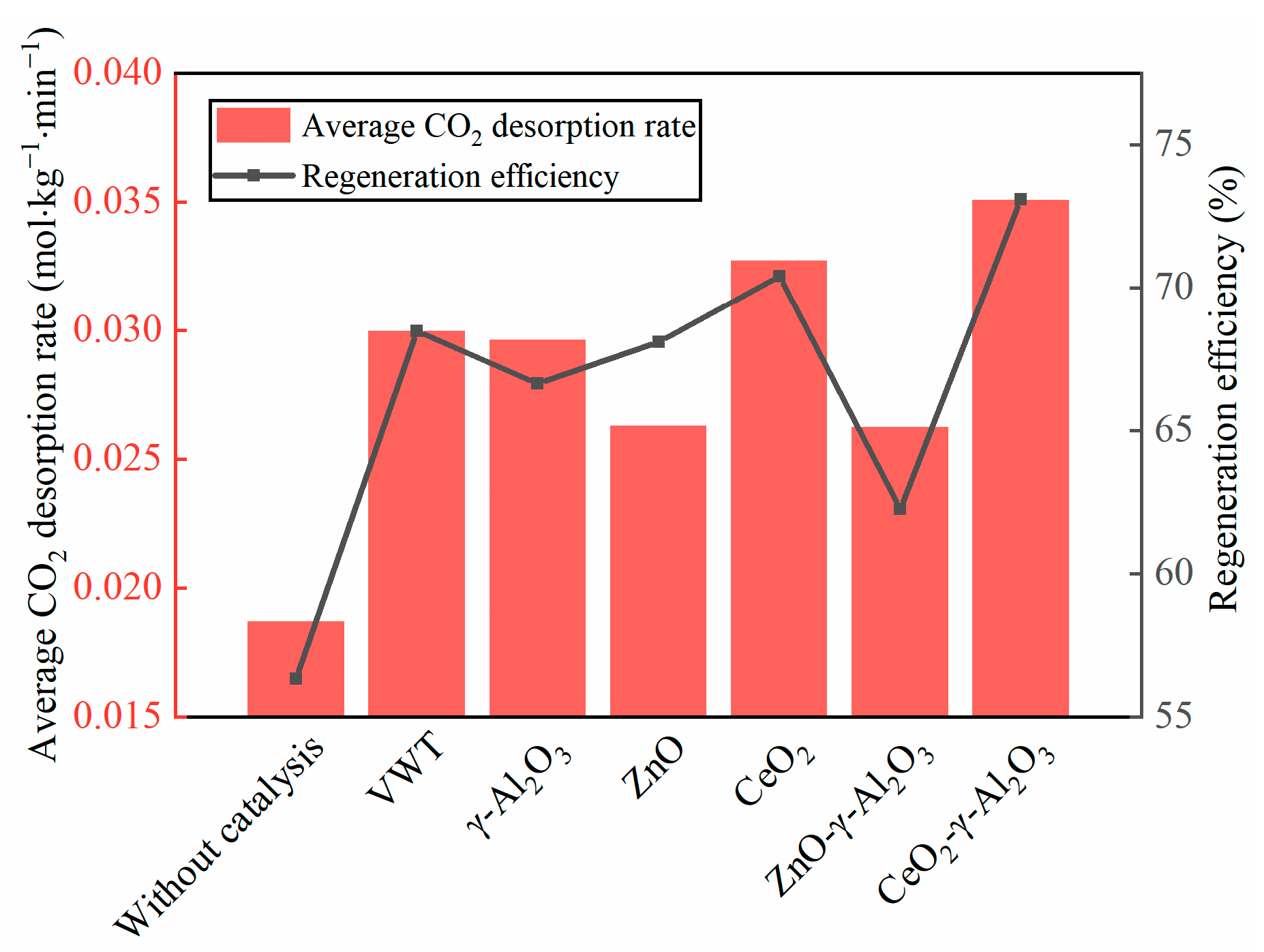
2.1. Catalytic Desorption Performance of CeO2-γ-Al2O3
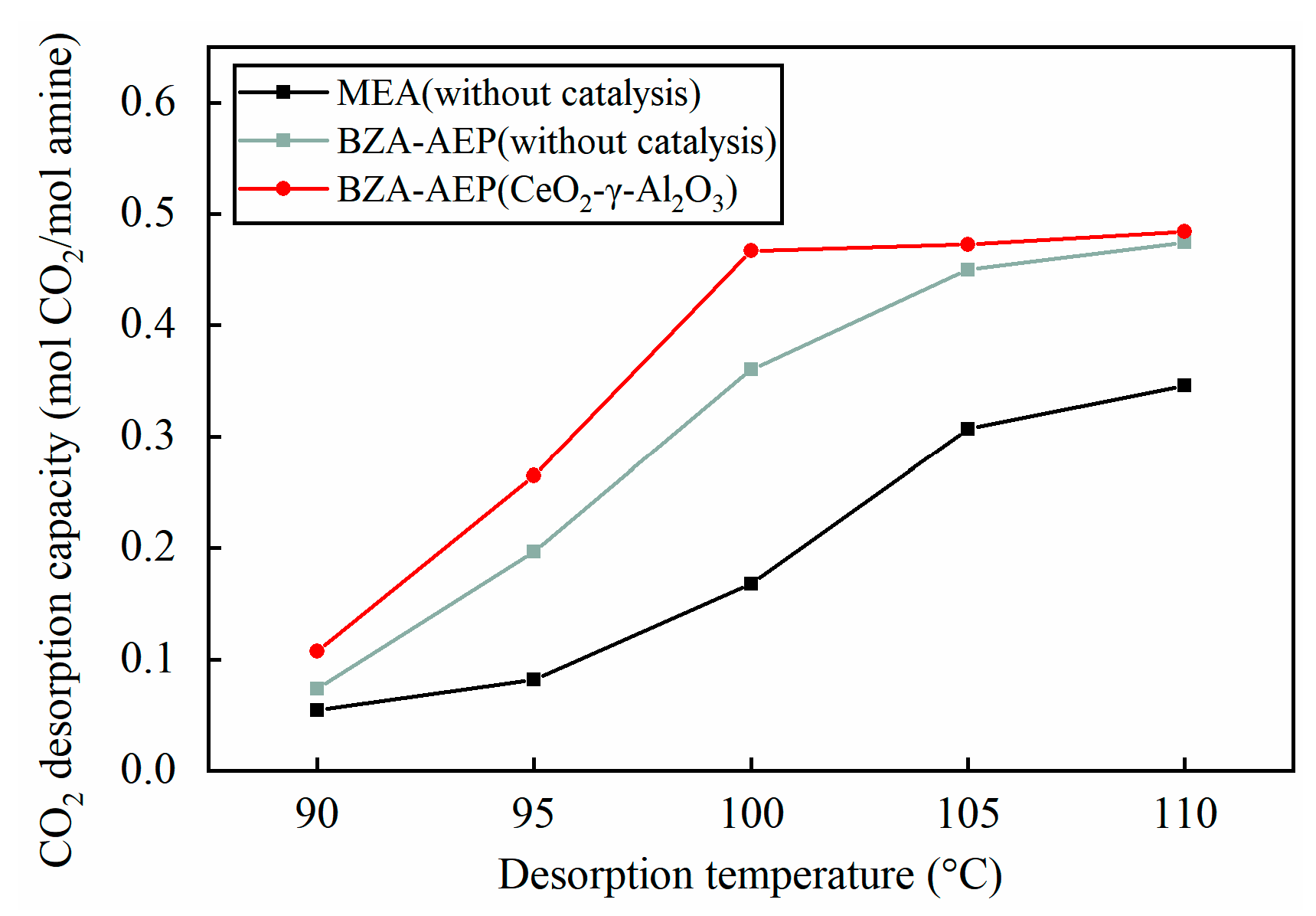
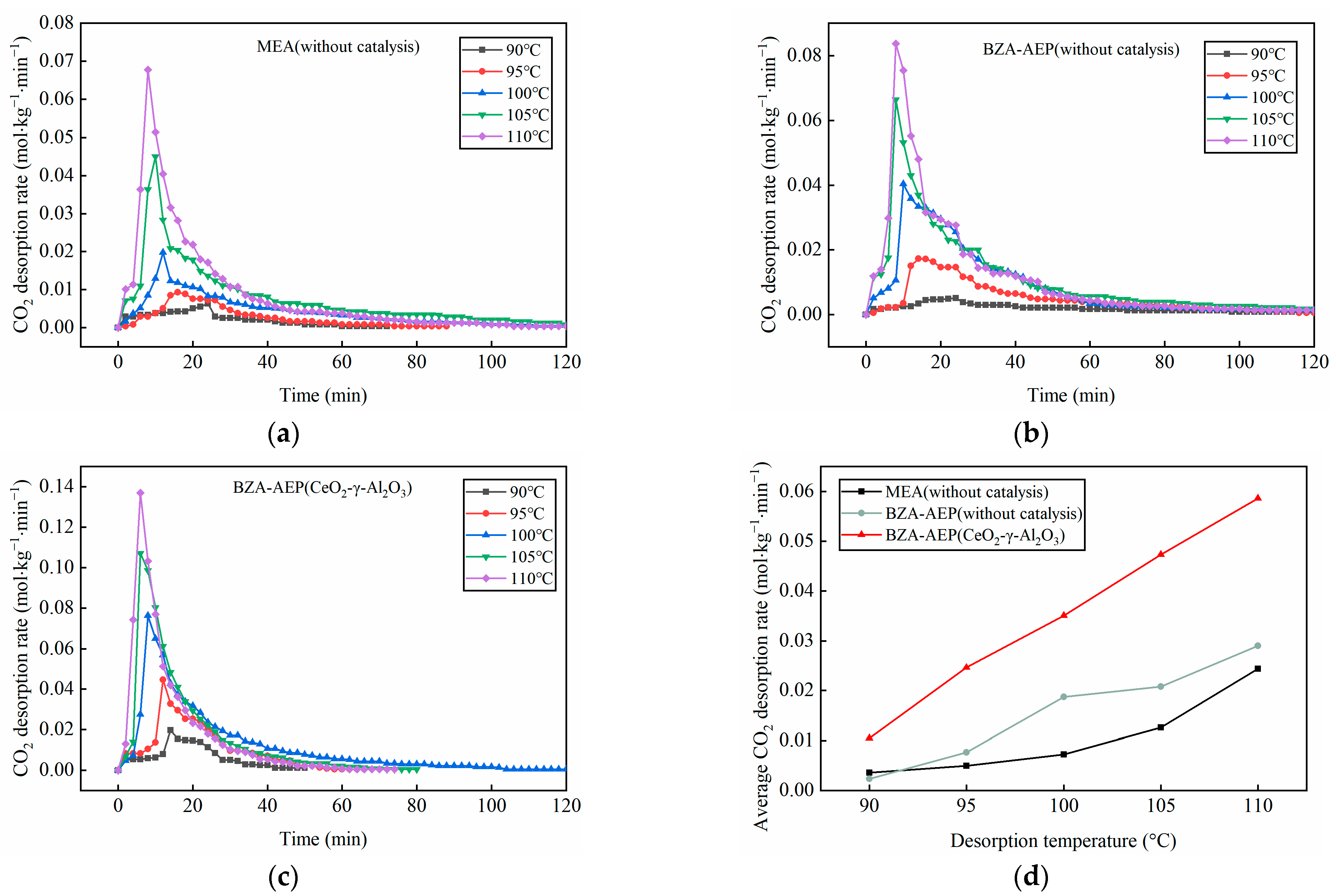
2.2. Effect of Temperature on Catalytic Desorption Performance of CeO2-γ-Al2O3
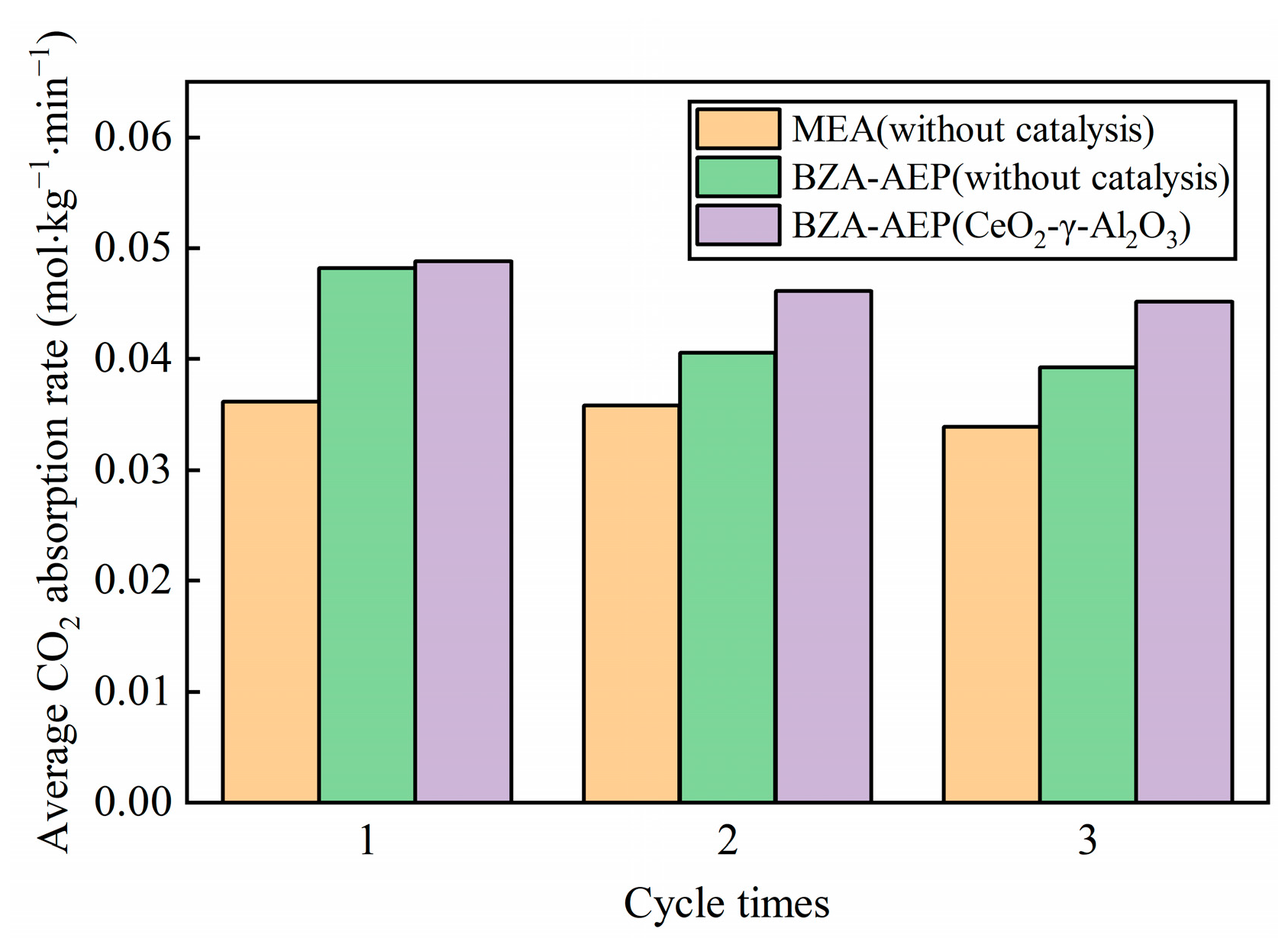
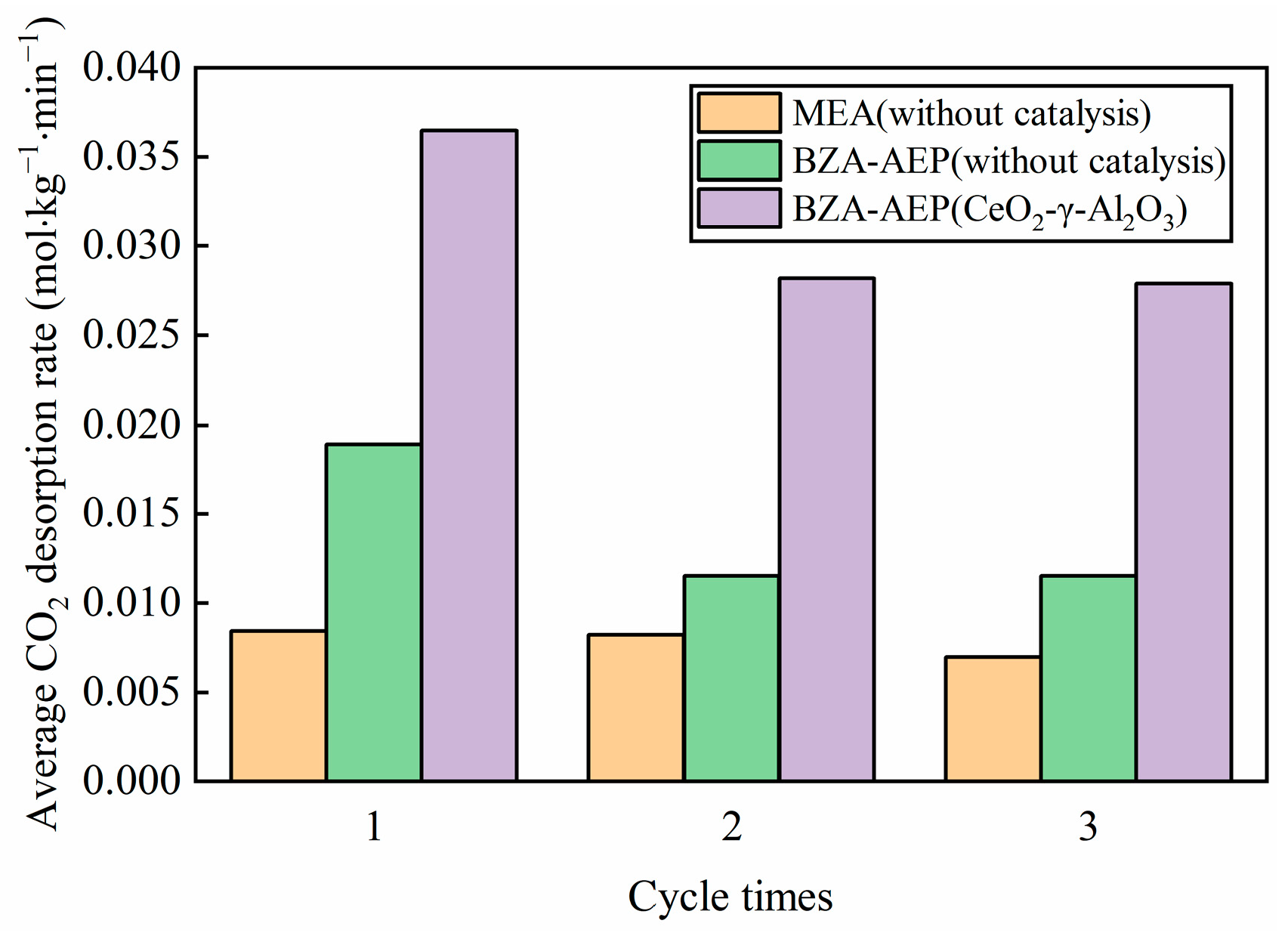
2.3. Cyclic Catalytic Desorption Performance of CeO2-γ-Al2O3
2.4. Characterization of CeO2-γ-Al2O3 Catalyst
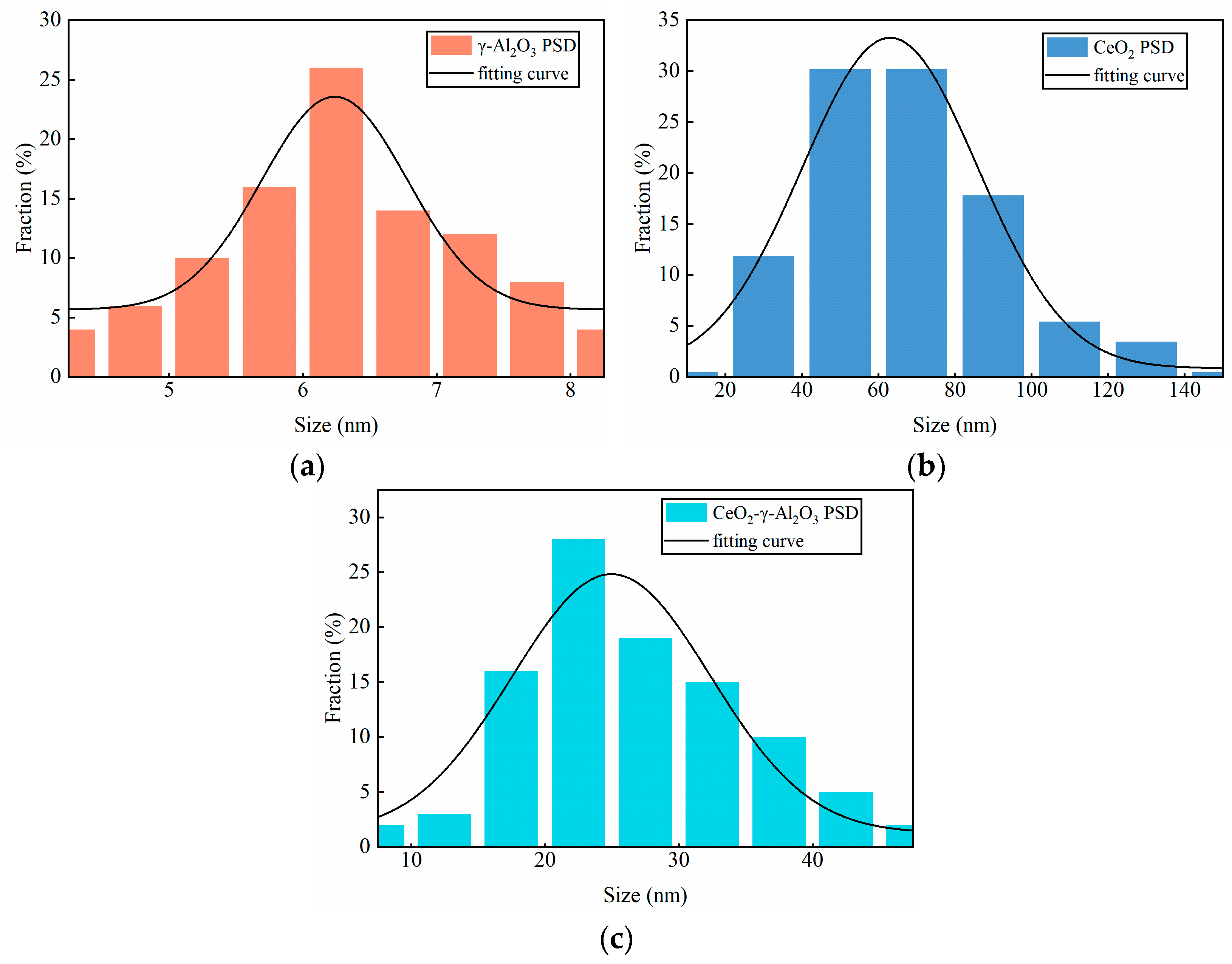
2.4.1. SEM Characterization
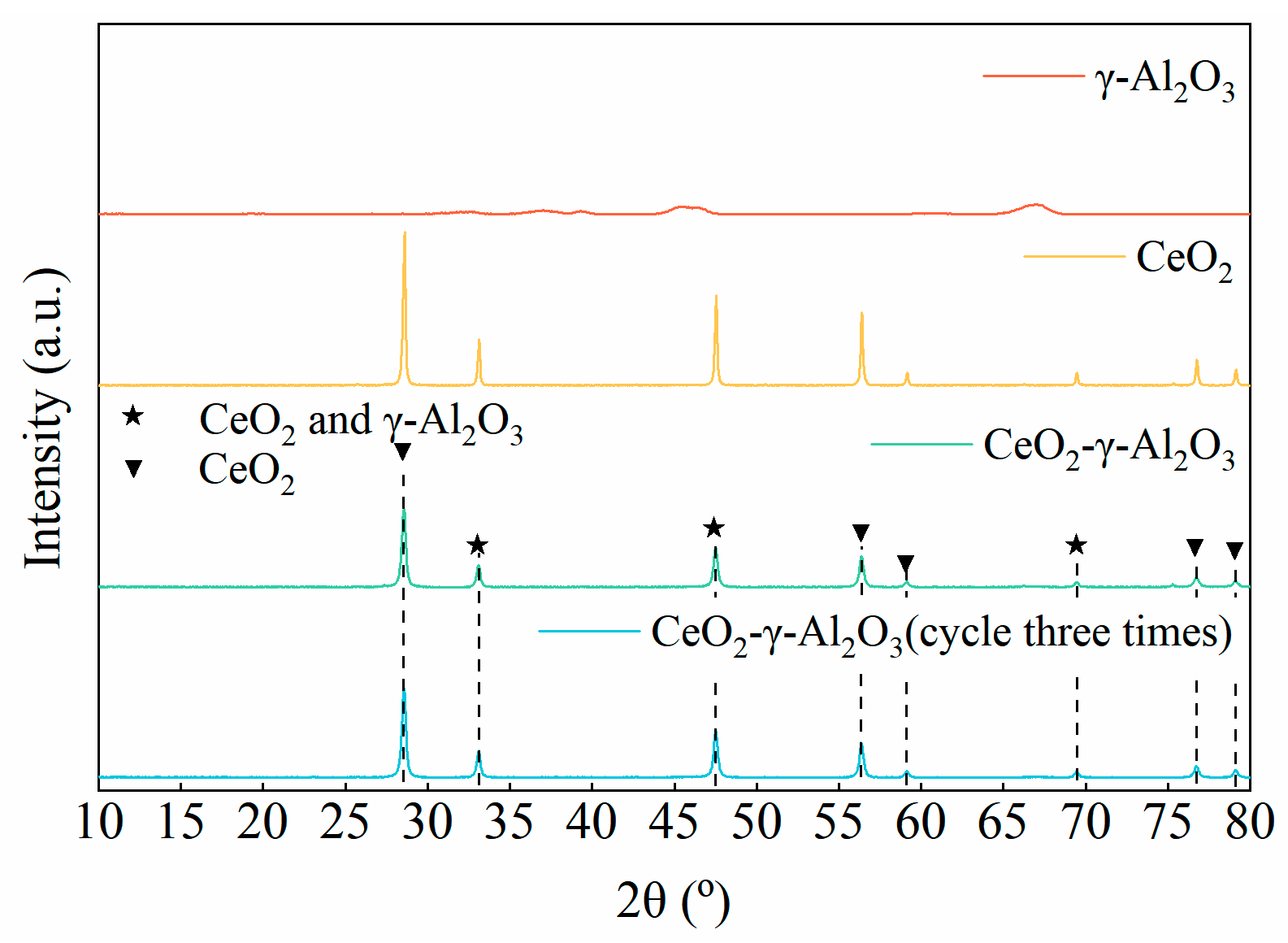
2.4.2. XRD Characterization
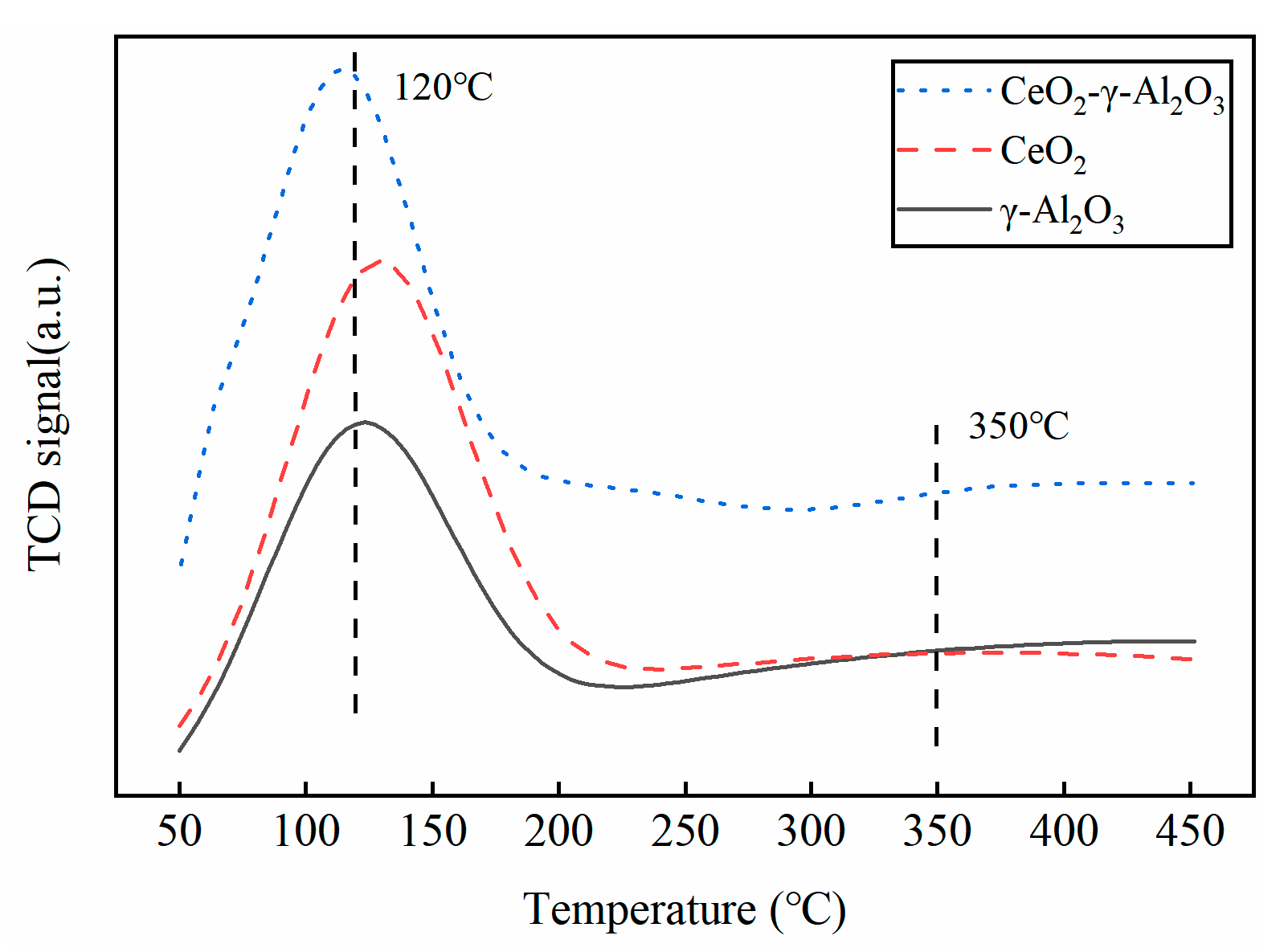
2.4.3. BET and NH3-TPD Characterization
2.5. Catalytic Mechanism of CeO2-γ-Al2O3
3. Materials and Methods
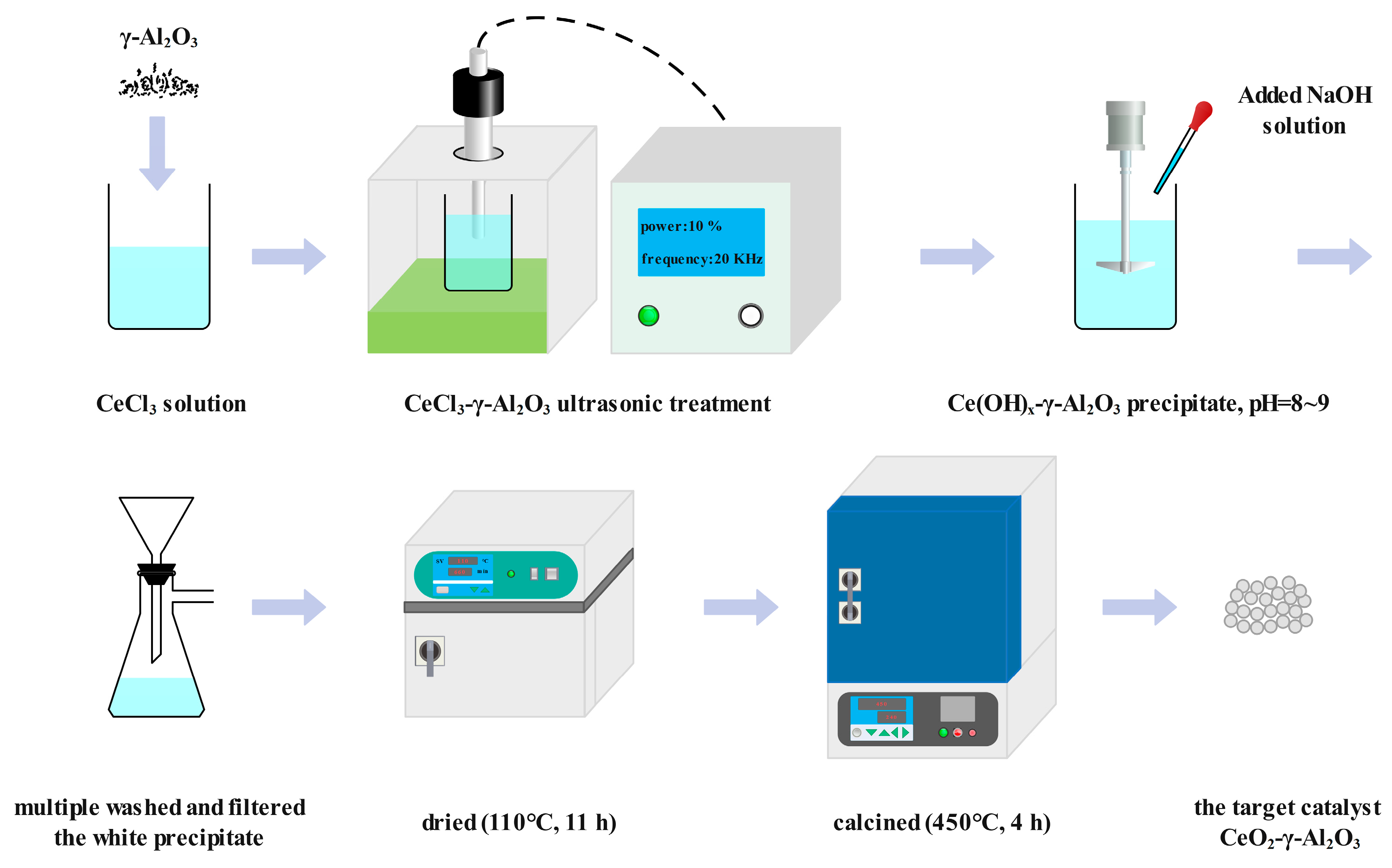
3.1. Catalyst Preparation Materials and Methods
3.2. Desorption Experimental Materials and Steup
4. Conclusions
- Through a comparison of the catalytic desorption performance of two Lewis acid catalysts, three precursor catalysts, and one commercial catalyst (VWT), CeO2-γ-Al2O3 exhibited superior catalytic desorption effect over other catalysts. In comparison to no catalyst, CeO2-γ-Al2O3 Lewis acid catalyst increased the desorption capacity of BZA-AEP by 30%, the average desorption rate by 87%, and the regeneration efficiency by 30%.
- The catalytic desorption effect of CeO2-γ-Al2O3 is particularly significant at low temperatures, reducing the desorption temperature of BZA-AEP by approximately 10 °C.
- CeO2-γ-Al2O3 cyclic catalytic desorption results show that CeO2-γ-Al2O3 has good catalytic stability. At the same time, through the characterization of the surface structure and crystal structure of the catalyst, there is no change in the structure of the catalyst before and after cycling.
- The catalytic mechanism of CeO2-γ-Al2O3 varies in different alkaline environments. The interaction between CeO2 and γ-Al2O3 provides the catalyst with a strong catalytic effect in both rich and poor solution.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Zhai, P.; Pörtner, H.O.; Roberts, D.; Skea, J.; Shukla, P.; Pirani, A.; Moufouma-Okia, W.; Péan, C.; Pidcock, R.; Connors, S. Global Warming of 1.5 °C. IPCC Spec. Rep. Impacts Glob. Warm. 2018, 1, 43–50. [Google Scholar]
- Wilberforce, T.; Olabi, A.G.; Sayed, E.T.; Elsaid, K.; Abdelkareem, M.A. Progress in Carbon Capture Technologies. Sci. Total Environ. 2021, 761, 143203. [Google Scholar] [CrossRef] [PubMed]
- Liang, Z.; Idem, R.; Tontiwachwuthikul, P.; Yu, F.; Liu, H.; Rongwong, W. Experimental Study on the Solvent Regeneration of a CO2-Loaded MEA Solution Using Single and Hybrid Solid Acid Catalysts. AIChE J. 2016, 62, 753–765. [Google Scholar] [CrossRef]
- Idem, R.; Shi, H.; Gelowitz, D.; Tontiwachwuthikul, P. Catalytic Method and Apparatus for Separating a Gaseous Component from an Incoming Gas Stream. WO2011120138A1, 6 October 2011. [Google Scholar]
- Shi, H.; Naami, A.; Idem, R.; Tontiwachwuthikul, P. Catalytic and Non Catalytic Solvent Regeneration during Absorption-Based CO2 Capture with Single and Blended Reactive Amine Solvents. Int. J. Greenh. Gas Control 2014, 26, 39–50. [Google Scholar] [CrossRef]
- Bhatti, U.H.; Shah, A.K.; Kim, J.N.; You, J.K.; Choi, S.H.; Lim, D.H.; Nam, S.; Park, Y.H.; Baek, I.H. Effects of Transition Metal Oxide Catalysts on MEA Solvent Regeneration for the Post-Combustion Carbon Capture Process. ACS Sustain. Chem. Eng. 2017, 5, 5862–5868. [Google Scholar] [CrossRef]
- Zhang, X.; Hong, J.; Liu, H.; Luo, X.; Olson, W.; Tontiwachwuthikul, P.; Liang, Z. SO42−/ZrO2 Supported on γ-Al2O3 as a Catalyst for CO2 Desorption from CO2-Loaded Monoethanolamine Solutions. AIChE J. 2018, 64, 3988–4001. [Google Scholar] [CrossRef]
- Zhang, X.; Liu, H.; Liang, Z.; Idem, R.; Tontiwachwuthikul, P.; Jaber Al-Marri, M.; Benamor, A. Reducing Energy Consumption of CO2 Desorption in CO2-Loaded Aqueous Amine Solution Using Al2O3/HZSM-5 Bifunctional Catalysts. Appl. Energy 2018, 229, 562–576. [Google Scholar] [CrossRef]
- Bhatti, U.H.; Kazmi, W.W.; Min, G.H.; Haider, J.; Nam, S.; Baek, I.H. Facilely Synthesized M-Montmorillonite (M = Cr, Fe, and Co) as Efficient Catalysts for Enhancing CO2 Desorption from Amine Solution. Ind. Eng. Chem. Res. 2021, 60, 13318–13325. [Google Scholar] [CrossRef]
- Wei, K.; Xing, L.; Li, Y.; Xu, T.; Li, Q.; Wang, L. Heteropolyacid Modified Cerium-Based MOFs Catalyst for Amine Solution Regeneration in CO2 Capture. Sep. Purif. Technol. 2022, 293, 121144. [Google Scholar] [CrossRef]
- WILEY-VCH. Ullmann’s Encyclopedia of Industrial Chemistry: Electronic Release 2007; Verlag Chemie: Hoboken, NJ, USA, 2007. [Google Scholar]
- Hart, L.D.; Lense, E. Alumina Chemicals: Science and Technology Handbook; John Wiley & Sons: Hoboken, NJ, USA, 1990. [Google Scholar]
- Shi, D.; Wang, H.; Kovarik, L.; Gao, F.; Wan, C.; Hu, J.Z.; Wang, Y. WOx supported on γ-Al2O3 with different morphologies as model catalysts for alkanol dehydration. J. Catal. 2018, 363, 1–8. [Google Scholar] [CrossRef]
- Khivantsev, K.; Jaegers, N.R.; Kwak, J.H.; Szanyi, J.; Kovarik, L. Precise Identification and Characterization of Catalytically Active Sites on the Surface of γ-Alumina. Angew. Chem. Int. Ed. 2021, 60, 17522–17530. [Google Scholar] [CrossRef] [PubMed]
- Mir, N.A.; Khan, A.; Umar, K.; Muneer, M. Photocatalytic Study of a Xanthene Dye Derivative, Phloxine B in Aqueous Suspension of TiO2: Adsorption Isotherm and Decolourization Kinetics. Energy Environ. Focus 2013, 2, 208–216. [Google Scholar] [CrossRef]
- Bushra, R.; Shahadat, M.; Ahmad, A.; Nabi, S.A.; Umar, K.; Oves, M.; Raeissi, A.S.; Muneer, M. Synthesis, Characterization, Antimicrobial Activity and Applications of PolyanilineTi(IV)Arsenophosphate Adsorbent for the Analysis of Organic and Inorganic Pollutants. J. Hazard. Mater. 2014, 264, 481–489. [Google Scholar] [CrossRef] [PubMed]
- Yaqoob, A.A.; binti Mohd Noor, N.H.; Umar, K.; Adnan, R.; Ibrahim, M.N.M.; Rashid, M. Graphene Oxide–ZnO Nanocomposite: An Efficient Visible Light Photocatalyst for Degradation of Rhodamine B. Appl. Nanosci. 2021, 11, 1291–1302. [Google Scholar] [CrossRef]
- Mao, X.; Chen, H.; Wang, Y.; Zhu, X.; Yang, G. Study on Benzylamine(BZA) and Aminoethylpiperazine(AEP) Mixed Absorbent on Ship-Based Carbon Capture. Molecules 2023, 28, 2661. [Google Scholar] [CrossRef] [PubMed]
- Jha, S.K.; Kumar, C.N.; Raj, R.P.; Jha, N.S.; Mohan, S. Synthesis of 3D Porous CeO2/Reduced Graphene Oxide Xerogel Composite and Low Level Detection of H2O2. Electrochim. Acta 2014, 120, 308–313. [Google Scholar] [CrossRef]
- Balakumar, V.; Kim, H.; Manivannan, R.; Kim, H.; Ryu, J.W.; Heo, G.; Son, Y.-A. Ultrasound-Assisted Method to Improve the Structure of CeO2@polyprrole Core-Shell Nanosphere and Its Photocatalytic Reduction of Hazardous Cr6+. Ultrason. Sonochem. 2019, 59, 104738. [Google Scholar] [CrossRef]
- Wang, D.; Peng, Y.; Yang, Q.; Hu, F.; Li, J.; Crittenden, J. NH3-SCR Performance of WO3 Blanketed CeO2 with Different Morphology: Balance of Surface Reducibility and Acidity. Catal. Today 2019, 332, 42–48. [Google Scholar] [CrossRef]
- Yao, X.; Chen, L.; Cao, J.; Yang, F.; Tan, W.; Dong, L. Morphology and Crystal-Plane Effects of CeO2 on TiO2/CeO2 Catalysts during NH3-SCR Reaction. Ind. Eng. Chem. Res. 2018, 57, 12407–12419. [Google Scholar] [CrossRef]
- Li, L.; Zhang, L.; Ma, K.; Zou, W.; Cao, Y.; Xiong, Y.; Tang, C.; Dong, L. Ultra-Low Loading of Copper Modified TiO2/CeO2 Catalysts for Low-Temperature Selective Catalytic Reduction of NO by NH3. Appl. Catal. B Environ. 2017, 207, 366–375. [Google Scholar] [CrossRef]
- Zeng, M.; Li, Y.; Mao, M.; Bai, J.; Ren, L.; Zhao, X. Synergetic Effect between Photocatalysis on TiO2 and Thermocatalysis on CeO2 for Gas-Phase Oxidation of Benzene on TiO2/CeO2 Nanocomposites. Acs Catal. 2015, 5, 3278–3286. [Google Scholar] [CrossRef]
- Zhou, R.-S.; Snyder, R.L. Structures and Transformation Mechanisms of the η, γ and θ Transition Aluminas. Acta Cryst. B 1991, 47, 617–630. [Google Scholar] [CrossRef]
- Grier, D.; McCarthy, G. North Dakota State University, Fargo, North Dakota, USA, ICDD Grant-in-Aid 1991; Powder Diffraction File; International Center for Diffraction Data: Newtown Square, PA, USA, 1994. [Google Scholar]
- Okada, M.; Fujiwara, K.; Uehira, M.; Matsumoto, N.; Takeda, S. Expansion of Nanosized Pores in Low-Crystallinity Nanoparticle-Assembled Plates via a Thermally Induced Increase in Solid-State Density. J. Colloid Interface Sci. 2013, 405, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Karamian, E.; Khandan, A.; Eslami, M.; Gheisari, H.; Rafiaei, N. Investigation of HA Nanocrystallite Size Crystallographic Characterizations in NHA, BHA and HA Pure Powders and Their Influence on Biodegradation of HA. In Advanced Materials Research; Trans Tech Publications Ltd.: Stafa-Zurich, Switzerland, 2014; Volume 829, pp. 314–318. [Google Scholar]
- Locardi, B.; Pazzaglia, U.E.; Gabbi, C.; Profilo, B. Thermal Behaviour of Hydroxyapatite Intended for Medical Applications. Biomaterials 1993, 14, 437–441. [Google Scholar] [CrossRef]
- Scherrer, P. Bestimmung Der Größe Und Der Inneren Struktur von Kolloidteilchen Mittels Röntgenstrahlen. Nachr. Ges. Wiss. Göttingen Math.-Phys. Kl. 1918, 1918, 98–100. [Google Scholar]
- Taylor, A.; Sinclair, H. On the Determination of Lattice Parameters by the Debye-Scherrer Method. Proc. Phys. Soc. 1945, 57, 126. [Google Scholar] [CrossRef]
- Pope, C.G. X-ray Diffraction and the Bragg Equation. J. Chem. Educ. 1997, 74, 129. [Google Scholar] [CrossRef]
- Wen, X.; Li, C.; Fan, X.; Gao, H.; Zhang, W.; Chen, L.; Zeng, G.; Zhao, Y. Experimental Study of Gaseous Elemental Mercury Removal with CeO2/γ-Al2O3. Energy Fuels 2011, 25, 2939–2944. [Google Scholar] [CrossRef]
- Yadav, G.D.; Sharma, R.V. Synthesis, Characterization and Applications of Highly Active and Robust Sulfated Fe–TiO2 Catalyst (ICT-3) with Superior Redox and Acidic Properties. J. Catal. 2014, 311, 121–128. [Google Scholar] [CrossRef]
- Caplow, M. Kinetics of Carbamate Formation and Breakdown. J. Am. Chem. Soc. 1968, 90, 6795–6803. [Google Scholar] [CrossRef]
- Donaldson, T.L.; Nguyen, Y.N. Carbon Dioxide Reaction Kinetics and Transport in Aqueous Amine Membranes. Ind. Eng. Chem. Fundam. 1980, 19, 260–266. [Google Scholar] [CrossRef]
- Prasongthum, N.; Natewong, P.; Reubroycharoen, P.; Idem, R. Solvent Regeneration of a CO2-Loaded BEA–AMP Bi-Blend Amine Solvent with the Aid of a Solid Brønsted Ce(SO4)2/ZrO2 Superacid Catalyst. Energy Fuels 2019, 33, 1334–1343. [Google Scholar] [CrossRef]
- Li, T.; Yu, Q.; Barzagli, F.; Li, C.; Che, M.; Zhang, Z.; Zhang, R. Energy Efficient Catalytic CO2 Desorption: Mechanism, Technological Progress and Perspective. Carbon Capture Sci. Technol. 2023, 6, 100099. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | Grain Size (nm) | 2θ (°) | Crystal Face (hkl) | Crystal Plane Spacing (Å) |
|---|---|---|---|---|
| γ-Al2O3 | 5.4 | 67.0543 | 440 | 1.3946 |
| CeO2 | 46.3 | 28.5500 | 111 | 3.1240 |
| γ-Al2O3-CeO2 | 25.6 | 28.5718 | 111 | 3.1216 |
| γ-Al2O3-CeO2 (after three times cycle) | 26.0 | 28.6107 | 111 | 3.1175 |
| Catalyst | Specific Surface Area (m2/g) | Acid Strength (mmol/g) | |
|---|---|---|---|
| Weak Acid | Total Acid | ||
| γ-Al2O3 | 149.3333 | 0.412 | 1.485 |
| CeO2 | 2.3596 | 1.332 | 1.801 |
| CeO2-γ-Al2O3 | 36.8573 | 1.594 | 2.098 |
| Reagent Name | Abbreviation | Specification |
|---|---|---|
| deionized water | DI | - |
| zinc sulfate heptahydrate | ZnSO4·7H2O | 99.5% |
| cerium chloride hexahydrate | CeCl3·6H2O | 99.99% |
| sodium hydroxide | NaOH | 95% |
| gamma alumina | γ-Al2O3 | 99.99% |
| carbon dioxide | CO2 | 99.9% |
| nitrogen | N2 | 99.9% |
| V2O5-WO3/TiO2 | VWT | TiO2:V2O5:WO3:SiO2 = 86:8.2:4:1 |
| Name | Parameter |
|---|---|
| absorbent | BZA-AEP |
| total amine concentration | 3 mol/kg |
| concentration ratio of BZA to AEP | 1.5 |
| amount of absorbent | 50 g |
| absorbent rich solution | Saturated solution under 5% CO2 + 95% N2 |
| amount of catalyst | 3 g |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, S.; Mao, X.; Chen, H.; Zhu, X.; Yang, G. Catalytic-CO2-Desorption Studies of BZA-AEP Mixed Absorbent by the Lewis Acid Catalyst CeO2-γ-Al2O3. Molecules 2023, 28, 4438. https://doi.org/10.3390/molecules28114438
Liu S, Mao X, Chen H, Zhu X, Yang G. Catalytic-CO2-Desorption Studies of BZA-AEP Mixed Absorbent by the Lewis Acid Catalyst CeO2-γ-Al2O3. Molecules. 2023; 28(11):4438. https://doi.org/10.3390/molecules28114438
Chicago/Turabian StyleLiu, Shenghua, Xudong Mao, Hao Chen, Xinbo Zhu, and Guohua Yang. 2023. "Catalytic-CO2-Desorption Studies of BZA-AEP Mixed Absorbent by the Lewis Acid Catalyst CeO2-γ-Al2O3" Molecules 28, no. 11: 4438. https://doi.org/10.3390/molecules28114438
APA StyleLiu, S., Mao, X., Chen, H., Zhu, X., & Yang, G. (2023). Catalytic-CO2-Desorption Studies of BZA-AEP Mixed Absorbent by the Lewis Acid Catalyst CeO2-γ-Al2O3. Molecules, 28(11), 4438. https://doi.org/10.3390/molecules28114438