
An In Silico Molecular Modelling-Based Prediction of Potential Keap1 Inhibitors from Hemidesmus indicus (L.) R.Br. against Oxidative-Stress-Induced Diseases


 , and
, and
Abstract
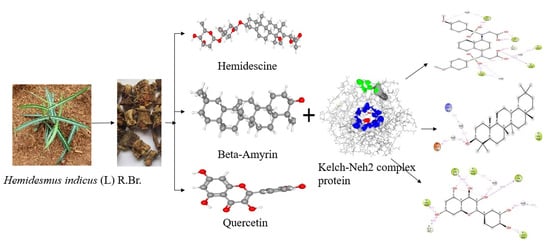
1. Introduction
2. Results
2.1. Antioxidant Power
2.1.1. DPPH Radical Scavenging Assay
2.1.2. ABTS Radical Scavenging Assay
2.1.3. FRAP Assay
2.2. Active Compounds Library
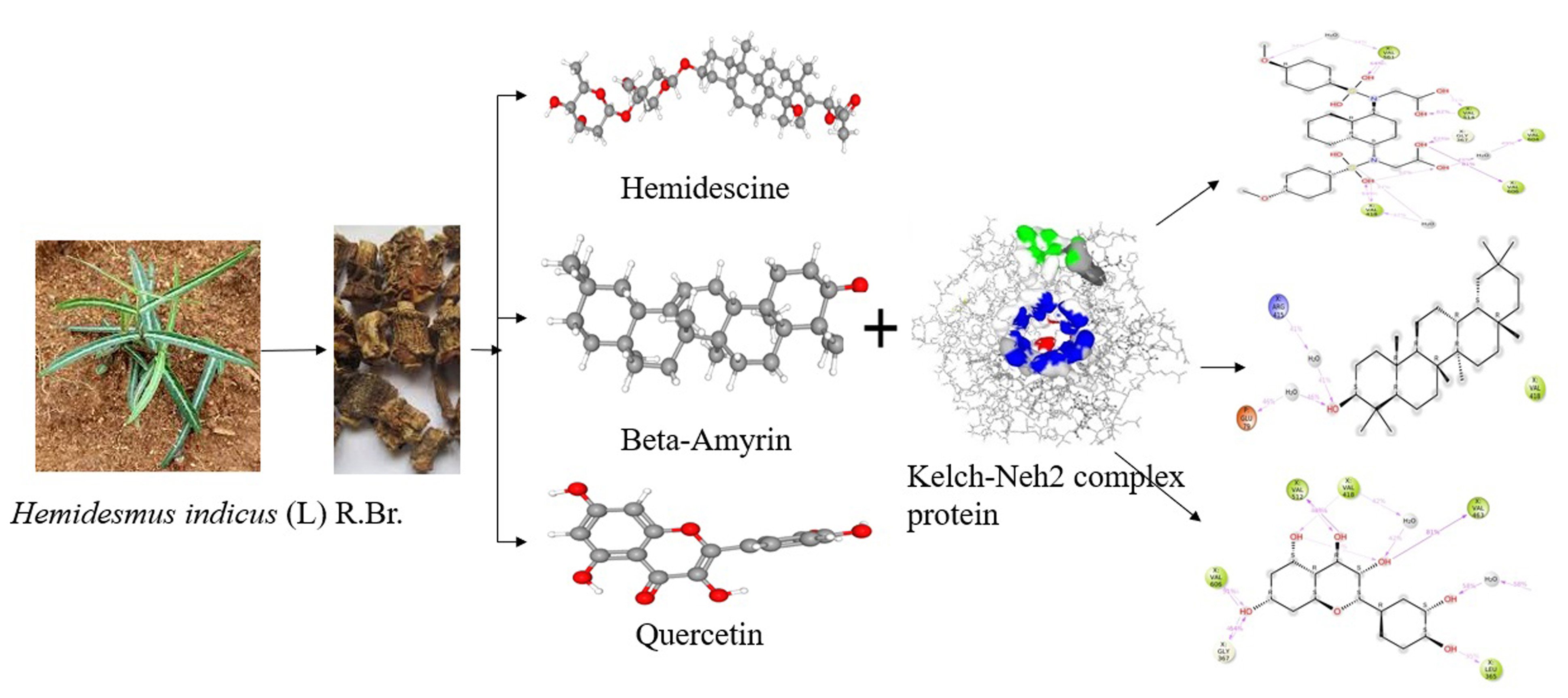
2.3. Active Binding Site Identification
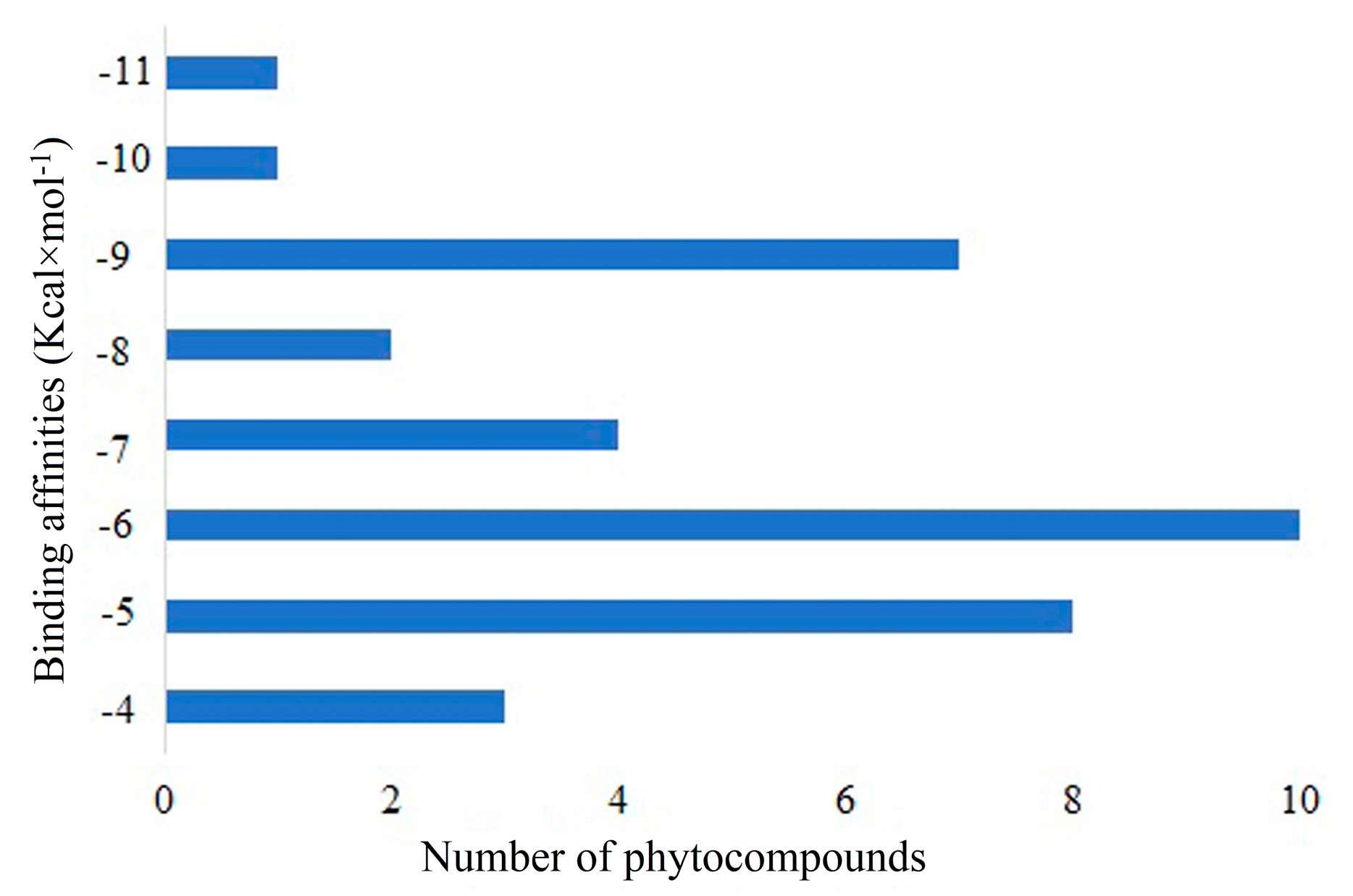
2.4. Molecular Docking
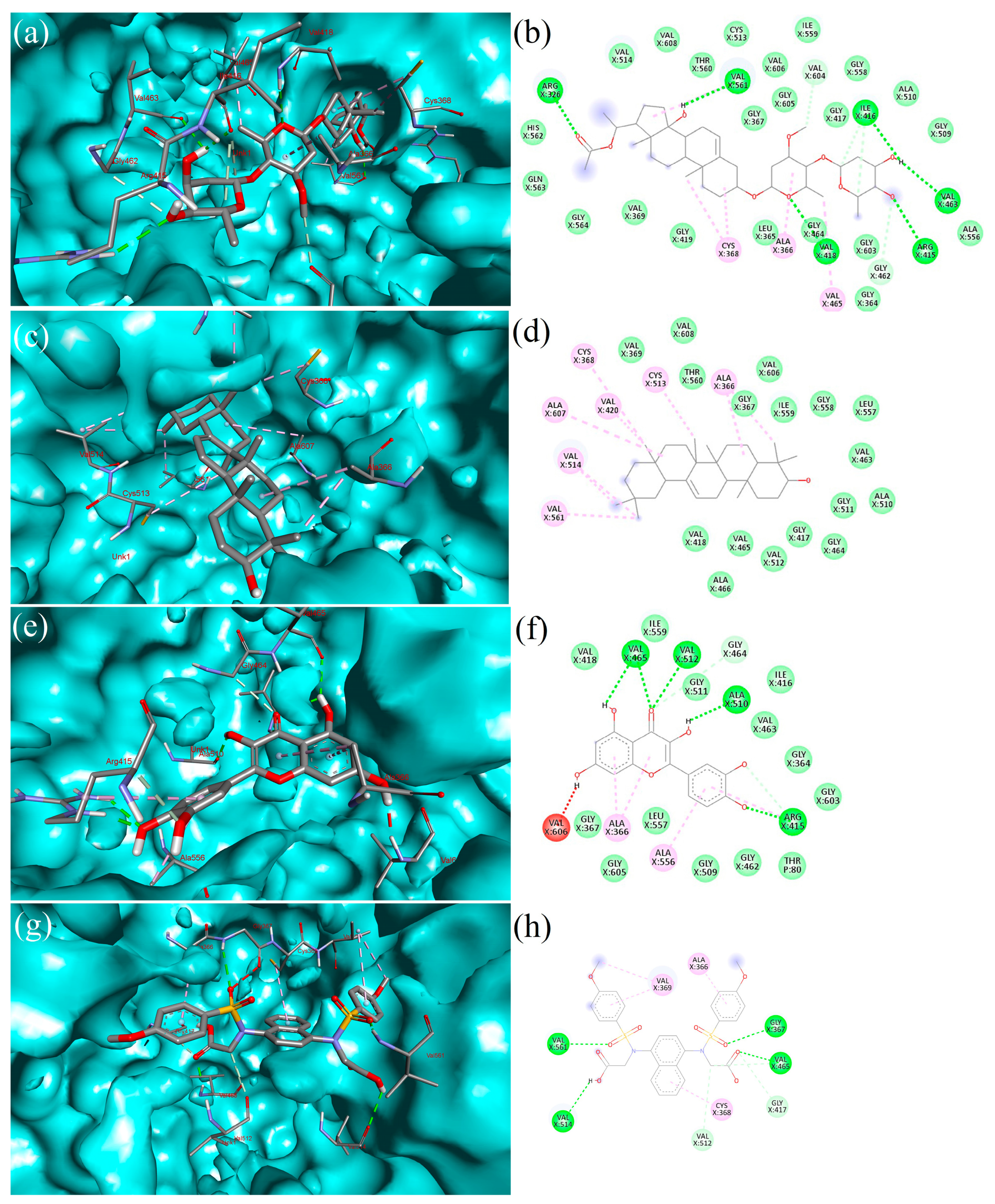
2.5. Interpretation of Receptor–Ligand Interactions
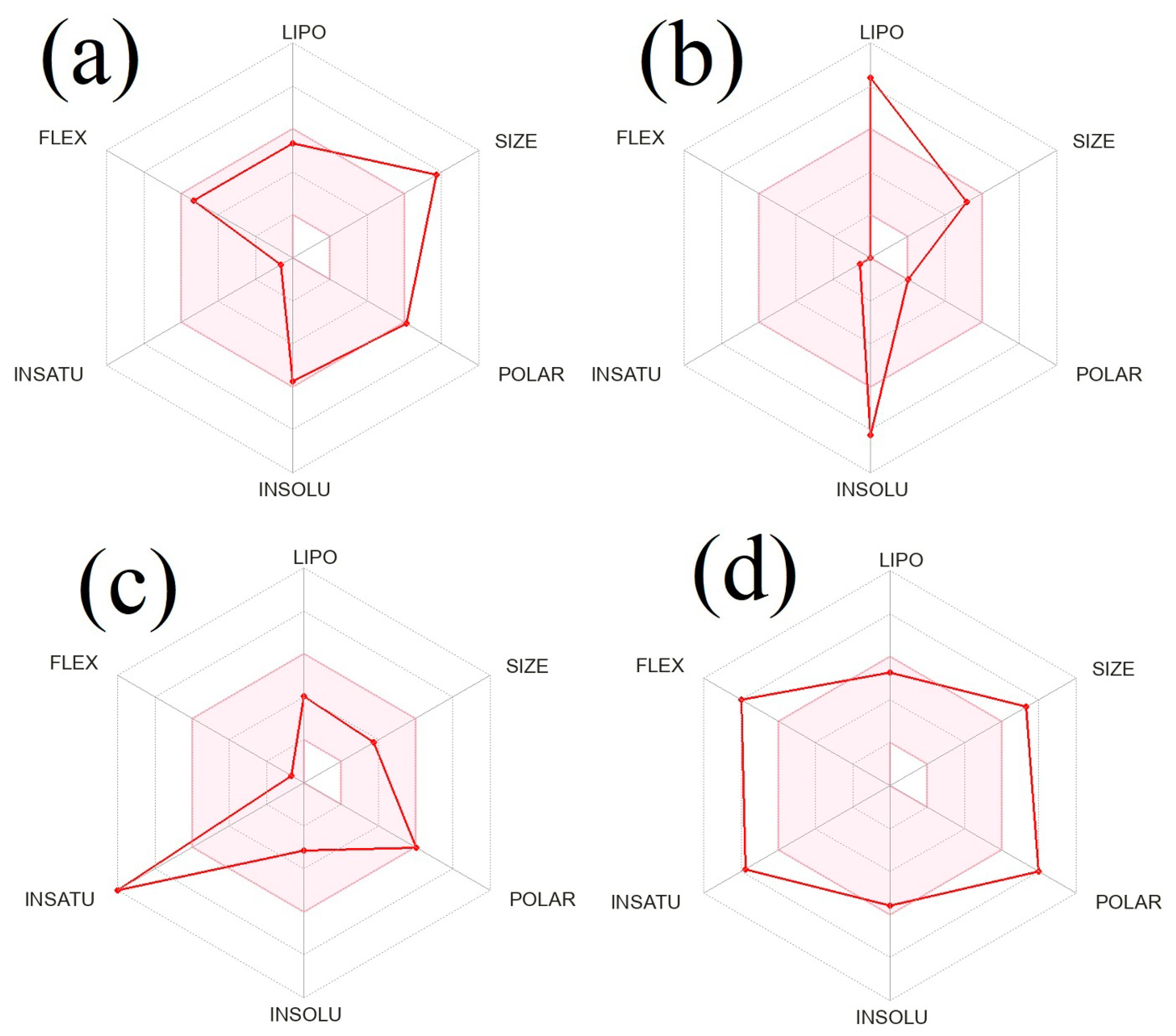
2.6. In silico Prediction of Physicochemical and ADME Properties
2.7. Analysis of Toxicity
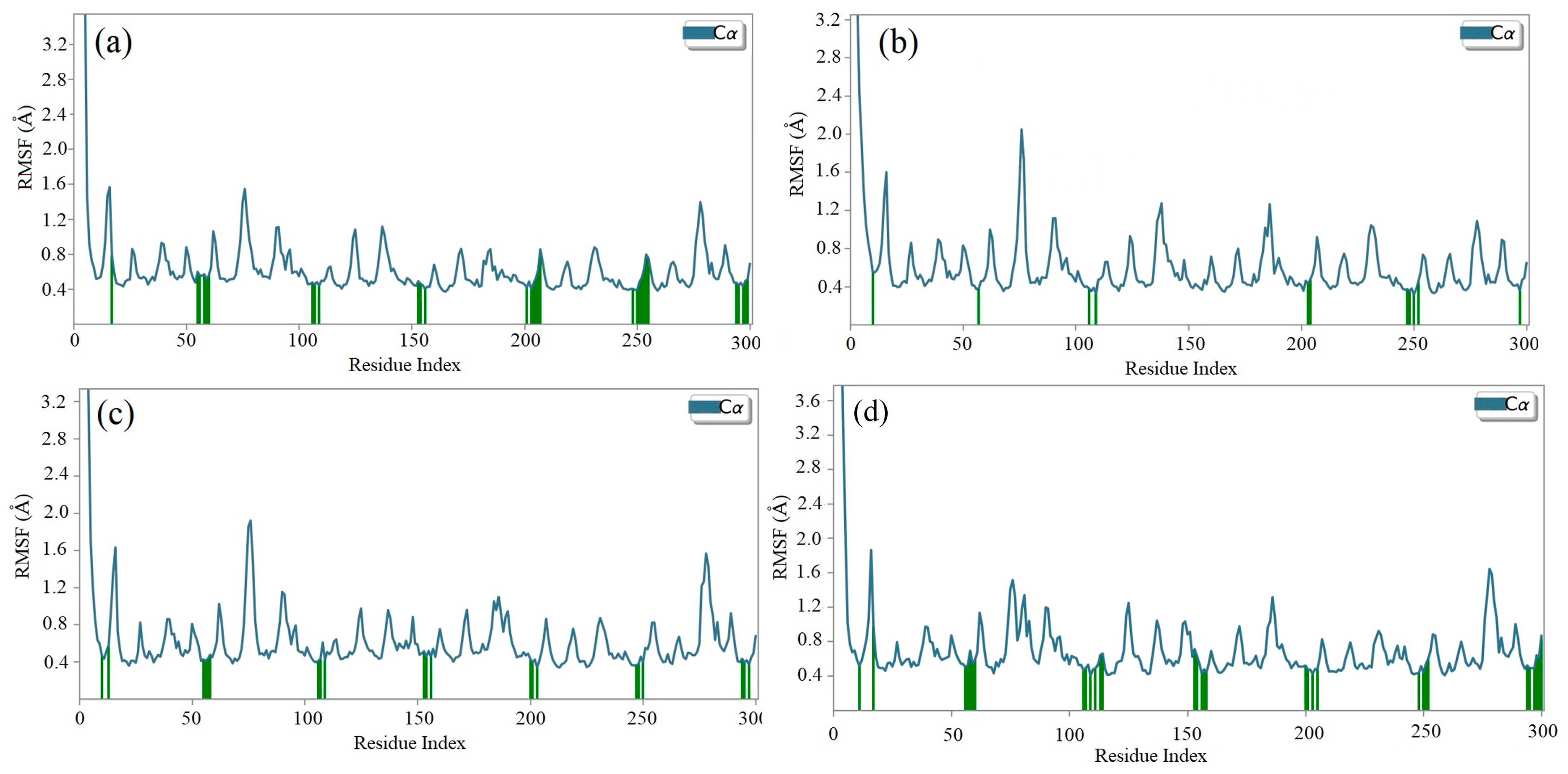
2.8. Molecular Dynamics (MD) Simulation
2.9. Density Functional Theory
3. Discussion
4. Materials and Methods
4.1. Plant Materials and Reagents
4.2. Antioxidant Activity
4.2.1. DPPH Radical Scavenging Activity
4.2.2. ABTS Radical Scavenging Activity
4.2.3. Ferric Reducing Antioxidant Potential (FRAP) Assay
4.3. Graph Theoretical Network Analysis
4.4. In silico Study
4.4.1. Ligand Library Preparation
4.4.2. Target Protein Preparation
4.4.3. Investigation of Protein–Ligand Interactions
Active Binding Site Prediction
4.5. Molecular Docking
4.6. Drug-Likeness Evaluation
4.7. Molecular Dynamics Simulation Studies
4.8. Density Functional Theory (DFT)
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Saccomano, S.C.; Cash, K.J. A near-infrared optical nanosensor for measuring aerobic respiration in microbial systems. Analyst 2022, 147, 120–129. [Google Scholar] [CrossRef] [PubMed]
- Koltover, V.K. Redox Signaling and Biomarkers in Ageing; Springer: Berlin/Heidelberg, Germany, 2022; pp. 21–44. [Google Scholar]
- Martemucci, G.; Costagliola, C.; Mariano, M.; D’andrea, L.; Napolitano, P.; D’Alessandro, A.G. Free radical properties, source and targets, antioxidant consumption and health. Oxygen 2022, 2, 48–78. [Google Scholar] [CrossRef]
- Mohan, U.P.; Sriram, B.; Panneerselvam, T.; Devaraj, S.; MubarakAli, D.; Parasuraman, P.; Palanisamy, P.; Premanand, A.; Arunachalam, S.; Kunjiappan, S. Utilization of plant-derived Myricetin molecule coupled with ultrasound for the synthesis of gold nanoparticles against breast cancer. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2020, 393, 1963–1976. [Google Scholar] [CrossRef] [PubMed]
- Kalimuthu, A.K.; Panneerselvam, T.; Pavadai, P.; Pandian, S.R.K.; Sundar, K.; Murugesan, S.; Ammunje, D.N.; Kumar, S.; Arunachalam, S.; Kunjiappan, S.; et al. Pharmacoinformatics-based investigation of bioactive compounds of Rasam (South Indian recipe) against human cancer. Sci. Rep. 2021, 11, 21488. [Google Scholar] [CrossRef]
- Aggarwal, V.; Tuli, H.S.; Varol, A.; Thakral, F.; Yerer, M.B.; Sak, K.; Varol, M.; Jain, A.; Khan, M.A.; Sethi, G. Role of reactive oxygen species in cancer progression: Molecular mechanisms and recent advancements. Biomolecules 2019, 9, 735. [Google Scholar] [CrossRef]
- Palanichamy, C.; Pavadai, P.; Panneerselvam, T.; Arunachalam, S.; Babkiewicz, E.; Pandian, S.R.K.; Jeyarajaguru, K.S.; Ammunje, D.N.; Kannan, S.; Chandrasekaran, J. Aphrodisiac Performance of Bioactive Compounds from Mimosa pudica Linn.: In Silico Molecular Docking and Dynamics Simulation Approach. Molecules 2022, 27, 3799. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Enriquez, F.; Torres, I.; Vina, D. Natural products for the treatment of Alzheimer’s disease: Present and future expectations. Front. Clin. Drug Res. -CNS Neurol. Disord. 2017, 5, 74–170. [Google Scholar]
- Adwas, A.A.; Elsayed, A.; Azab, A.; Quwaydir, F. Oxidative stress and antioxidant mechanisms in human body. J. Appl. Biotechnol. Bioeng. 2019, 6, 43–47. [Google Scholar]
- Santovito, G.; Trentin, E.; Gobbi, I.; Bisaccia, P.; Tallandini, L.; Irato, P. Non-enzymatic antioxidant responses of Mytilus galloprovincialis: Insights into the physiological role against metal-induced oxidative stress. Comp. Biochem. Physiol. Part C Toxicol. Pharmacol. 2021, 240, 108909. [Google Scholar] [CrossRef]
- Kunjiappan, S.; Bhattacharjee, C.; Chowdhury, R. Hepatoprotective and antioxidant effects of Azolla microphylla based gold nanoparticles against acetaminophen induced toxicity in a fresh water common carp fish (Cyprinus carpio L.). Nanomed. J. 2015, 2, 88–110. [Google Scholar]
- Mehla, N.; Chhajer, A.K.; Kumar, K.; Dahiya, S.; Mohindroo, V. Applications of Antioxidants: A Review 13. In Plant Antioxidants and Health; Springer: Berlin/Heidelberg, Germany, 2022; p. 313. [Google Scholar]
- Zhang, J.; Duan, D.; Song, Z.L.; Liu, T.; Hou, Y.; Fang, J. Small molecules regulating reactive oxygen species homeostasis for cancer therapy. Med. Res. Rev. 2021, 41, 342–394. [Google Scholar] [CrossRef] [PubMed]
- Pickering, R.J.; Rosado, C.J.; Sharma, A.; Buksh, S.; Tate, M.; de Haan, J.B. Recent novel approaches to limit oxidative stress and inflammation in diabetic complications. Clin. Transl. Immunol. 2018, 7, e1016. [Google Scholar] [CrossRef] [PubMed]
- Kang, Q.; Yang, C. Oxidative stress and diabetic retinopathy: Molecular mechanisms, pathogenetic role and therapeutic implications. Redox Biol. 2020, 37, 101799. [Google Scholar] [CrossRef]
- Tonelli, C.; Chio, I.I.C.; Tuveson, D.A. Transcriptional regulation by Nrf2. Antioxid. Redox Signal. 2018, 29, 1727–1745. [Google Scholar] [CrossRef]
- Baird, L.; Yamamoto, M. Molecular mechanisms regulating the pharmacological actions of icariin with special focus on PI3K-AKT and Nrf-2 signaling pathways. Mol. Cell. Biol. 2020, 40, e00099-20. [Google Scholar]
- Hayes, A.J.; Skouras, C.; Haugk, B.; Charnley, R.M. Keap1–Nrf2 signalling in pancreatic cancer. Int. J. Biochem. Cell Biol. 2015, 65, 288–299. [Google Scholar] [CrossRef]
- Tao, S.; Liu, P.; Luo, G.; de la Vega, M.R.; Chen, H.; Wu, T.; Tillotson, J.; Chapman, E.; Zhang, D.D. p97 negatively regulates NRF2 by extracting ubiquitylated NRF2 from the KEAP1-CUL3 E3 complex. Mol. Cell. Biol. 2017, 37, e00660-16. [Google Scholar] [CrossRef]
- Deshmukh, P.; Unni, S.; Krishnappa, G.; Padmanabhan, B. The Keap1–Nrf2 pathway: Promising therapeutic target to counteract ROS-mediated damage in cancers and neurodegenerative diseases. Biophys. Rev. 2017, 9, 41–56. [Google Scholar] [CrossRef] [PubMed]
- Vallejo, M.J.; Salazar, L.; Grijalva, M. Oxidative stress modulation and ROS-mediated toxicity in cancer: A review on in vitro models for plant-derived compounds. Oxidative Med. Cell. Longev. 2017, 2017, 4586068. [Google Scholar] [CrossRef]
- Kumar, Y.; Yadav, D.N.; Ahmad, T.; Narsaiah, K. Recent trends in the use of natural antioxidants for meat and meat products. Compr. Rev. Food Sci. Food Saf. 2015, 14, 796–812. [Google Scholar] [CrossRef]
- Srivastava, A.; Srivastava, P.; Pandey, A.; Khanna, V.; Pant, A. Phytomedicine: A Potential Alternative Medicine in Controlling Neurological Disorders, New Look to Phytomedicine; Elsevier: Amsterdam, The Netherlands, 2019; pp. 625–655. [Google Scholar]
- Kawlni, L.; Bora, M.; Upadhyay, S.N.; Mukherjee, K.; Hazra, J. Pharmacological and therapeutic profile of anantamula (hemidesmus indicus (l.) R. Br.): A comprehensive review. Int. J. Ayurveda Pharma Res. 2017, 5. [Google Scholar]
- Turrini, E.; Calcabrini, C.; Tacchini, M.; Efferth, T.; Sacchetti, G.; Guerrini, A.; Paganetto, G.; Catanzaro, E.; Greco, G.; Fimognari, C. In vitro study of the cytotoxic, cytostatic, and antigenotoxic profile of Hemidesmus indicus (L.) R. Br. (Apocynaceae) crude drug extract on Tlymphoblastic cells. Toxins 2018, 10, 70. [Google Scholar] [CrossRef] [PubMed]
- Valko, M.; Rhodes, C.; Moncol, J.; Izakovic, M.; Mazur, M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem. Biol. Interact. 2006, 160, 1–40. [Google Scholar] [CrossRef] [PubMed]
- Gladyshev, V.N. The free radical theory of aging is dead. Long live the damage theory! Antioxid. Redox Signal. 2014, 20, 727–731. [Google Scholar] [CrossRef]
- Di Mascio, P.; Martinez, G.R.; Miyamoto, S.; Ronsein, G.E.; Medeiros, M.H.; Cadet, J. Singlet molecular oxygen reactions with nucleic acids, lipids, and proteins. Chem. Rev. 2019, 119, 2043–2086. [Google Scholar] [CrossRef] [PubMed]
- Russell, W.; Duthie, G. Plant secondary metabolites and gut health: The case for phenolic acids. Proc. Nutr. Soc. 2011, 70, 389–396. [Google Scholar] [CrossRef]
- Raghunath, A.; Sundarraj, K.; Nagarajan, R.; Arfuso, F.; Bian, J.; Kumar, A.P.; Sethi, G.; Perumal, E. Antioxidant response elements: Discovery, classes, regulation and potential applications. Redox Biol. 2018, 17, 297–314. [Google Scholar] [CrossRef]
- Ediriweera, M.K.; Tennekoon, K.H.; Samarakoon, S.R. Vernolactone promotes apoptosis and autophagy in human teratocarcinomal (NTERA-2) cancer stem-like cells. J. Appl. Toxicol. 2019, 39, 38–71. [Google Scholar] [CrossRef]
- Selvaraj, K.; Chowdhury, R.; Bhattacharjee, C. Isolation and structural elucidation of flavonoids from aquatic fern Azolla microphylla and evaluation of free radical scavenging activity. Int. J. Pharm. Pharm. Sci. 2013, 5, 743–749. [Google Scholar]
- Cetin, A.; Geçibesler, I.H. Evaluation as antioxidant agents of 1, 2, 4-triazole derivatives: Effects of essential functional groups. J. Appl. Pharm. Sci. 2015, 5, 120–126. [Google Scholar] [CrossRef]
- Awika, J.M.; Rooney, L.W.; Wu, X.; Prior, R.L.; Cisneros-Zevallos, L. Assays for hydrophilic and lipophilic antioxidant capacity (oxygen radical absorbance capacity (ORACFL)) of plasma and other biological and food samples. J. Agric. Food Chem. 2003, 51, 6657–6662. [Google Scholar] [CrossRef]
- Olğaç, A.; Türe, A.; Olğaç, S.; Möller, S. High-Performance Modelling and Simulation for Big Data Applications; Springer: Cham, Switzerland, 2019; pp. 250–278. [Google Scholar]
- Khan, D.A.; Hamdani, S.D.A.; Iftikhar, S.; Malik, S.Z.; Zaidi, N.-U.-S.S.; Gul, A.; Babar, M.M.; Ozturk, M.; Unal, B.T.; Gonenc, T. Pharmacoinformatics approaches in the discovery of drug-like antimicrobials of plant origin. J. Biomol. Struct. Dyn. 2022, 40, 7612–7628. [Google Scholar] [CrossRef]
- Gechev, T.S.; Van Breusegem, F.; Stone, J.M.; Denev, I.; Laloi, C. Reactive oxygen species as signals that modulate plant stress responses and programmed cell death. Bioessays 2006, 28, 1091–1101. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.-Y.; Lu, M.-C.; Xu, L.L.; Yang, T.-T.; Xi, M.-Y.; Xu, X.-L.; Guo, X.-K.; Zhang, X.-J.; You, Q.-D.; Sun, H.-P. Discovery of potent Keap1–Nrf2 protein–protein interaction inhibitor based on molecular binding determinants analysis. J. Med. Chem. 2014, 57, 2736–2745. [Google Scholar] [CrossRef]
- Ahmad, K.; Bhat, A.R.; Athar, F. Antioxidant and apoptotic effects of Callistemon lanceolatus leaves and their compounds against human cancer cells. Lett. Drug Des. Discov. 2017, 14, 488–499. [Google Scholar] [CrossRef]
- Cosme, P.; Rodríguez, A.B.; Espino, J.; Garrido, M. Plant phenolics: Bioavailability as a key determinant of their potential health-promoting applications. Antioxidants 2020, 9, 1263. [Google Scholar] [CrossRef]
- Chen, X.; Li, H.; Tian, L.; Li, Q.; Luo, J.; Zhang, Y. Analysis of the physicochemical properties of acaricides based on Lipinski’s rule of five. J. Comput. Biol. 2020, 27, 1397–1406. [Google Scholar] [CrossRef]
- Zafrani, Y.; Yeffet, D.; Sod-Moriah, G.; Berliner, A.; Amir, D.; Marciano, D.; Gershonov, E.; Saphier, S. Difluoromethyl bioisostere: Examining the “lipophilic hydrogen bond donor” concept. J. Med. Chem. 2017, 60, 797–804. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-J.; Gan, R.-Y.; Li, S.; Zhou, Y.; Li, A.-N.; Xu, D.-P.; Li, H.-B. Antioxidant phytochemicals for the prevention and treatment of chronic diseases. Molecules 2015, 20, 21138–21156. [Google Scholar] [CrossRef] [PubMed]
- Di, L.; Kerns, E. Drug-Like Properties: Concepts, Structure Design and Methods from ADME to Toxicity Optimization; Academic Press: Cambridge, MA, USA, 2015. [Google Scholar]
- Deore, A.B.; Dhumane, J.R.; Wagh, R.; Sonawane, R. The stages of drug discovery and development process. Asian J. Pharm. Res. Dev. 2019, 7, 62–67. [Google Scholar] [CrossRef]
- Fermini, B.; Fossa, A.A. The impact of drug-induced QT interval prolongation on drug discovery and development. Nat. Rev. Drug Discov. 2003, 2, 439–447. [Google Scholar] [CrossRef] [PubMed]
- Rasool, N.; Akhtar, A.; Hussain, W. Insights into the inhibitory potential of selective phytochemicals against Mpro of 2019-nCoV: A computer-aided study. Struct. Chem. 2020, 31, 1777–1783. [Google Scholar] [CrossRef]
- Miar, M.; Shiroudi, A.; Pourshamsian, K.; Oliaey, A.R.; Hatamjafari, F. Theoretical investigations on the HOMO–LUMO gap and global reactivity descriptor studies, natural bond orbital, and nucleus-independent chemical shifts analyses of 3-phenylbenzo [d] thiazole-2 (3 H)-imine and its para-substituted derivatives: Solvent and substituent effects. J. Chem. Res. 2021, 45, 147–158. [Google Scholar]
- Jordaan, M.A.; Ebenezer, O.; Damoyi, N.; Shapi, M. Mapping scientific productivity trends and hotspots in remdesivir research publications: A bibliometric study from 2016 to 2021. Heliyon 2020, 6, e04642. [Google Scholar] [CrossRef] [PubMed]
- Kalimuthu, A.K.; Parasuraman, P.; Sivakumar, P.; Murugesan, S.; Arunachalam, S.; Pandian, S.R.K.; Ravishankar, V.; Ammunje, D.N.; Sampath, M.; Panneerselvam, T. In silico, in vitro screening of antioxidant and anticancer potentials of bioactive secondary metabolites from an endophytic fungus (Curvularia sp.) from Phyllanthus niruri L. Environ. Sci. Pollut. Res. 2022, 1–18. [Google Scholar] [CrossRef]
- Ganesan, V.; Gurumani, V.; Kunjiappan, S.; Panneerselvam, T.; Somasundaram, B.; Kannan, S.; Chowdhury, A.; Saravanan, G.; Bhattacharjee, C. Optimization and analysis of microwave-assisted extraction of bioactive compounds from Mimosa pudica L. using RSM & ANFIS modeling. J. Food Meas. Charact. 2018, 12, 228–242. [Google Scholar]
- Benzie, I.F.; Strain, J.J. The ferric reducing ability of plasma (FRAP) as a measure of “antioxidant power”: The FRAP assay. Anal. Biochem. 1996, 239, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Pulido, R.; Bravo, L.; Saura-Calixto, F. Antioxidant activity of dietary polyphenols as determined by a modified ferric reducing/antioxidant power assay. J. Agric. Food Chem. 2000, 48, 3396–3402. [Google Scholar] [CrossRef] [PubMed]
- Dessai, P.G.; Dessai, S.P.; Dabholkar, R.; Pednekar, P.; Naik, S.; Mamledesai, S.; Gopal, M.; Pavadai, P.; Kumar, B.K.; Murugesan, S. Design, synthesis, graph theoretical analysis and molecular modelling studies of novel substituted quinoline analogues as promising anti-breast cancer agents. Mol. Divers. 2022, 1–20. [Google Scholar] [CrossRef]
- Mohanraj, K.; Karthikeyan, B.S.; Vivek-Ananth, R.; Chand, R.; Aparna, S.; Mangalapandi, P.; Samal, A. IMPPAT: A curated database of Indian Medicinal Plants, Phytochemistry and Therapeutics. Sci. Rep. 2018, 8, 4329. [Google Scholar] [CrossRef]
- Burley, S.K.; Berman, H.M.; Bhikadiya, C.; Bi, C.; Chen, L.; Di Costanzo, L.; Christie, C.; Dalenberg, K.; Duarte, J.M.; Dutta, S. RCSB Protein Data Bank: Biological macromolecular structures enabling research and education in fundamental biology, biomedicine, biotechnology and energy. Nucleic Acids Res. 2019, 47, D464–D474. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, W.; Littlejohn, T.G. Swiss-PDB viewer (deep view). Brief. Bioinform. 2001, 2, 195–197. [Google Scholar] [CrossRef] [PubMed]
- Jendele, L.; Krivak, R.; Skoda, P.; Novotny, M.; Hoksza, D. Proteins Plus: Interactive analysis of protein–ligand binding interfaces. Nucleic Acids Res. 2019, 47, W345–W349. [Google Scholar] [CrossRef] [PubMed]
- Kumar, B.; Parasuraman, P.; Murthy, T.P.K.; Murahari, M.; Chandramohan, V. In silico screening of therapeutic potentials from Strychnos nux-vomica against the dimeric main protease (Mpro) structure of SARS-CoV-2. J. Biomol. Struct. Dyn. 2022, 40, 7796–7814. [Google Scholar] [CrossRef] [PubMed]
- Alamri, M.A.; Qamar, M.T.U.; Mirza, M.U.; Bhadane, R.; Alqahtani, S.M.; Muneer, I.; Froeyen, M.; Salo-Ahen, O.M. Pharmacoinformatics and molecular dynamics simulation studies reveal potential covalent and FDA-approved inhibitors of SARS-CoV-2 main protease 3CLpro. J. Biomol. Struct. Dyn. 2021, 39, 4936–4948. [Google Scholar] [CrossRef]
- Mishra, S.; Dahima, R. Identification of potential antiviral inhibitors from hydroxychloroquine and 1, 2, 4, 5-tetraoxanes analogues and investigation of the mechanism of action in SARS-CoV-2. J. Drug Deliv. Ther. 2019, 9, 366–369. [Google Scholar]
- Vijayalakshmi, M.; Dhanapradeeba, V.; Kunjiappan, S.; Sundar, K.; Pandian, S.R.K. Targeting TLRs with the Derivatives of Mimosa Pudica: An In Silico Approach. Biointerface Res. Appl. Chem. 2022, 2023, 3. [Google Scholar]
- Shaw, D.E.; Deneroff, M.M.; Dror, R.O.; Kuskin, J.S.; Larson, R.H.; Salmon, J.K.; Young, C.; Batson, B.; Bowers, K.J.; Chao, J.C. Anton, a special-purpose machine for molecular dynamics simulation. ACM SIGARCH Comput. Archit. News 2007, 35, 1–12. [Google Scholar] [CrossRef]
- Mark, P.; Nilsson, L. Structure and dynamics of the TIP3P, SPC, and SPC/E water models at 298 K. J. Phys. Chem. A 2001, 105, 9954–9960. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Maxwell, D.S.; Tirado-Rives, J. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J. Am. Chem. Soc. 1996, 118, 11225–11236. [Google Scholar] [CrossRef]
- Tuckerman, M.; Berne, B.J.; Martyna, G.J. Reversible multiple time scale molecular dynamics. J. Chem. Phys. 1992, 97, 1990–2001. [Google Scholar] [CrossRef]
- Cheng, A.; Merz, K.M. Application of the Nosé− Hoover chain algorithm to the study of protein dynamics. J. Phys. Chem. 1996, 100, 1927–1937. [Google Scholar] [CrossRef]
- Kalibaeva, G.; Ferrario, M.; Ciccotti, G. Constant pressure-constant temperature molecular dynamics: A correct constrained NPT ensemble using the molecular virial. Mol. Phys. 2003, 101, 765–778. [Google Scholar] [CrossRef]
- Saravanan, G.; Panneerselvam, T.; Kunjiappan, S.; Parasuraman, P.; Alagarsamy, V.; Udayakumar, P.; Soundararajan, M.; Joshi, S.D.; Ramalingam, S.; Ammunje, D.N. Graph theoretical analysis, in silico modeling, prediction of toxicity, metabolism and synthesis of novel 2-(methyl/phenyl)-3-(4-(5-substituted-1, 3, 4-oxadiazol-2-yl) phenyl) quinazolin-4 (3H)-ones as NMDA receptor inhibitor. Drug Dev. Res. 2019, 80, 368–385. [Google Scholar] [CrossRef] [PubMed]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S. No | Compound ID (CID) | Active Compound | Docking Score (Kcal × mol−1) |
|---|---|---|---|
| 1. | 379 | Octanoic acid | −4.6 |
| 2. | 643731 | trans-2,cis-6-Nonadienal | −4.7 |
| 3. | 2969 | Decanoic acid | −4.8 |
| 4. | 785 | Hydroquinone | −5.0 |
| 5. | 12412 | Hexatriacontane | −5.0 |
| 6. | 21146488 | (1S,3aR,5aR,5bR,7aS,11aS,11bR,13aR,13bR)-3a,5a,5b,8,8,11a-hexamethyl-1-propan-2-yl-1,2,3,4,5,6,7,7a,9,10,11,11b,12,13,13a,13b-hexadecahydrocyclopenta[a]chrysene | −5.0 |
| 7. | 460 | Guaiacol | −5.1 |
| 8. | 6998 | Salicylaldehyde | −5.1 |
| 9. | 31244 | 4-Methoxybenzaldehyde | −5.1 |
| 10. | 3893 | Lauric acid | −5.2 |
| 11. | 22311 | Limonene | −5.2 |
| 12. | 9007 | 3-Methoxyphenol | −5.3 |
| 13. | 985 | Palmitic acid | −5.6 |
| 14. | 2758 | Eucalyptol | −5.7 |
| 15. | 4133 | Methyl salicylate | −5.7 |
| 16. | 69600 | 2-Hydroxy-4-methoxybenzaldehyde | −5.7 |
| 17. | 1183 | Vanillin | −5.7 |
| 18. | 444539 | Cinnamic acid | −5.8 |
| 19. | 8294 | Linalyl acetate | −5.8 |
| 20. | 12127 | Isovanillin | −5.9 |
| 21. | 11230 | 4-Carvomenthenol | −5.9 |
| 22. | 61130 | Myrtenal | −5.9 |
| 23. | 29025 | Verbenone | −5.9 |
| 24. | 93046 | 2,10-Epoxypinane | −5.9 |
| 25. | 6552009 | d-Borneol | −6.0 |
| 26. | 10582 | Myrtenol | −6.0 |
| 27. | 6989 | Thymol | −6.0 |
| 28. | 17100 | Alpha-Terpineol | −6.0 |
| 29. | 121719 | Pinocarvone | −6.1 |
| 30. | 5355854 | Pentyl cinnamate | −6.2 |
| 31. | 5284507 | Nerolidol | −6.2 |
| 32. | 5281522 | Isocaryophyllene | −6.3 |
| 33. | 2537 | Camphor | −6.3 |
| 34. | 3469 | 2,5-Dihydroxybenzoic acid | −6.4 |
| 35. | 6448 | Bornyl acetate | −6.4 |
| 36. | 30248 | Dihydrocarvyl acetate | −6.4 |
| 37. | 75231 | 4-Methoxysalicylic acid | −6.4 |
| 38. | 8468 | Vanillic acid | −6.4 |
| 39. | 637542 | 4-Hydroxycinnamic acid | −6.5 |
| 40. | 6918391 | Beta-Elemene | −6.6 |
| 41. | 6950273 | Isobornyl acetate | −6.6 |
| 42. | 72 | 3,4-Dihydroxybenzoic acid | −6.6 |
| 43. | 10742 | Syringic acid | −6.6 |
| 44. | 3102 | Benzophenone | −6.7 |
| 45. | 370 | Gallic acid | −6.8 |
| 46. | 111037 | Alpha-Terpinyl acetate | −6.8 |
| 47. | 689043 | Caffeic acid | −6.8 |
| 48. | 445858 | Ferulic acid | −6.9 |
| 49. | 91354 | Aromadendrene | −6.9 |
| 50. | 92812 | Ledol | −7.0 |
| 51. | 5369459 | Phenethyl cinnamate | −7.1 |
| 52. | 2345 | Benzyl benzoate | −7.1 |
| 53. | 442343 | Levomenol | −7.3 |
| 54. | 100949538 | Alpha-Muurolol | −7.5 |
| 55. | 442393 | Beta-Selinene | −7.5 |
| 56. | 5280804 | Isoquercitrin | −8.8 |
| 57. | 5281643 | Hyperoside | −8.8 |
| 58. | 9548870 | Ursane | −8.9 |
| 59. | 92157 | Lupeol acetate | −9.0 |
| 60. | 5280805 | Rutin | −9.1 |
| 61. | 259846 | Lupeol | −9.1 |
| 62. | 222284 | Beta-Sitosterol | −9.3 |
| 63. | 9548717 | Oleanane | −9.5 |
| 64. | 92156 | Beta-Amyrin acetate | −9.6 |
| 65. | 16129778 | Tannic acid | −9.6 |
| 66. | 73170 | Alpha-Amyrin | −9.7 |
| 67. | 5280343 | Quercetin | −9.8 |
| 68. | 73145 | Beta-Amyrin | −10 |
| 69. | 101664025 | Hemidescine | −11.3 |
| Standard Drug | |||
| 70. | 73330369 | CPUY192018 | −9.10 |
| Parameter | Hemidescine (CID: 101664025) | Beta-Amyrin (CID: 73145) | Quercetin (CID: 5280343) | CPUY192018 (CID: 73330369) |
|---|---|---|---|---|
| Formula | C36H58O10 | C30H50O | C15H10O7 | C28H26N2O10S2 |
| MW (g × mol−1) | 650.84 | 426.72 | 302.24 | 614.64 |
| Num. heavy atoms | 46 | 0 | 22 | 42 |
| Num. arom. heavy atoms | 0 | 12 | 16 | 22 |
| Fraction Csp3 | 0.92 | 0.93 | 0.00 | 0.14 |
| Num. rotatable bonds | 8 | 0 | 1 | 12 |
| Num. H-bond acceptors | 10 | 1 | 7 | 10 |
| Num. H-bond donors | 3 | 1 | 5 | 02 |
| Molar Refractivity | 171.72 | 134.88 | 78.04 | 154.12 |
| TPSA (Å2) | 131.14 | 20.23 | 131.36 | 184.58 |
| Solubility class | Moderately soluble | Poorly Soluble | Soluble | Moderately soluble |
| GI absorption | Low | Low | High | Low |
| BBB permeation | No | No | No | No |
| Violation of Lipinski’s rule of five | 1 | 1 | 0 | 2 |
| Violation of Veber rule | Yes | Yes | Yes | 2 |
| Bioavailability Score | 0.55 | 0.55 | 0.55 | 0.11 |
| Synthetic accessibility | 8.01 | 6.04 | 3.23 | 3.83 |
| Compound | AMES Toxicity | Max. Tolerated Dose (Human) | hERG Inhibition | LD50 | Hepatotoxicity | Carcinogenicity | Skin Sensitisation | T. pyriformis Toxicity | Minnow Toxicity |
|---|---|---|---|---|---|---|---|---|---|
| Hemidescine (CID: 101664025) | No | −1.41 | No | 2.442 | No | No | No | 0.286 | 0.714 |
| Beta-Amyrin (CID: 73145) | No | +0.33 | No | 2.139 | No | No | No | 0.599 | −2.344 |
| Quercetin (CID: 5280343) | Yes | +0.984 | No | 2.251 | No | No | No | 0.418 | 2.487 |
| CPUY192018 (CID: 73330369) | No | +0.52 | No | 1.841 | Yes | No | No | 0.286 | −0.527 |
| Compound Name | HOMO | EHOMO (ev) | LUMO | ELUMO (ev) | Energy Gap (Δev) |
|---|---|---|---|---|---|
| Hemidescine |  | −9.5378 |  | −4.4741 | 5.0637 |
| Beta Amyrin |  | −9.8576 |  | −4.6115 | 5.2460 |
| Quercetin |  | −8.4817 |  | −5.3682 | 3.1135 |
| Standard drug (CPUY192018) |  | −8.5658 |  | −5.9731 | 2.5927 |
| Gene | Betweenness | Closeness | Degree | Eccentricity | EigenVector | Radiality | Stress |
|---|---|---|---|---|---|---|---|
| SHC2 | 33 | 2.45 | 3 | 0.17 | 3.61E-16 | 28.74312 | 36 |
| IGF1R | 8 | 5.33 | 3 | 0.14 | 1.36E-16 | 29.22018 | 14 |
| LEF1 | 22 | 2.00 | 3 | 1.01 | 9.65E-30 | 24.22936 | 22 |
| CSNK1A1L | 79.5 | 5.16 | 3 | 0.33 | 4.39E-30 | 25.68807 | 81 |
| DVL1 | 67.5 | 4.60 | 3 | 0.14 | 2.53E-30 | 27.99083 | 70 |
| PLCG1 | 32 | 3.95 | 3 | 0.17 | −1.74E-17 | 28.23853 | 32 |
| PRKCA | 32 | 2.08 | 3 | 0.25 | 1.84E-15 | 27.2844 | 32 |
| TERC | 12.5 | 6.45 | 3 | 0.17 | −3.22E-14 | 24.2844 | 13 |
| EGFR | 69 | 8.75 | 4 | 0.14 | −3.04E-25 | 28.40367 | 69 |
| HRAS | 122 | 4.99 | 4 | 0.39 | 1.63E-30 | 54.9633 | 167 |
| ARAF | 122 | 4.06 | 4 | 0.55 | 2.11E-14 | 53.21101 | 172 |
| GRB2 | 110 | 5.46 | 4 | 0.25 | 2.55E-15 | 57.21101 | 145 |
| WNT16 | 81 | 10.03 | 4 | 0.04 | 1.23E-14 | 29.49541 | 102 |
| RB1 | 16.5 | 2.00 | 4 | 2.00 | −9.90E-30 | 47.02752 | 17 |
| MET | 92 | 7.28 | 4 | 0.04 | −2.68E-15 | 29.10092 | 122 |
| PIK3CA | 61 | 5.08 | 5 | 0.5 | −6.01E-15 | 57.19266 | 66 |
| C05981 | 64 | 5.00 | 5 | 0.67 | −1.71E-15 | 56.22936 | 68 |
| NFE2L2 | 40 | 4.00 | 5 | 1.00 | 8.10E-15 | 25.58716 | 40 |
| AKT3 | 57 | 5.5 | 6 | 1.00 | −1.82E-15 | 53.80734 | 60 |
| MAP2K1 | 97 | 9.5 | 10 | 2.00 | 6.27E-15 | 79.3945 | 100 |
| TP53 | 21.5 | 8.28 | 8 | 0.20 | −5.49E-14 | 23.59633 | 22 |
| KEAP1 | 10.5 | 1.00 | 11 | 1.00 | 6.88E-31 | 29.38532 | 12 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vellur, S.; Pavadai, P.; Babkiewicz, E.; Ram Kumar Pandian, S.; Maszczyk, P.; Kunjiappan, S. An In Silico Molecular Modelling-Based Prediction of Potential Keap1 Inhibitors from Hemidesmus indicus (L.) R.Br. against Oxidative-Stress-Induced Diseases. Molecules 2023, 28, 4541. https://doi.org/10.3390/molecules28114541
Vellur S, Pavadai P, Babkiewicz E, Ram Kumar Pandian S, Maszczyk P, Kunjiappan S. An In Silico Molecular Modelling-Based Prediction of Potential Keap1 Inhibitors from Hemidesmus indicus (L.) R.Br. against Oxidative-Stress-Induced Diseases. Molecules. 2023; 28(11):4541. https://doi.org/10.3390/molecules28114541
Chicago/Turabian StyleVellur, Senthilkumar, Parasuraman Pavadai, Ewa Babkiewicz, Sureshbabu Ram Kumar Pandian, Piotr Maszczyk, and Selvaraj Kunjiappan. 2023. "An In Silico Molecular Modelling-Based Prediction of Potential Keap1 Inhibitors from Hemidesmus indicus (L.) R.Br. against Oxidative-Stress-Induced Diseases" Molecules 28, no. 11: 4541. https://doi.org/10.3390/molecules28114541
APA StyleVellur, S., Pavadai, P., Babkiewicz, E., Ram Kumar Pandian, S., Maszczyk, P., & Kunjiappan, S. (2023). An In Silico Molecular Modelling-Based Prediction of Potential Keap1 Inhibitors from Hemidesmus indicus (L.) R.Br. against Oxidative-Stress-Induced Diseases. Molecules, 28(11), 4541. https://doi.org/10.3390/molecules28114541