
Structure-Guided Development of Bivalent Aptamers Blocking SARS-CoV-2 Infection
 , , ,
, , ,
Abstract
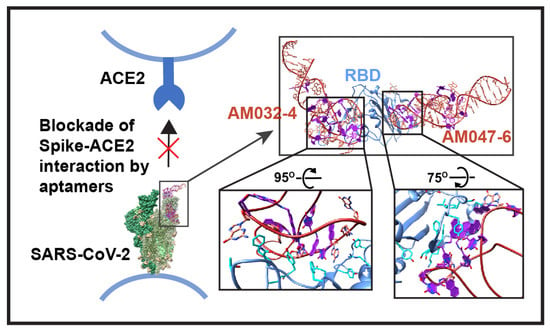
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Selection and Characterization of Aptamers Binding to Spike Proteins of SARS-CoV-2
2.2. Aptamers AM032-0 and AM047-0 Block SARS-CoV-2 Infection through Inhibition of the Interaction between RBD and ACE2
2.3. Determination of the Three-Dimensional Structure of RBD–Aptamer Complex by Cryo-EM
2.4. Aptamers AM032-0 and AM047-0 Block the Association of ACE2 to RBD through Distinct Mechanisms
2.5. Optimization of Aptamers AM032-0 and AM047-0
2.6. Structure of RBD–Fab–AM032-4–AM047-6 Complex
2.7. Generation of a Bivalent Aptamer That Strongly Inhibits SARS-CoV-2 Pseudovirus Infection
3. Discussion
4. Materials and Methods
4.1. Cloning, Expression, and Purification of Proteins
4.2. Preparation of Surrogate Virus
4.3. Systematic Evolution of Ligands by Exponential Enrichment (SELEX)
4.4. SARS-CoV-2 Spike Aptamer Synthesis
4.5. Measurement of the KD Values of Aptamers
4.6. Protein-Binding Competition Assay
4.7. Enzyme Linked Immunosorbent Assay (ELISA)
4.8. Measurement of Antiviral Activities of Aptamers
4.9. Cryo-EM Grid Preparation
4.10. Data Processing for RBD–Fab–AM032-0–AM047-0 Complex
4.11. Data Processing for RBD–Fab–AM032-4–AM047-6 Complex
4.12. Model Building and Refinement
4.13. Accession Numbers
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [PubMed]
- Coronavirus Disease 2019 (COVID-19). WHO. Available online: https://covid19.who.int/ (accessed on 10 May 2023).
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef] [PubMed]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Kruger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e278. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.F.; Kok, K.H.; Zhu, Z.; Chu, H.; To, K.K.; Yuan, S.; Yuen, K.Y. Genomic characterization of the 2019 novel human-pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan. Emerg. Microbes Infect. 2020, 9, 221–236. [Google Scholar] [CrossRef]
- Piccoli, L.; Park, Y.J.; Tortorici, M.A.; Czudnochowski, N.; Walls, A.C.; Beltramello, M.; Silacci-Fregni, C.; Pinto, D.; Rosen, L.E.; Bowen, J.E.; et al. Mapping Neutralizing and Immunodominant Sites on the SARS-CoV-2 Spike Receptor-Binding Domain by Structure-Guided High-Resolution Serology. Cell 2020, 183, 1024–1042.e102. [Google Scholar] [CrossRef]
- Polack, F.P.; Thomas, S.J.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Perez, J.L.; Perez Marc, G.; Moreira, E.D.; Zerbini, C.; et al. Safety and Efficacy of the BNT162b2 mRNA COVID-19 Vaccine. N. Engl. J. Med. 2020, 383, 2603–2615. [Google Scholar] [CrossRef]
- Baden, L.R.; El Sahly, H.M.; Essink, B.; Kotloff, K.; Frey, S.; Novak, R.; Diemert, D.; Spector, S.A.; Rouphael, N.; Creech, C.B.; et al. Efficacy and Safety of the mRNA-1273 SARS-CoV-2 Vaccine. N. Engl. J. Med. 2021, 384, 403–416. [Google Scholar] [CrossRef]
- Su, S. Learning from the past: Development of safe and effective COVID-19 vaccines. Nat. Rev. Microbiol. 2021, 19, 211–219. [Google Scholar] [CrossRef]
- Hassine, I.H. COVID-19 vaccines and variants of concern: A review. Rev. Med. Virol. 2022, 32, e2313. [Google Scholar] [CrossRef] [PubMed]
- Bernal, A.J.; da Silva, M.M.G.; Musungaie, D.B.; Kovalchuk, E.; Gonzalez, A.; Delos Reyes, V.; Martin-Quiros, A.; Caraco, Y.; Williams-Diaz, A.; Brown, M.L.; et al. Molnupiravir for Oral Treatment of COVID-19 in Nonhospitalized Patients. N. Engl. J. Med. 2022, 386, 509–520. [Google Scholar] [CrossRef] [PubMed]
- Najjar-Debbiny, R.; Gronich, N.; Weber, G.; Khoury, J.; Amar, M.; Stein, N.; Goldstein, L.H.; Saliba, W. Effectiveness of Paxlovid in Reducing Severe Coronavirus Disease 2019 and Mortality in High-Risk Patients. Clin. Infect. Dis. 2022, 76, 1158–1159. [Google Scholar] [CrossRef] [PubMed]
- Barnes, C.O.; Jette, C.A.; Abernathy, M.E.; Dam, K.A.; Esswein, S.R.; Gristick, H.B.; Malyutin, A.G.; Sharaf, N.G.; Huey-Tubman, K.E.; Lee, Y.E.; et al. SARS-CoV-2 neutralizing antibody structures inform therapeutic strategies. Nature 2020, 588, 682–687. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.S.; Wheatley, A.K.; Kent, S.J.; DeKosky, B.J. Antibody-dependent enhancement and SARS-CoV-2 vaccines and therapies. Nat. Microbiol. 2020, 5, 1185–1191. [Google Scholar] [CrossRef]
- Dejnirattisai, W.; Jumnainsong, A.; Onsirisakul, N.; Fitton, P.; Vasanawathana, S.; Limpitikul, W.; Puttikhunt, C.; Edwards, C.; Duangchinda, T.; Supasa, S.; et al. Cross-Reacting Antibodies Enhance Dengue Virus Infection in Humans. Science 2010, 328, 745–748. [Google Scholar] [CrossRef] [PubMed]
- Aleem, A.; Akbar Samad, A.B.; Slenker, A.K. Emerging Variants of SARS-CoV-2 And Novel Therapeutics Against Coronavirus (COVID-19). In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Hoffmann, M.; Arora, P.; Gross, R.; Seidel, A.; Hornich, B.F.; Hahn, A.S.; Kruger, N.; Graichen, L.; Hofmann-Winkler, H.; Kempf, A.; et al. SARS-CoV-2 variants B.1.351 and P.1 escape from neutralizing antibodies. Cell 2021, 184, 2384–2393. [Google Scholar] [CrossRef]
- Korber, B.; Fischer, W.M.; Gnanakaran, S.; Yoon, H.; Theiler, J.; Abfalterer, W.; Hengartner, N.; Giorgi, E.E.; Bhattacharya, T.; Foley, B.; et al. Tracking Changes in SARS-CoV-2 Spike: Evidence that D614G Increases Infectivity of the COVID-19 Virus. Cell 2020, 182, 812–827. [Google Scholar] [CrossRef]
- Tuerk, C.; Gold, L. Systematic Evolution of Ligands by Exponential Enrichment—Rna Ligands to Bacteriophage-T4 DNA-Polymerase. Science 1990, 249, 505–510. [Google Scholar] [CrossRef]
- Keefe, A.D.; Pai, S.; Ellington, A. Aptamers as therapeutics. Nat. Rev. Drug. Discov. 2010, 9, 537–550. [Google Scholar] [CrossRef]
- Xu, X.; Zhang, C.; Denton, D.T.; O’Connell, D.; Drolet, D.W.; Geisbrecht, B.V. Inhibition of the Complement Alternative Pathway by Chemically Modified DNA Aptamers That Bind with Picomolar Affinity to Factor B. J. Immunol. 2021, 206, 861–873. [Google Scholar] [CrossRef] [PubMed]
- Vaught, J.D.; Bock, C.; Carter, J.; Fitzwater, T.; Otis, M.; Schneider, D.; Rolando, J.; Waugh, S.; Wilcox, S.K.; Eaton, B.E. Expanding the Chemistry of DNA for in Vitro Selection. J. Am. Chem. Soc. 2010, 132, 4141–4151. [Google Scholar] [CrossRef]
- Gelinas, A.D.; Tan, T.K.; Liu, S.; Jaramillo, J.G.; Chadwick, J.; Harding, A.C.; Zhang, C.; Ream, B.E.; Chase, C.N.; Otis, M.R.; et al. Broadly neutralizing aptamers to SARS-CoV-2: A diverse panel of modified DNA antiviral agents. Mol. Ther. Nucleic Acids 2023, 31, 370–382. [Google Scholar] [CrossRef]
- Gold, L.; Ayers, D.; Bertino, J.; Bock, C.; Bock, A.; Brody, E.N.; Carter, J.; Dalby, A.B.; Eaton, B.E.; Fitzwater, T.; et al. Aptamer-Based Multiplexed Proteomic Technology for Biomarker Discovery. PLoS ONE 2010, 5, e15004. [Google Scholar] [CrossRef]
- Sun, M.; Wu, Z.J.; Zhang, J.L.; Chen, M.Y.; Lu, Y.; Yang, C.Y.; Song, Y.L. Spherical neutralizing aptamer suppresses SARS-CoV-2 Omicron escape. Nano Today 2022, 44, e101499. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Liu, S.W.; Wei, X.Y.; Wan, S.; Huang, M.J.; Song, T.; Lu, Y.; Weng, X.N.; Lin, Z.; Chen, H.L.; et al. Aptamer Blocking Strategy Inhibits SARS-CoV-2 Virus Infection. Angew. Chem. Int. Ed. 2021, 60, 10266–10272. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Song, J.; Wei, X.; Huang, M.; Sun, M.; Zhu, L.; Lin, B.; Shen, H.; Zhu, Z.; Yang, C. Discovery of Aptamers Targeting the Receptor-Binding Domain of the SARS-CoV-2 Spike Glycoprotein. Anal. Chem. 2020, 92, 9895–9900. [Google Scholar] [CrossRef]
- Liu, X.H.; Wang, Y.L.; Wu, J.; Qi, J.J.; Zeng, Z.H.; Wan, Q.Y.; Chen, Z.H.; Manandhar, P.; Cavener, V.S.; Boyle, N.R.; et al. Neutralizing Aptamers Block S/RBD-ACE2 Interactions and Prevent Host Cell Infection. Angew. Chem. Int. Ed. 2021, 60, 10273–10278. [Google Scholar] [CrossRef]
- Valero, J.; Civit, L.; Dupont, D.M.; Selnihhin, D.; Reinert, L.S.; Idorn, M.; Israels, B.A.; Bednarz, A.M.; Bus, C.; Asbach, B.; et al. A serum-stable RNA aptamer specific for SARS-CoV-2 neutralizes viral entry. Proc. Natl. Acad. Sci. USA 2021, 118, e2112942118. [Google Scholar] [CrossRef]
- Schmitz, A.; Weber, A.; Bayin, M.; Breuers, S.; Fieberg, V.; Famulok, M.; Mayer, G. A SARS-CoV-2 Spike Binding DNA Aptamer that Inhibits Pseudovirus Infection by an RBD-Independent Mechanism. Angew. Chem. Int. Ed. 2021, 60, 10279–10285. [Google Scholar] [CrossRef]
- Zhang, N.; Chen, Z.; Liu, D.; Jiang, H.; Zhang, Z.K.; Lu, A.; Zhang, B.T.; Yu, Y.; Zhang, G. Structural Biology for the Molecular Insight between Aptamers and Target Proteins. Int. J. Mol. Sci. 2021, 22, 4093. [Google Scholar] [CrossRef] [PubMed]
- Kacherovsky, N.; Yang, L.F.; Dang, H.V.; Cheng, E.L.; Cardle, I.I.; Walls, A.C.; McCallum, M.; Sellers, D.L.; DiMaio, F.; Salipante, S.J.; et al. Discovery and Characterization of Spike N-Terminal Domain-Binding Aptamers for Rapid SARS-CoV-2 Detection. Angew. Chem. Int. Ed. 2021, 60, 21211–21215. [Google Scholar] [CrossRef]
- Kwon, J.; Narayan, C.; Kim, C.; Han, M.J.; Kim, M.; Jang, S.K. Development of a Subtype-Specific Diagnostic System for Influenza Virus H3N2 Using a Novel Virus-Based Systematic Evolution of Ligands by Exponential Enrichment (Viro-SELEX). J. Biomed. Nanotechnol. 2019, 15, 1609–1621. [Google Scholar] [CrossRef] [PubMed]
- Narayan, C.; Kwon, J.; Kim, C.; Kim, S.J.; Jang, S.K. Virus-based SELEX (viro-SELEX) allows development of aptamers targeting knotty proteins. Analyst 2020, 145, 1473–1482. [Google Scholar] [CrossRef] [PubMed]
- Crawford, K.H.D.; Eguia, R.; Dingens, A.S.; Loes, A.N.; Malone, K.D.; Wolf, C.R.; Chu, H.L.Y.; Tortorici, M.A.; Veesler, D.; Murphy, M.; et al. Protocol and Reagents for Pseudotyping Lentiviral Particles with SARS-CoV-2 Spike Protein for Neutralization Assays. Viruses 2020, 12, 513. [Google Scholar] [CrossRef]
- Hansen, J.; Baum, A.; Pascal, K.E.; Russo, V.; Giordano, S.; Wloga, E.; Fulton, B.O.; Yan, Y.; Koon, K.; Patel, K.; et al. Studies in humanized mice and convalescent humans yield a SARS-CoV-2 antibody cocktail. Science 2020, 369, 1010–1014. [Google Scholar] [CrossRef]
- Punjani, A.; Rubinstein, J.L.; Fleet, D.J.; Brubaker, M.A. cryoSPARC: Algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 2017, 14, 290–296. [Google Scholar] [CrossRef]
- Ahmad, J.; Jiang, J.; Boyd, L.F.; Zeher, A.; Huang, R.; Xia, D.; Natarajan, K.; Margulies, D.H. Structures of synthetic nanobody-SARS-CoV-2 receptor-binding domain complexes reveal distinct sites of interaction. J. Biol. Chem. 2021, 297, 101202. [Google Scholar] [CrossRef]
- Emsley, P.; Cowtan, K. Coot: Model-building tools for molecular graphics. Acta Crystallogr. D 2004, 60, 2126–2132. [Google Scholar] [CrossRef]
- Xu, G.H.; Zhao, J.J.; Yu, H.; Wang, C.; Huang, Y.Y.; Zhao, Q.; Zhou, X.; Li, C.G.; Liu, M.L. Structural Insights into the Mechanism of High-Affinity Binding of Ochratoxin A by a DNA Aptamer. J. Am. Chem. Soc. 2022, 144, 7731–7740. [Google Scholar] [CrossRef]
- Riccardi, C.; Napolitano, E.; Musumeci, D.; Montesarchio, D. Dimeric and Multimeric DNA Aptamers for Highly Effective Protein Recognition. Molecules 2020, 25, 5227. [Google Scholar] [CrossRef] [PubMed]
- Tian, L.; Heyduk, T. Bivalent Ligands with Long Nanometer-Scale Flexible Linkers. Biochemistry 2009, 48, 264–275. [Google Scholar] [CrossRef] [PubMed]
- Odeh, F.; Nsairat, H.; Alshaer, W.; Ismail, M.A.; Esawi, E.; Qaqish, B.; Al Bawab, A.; Ismail, S.I. Aptamers Chemistry: Chemical Modifications and Conjugation Strategies. Molecules 2020, 25, 3. [Google Scholar] [CrossRef] [PubMed]
- Obika, S.; Nanbu, D.; Hari, Y.; Morio, K.-I.; In, Y.; Ishida, T.; Imanishi, T. Synthesis of 2′-O,4′-C-methyleneuridine and -cytidine. Novel bicyclic nucleosides having a fixed C3, -endo sugar puckering. Tetrahedron Lett. 1997, 38, 8735–8738. [Google Scholar] [CrossRef]
- Ni, S.J.; Yao, H.Z.; Wang, L.L.; Lu, J.; Jiang, F.; Lu, A.P.; Zhang, G. Chemical Modifications of Nucleic Acid Aptamers for Therapeutic Purposes. Int. J. Mol. Sci. 2017, 18, 1683. [Google Scholar] [CrossRef] [PubMed]
- Zhu, R.; Canena, D.; Sikora, M.; Klausberger, M.; Seferovic, H.; Mehdipour, A.R.; Hain, L.; Laurent, E.; Monteil, V.; Wirnsberger, G.; et al. Force-tuned avidity of spike variant-ACE2 interactions viewed on the single-molecule level. Nat. Commun. 2022, 13, 7926. [Google Scholar] [CrossRef] [PubMed]
- Iketani, S.; Liu, L.H.; Guo, Y.C.; Liu, L.Y.; Chan, J.F.W.; Huang, Y.M.; Wang, M.; Luo, Y.; Yu, J.; Chu, H.; et al. Antibody evasion properties of SARS-CoV-2 Omicron sublineages. Nature 2022, 604, 553–556. [Google Scholar] [CrossRef]
- Magazine, N.; Zhang, T.Y.; Wu, Y.Y.; McGee, M.C.; Veggiani, G.; Huang, W.S. Mutations and Evolution of the SARS-CoV-2 Spike Protein. Viruses 2022, 14, 640. [Google Scholar] [CrossRef]
- McCallum, M.; Walls, A.C.; Sprouse, K.R.; Bowen, J.E.; Rosen, L.; Dang, H.; DeMarco, A.; Franko, N.; Tilles, S.W.; Logue, J.; et al. Molecular basis of immune evasion by the Delta and Kappa SARS-CoV-2 variants. Science 2021, 374, 1621–1626. [Google Scholar] [CrossRef]
- Cao, Y.L.; Wang, J.; Jian, F.C.; Xiao, T.H.; Song, W.L.; Yisimayi, A.; Huang, W.J.; Li, Q.Q.; Wang, P.; An, R.; et al. Omicron escapes the majority of existing SARS-CoV-2 neutralizing antibodies. Nature 2022, 602, 657–663. [Google Scholar] [CrossRef]
- Chakraborty, C.; Bhattacharya, M.; Sharma, A.R.; Mallik, B. Omicron (B.1.1.529)—A new heavily mutated variant: Mapped location and probable properties of its mutations with an emphasis on S-glycoprotein. Int. J. Biol. Macromol. 2022, 219, 980–997. [Google Scholar] [CrossRef] [PubMed]
- Tegally, H.; Moir, M.; Everatt, J.; Giovanetti, M.; Scheepers, C.; Wilkinson, E.; Subramoney, K.; Makatini, Z.; Moyo, S.; Amoako, D.G.; et al. Emergence of SARS-CoV-2 Omicron lineages BA.4 and BA.5 in South Africa. Nat. Med. 2022, 28, 1785–1790. [Google Scholar] [CrossRef] [PubMed]
- Lai, X.L.; Soler-Lopez, M.; Wichers, H.J.; Dijkstra, B.W. Large-Scale Recombinant Expression and Purification of Human Tyrosinase Suitable for Structural Studies. PLoS ONE 2016, 11, e016169. [Google Scholar] [CrossRef] [PubMed]
- Gotzke, H.; Kilisch, M.; Martinez-Carranza, M.; Sograte-Idrissi, S.; Rajavel, A.; Schlichthaerle, T.; Engels, N.; Jungmann, R.; Stenmark, P.; Opazo, F.; et al. The ALFA-tag is a highly versatile tool for nanobody-based bioscience applications. Nat. Commun. 2019, 10, 4403. [Google Scholar] [CrossRef]
- Lim, C.S.; Jang, Y.H.; Lee, G.Y.; Han, G.M.; Jeong, H.J.; Kim, J.W.; Lee, J.O. TLR3 forms a highly organized cluster when bound to a poly(I:C) RNA ligand. Nat. Commun. 2022, 13, 6876. [Google Scholar] [CrossRef]
- Rodriguez-Hernandez, A.P.; Martinez-Flores, D.; Cruz-Resendiz, A.; Padilla-Flores, T.; Gonzalez-Flores, R.; Estrada, K.; Sampieri, A.; Camacho-Zarco, A.R.; Vaca, L. Baculovirus Display of Peptides and Proteins for Medical Applications. Viruses 2023, 15, 411. [Google Scholar] [CrossRef]
- Bepler, T.; Morin, A.; Rapp, M.; Brasch, J.; Shapiro, L.; Noble, A.J.; Berger, B. Positive-unlabeled convolutional neural networks for particle picking in cryo-electron micrographs. Nat. Methods 2019, 16, 1153–1160. [Google Scholar] [CrossRef]
- Punjani, A.; Fleet, D. 3D Flexible Refinement: Structure and Motion of Flexible Proteins from Cryo-EM. Microsc. Microanal. 2022, 288, 1218. [Google Scholar] [CrossRef]
- Kappel, K.; Zhang, K.; Su, Z.; Watkins, A.M.; Kladwang, W.; Li, S.; Pintilie, G.; Topkar, V.V.; Rangan, R.; Zheludev, I.N.; et al. Accelerated cryo-EM-guided determination of three-dimensional RNA-only structures. Nat. Methods 2020, 17, 699–707. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Liebschner, D.; Afonine, P.V.; Baker, M.L.; Bunkoczi, G.; Chen, V.B.; Croll, T.I.; Hintze, B.; Hung, L.W.; Jain, S.; McCoy, A.J.; et al. Macromolecular structure determination using X-rays, neutrons and electrons: Recent developments in Phenix. Acta Crystallogr. D Struct. Biol. 2019, 75, 861–877. [Google Scholar] [CrossRef] [PubMed]
- Dhawan, M.; Sharma, A.; Priyanka; Thakur, N.; Rajkhowa, T.K.; Choudhary, O.P. Delta variant (B.1.617.2) of SARS-CoV-2: Mutations, impact, challenges and possible solutions. Hum. Vacc. Immunother. 2022, 18, 2068883. [Google Scholar] [CrossRef] [PubMed]
- Li, S.X.; Olson, W.K.; Lu, X.J. Web 3DNA 2.0 for the analysis, visualization, and modeling of 3D nucleic acid structures. Nucleic Acids Res. 2019, 47, W26–W34. [Google Scholar] [CrossRef] [PubMed]
- Mills, J.E.J.; Dean, P.M. Three-dimensional hydrogen-bond geometry and probability information from a crystal survey. J. Comput. Aid. Mol. Des. 1996, 10, 607–622. [Google Scholar] [CrossRef] [PubMed]
- Saenger, W. Principles of Nucliec Acid Structure; Springer: New York, NY, USA, 1984; pp. 120–121. [Google Scholar]
- Wang, D.L.; Zhang, S.Y.; Li, L.A.; Liu, X.; Mei, K.R.; Wang, X.Q. Structural insights into the assembly and activation of IL-1 beta with its receptors. Nat. Immunol. 2010, 11, 905–911. [Google Scholar] [CrossRef]
- Bao, L.; Zhang, X.; Jin, L.; Tan, Z.J. Flexibility of nucleic acids: From DNA to RNA. Chinese Phys. B 2016, 25. [Google Scholar] [CrossRef]
- Zuker, M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003, 31, 3406–3415. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rahman, M.S.; Han, M.J.; Kim, S.W.; Kang, S.M.; Kim, B.R.; Kim, H.; Lee, C.J.; Noh, J.E.; Kim, H.; Lee, J.-O.; et al. Structure-Guided Development of Bivalent Aptamers Blocking SARS-CoV-2 Infection. Molecules 2023, 28, 4645. https://doi.org/10.3390/molecules28124645
Rahman MS, Han MJ, Kim SW, Kang SM, Kim BR, Kim H, Lee CJ, Noh JE, Kim H, Lee J-O, et al. Structure-Guided Development of Bivalent Aptamers Blocking SARS-CoV-2 Infection. Molecules. 2023; 28(12):4645. https://doi.org/10.3390/molecules28124645
Chicago/Turabian StyleRahman, Md Shafiqur, Min Jung Han, Sang Won Kim, Seong Mu Kang, Bo Ri Kim, Heesun Kim, Chang Jun Lee, Jung Eun Noh, Hanseong Kim, Jie-Oh Lee, and et al. 2023. "Structure-Guided Development of Bivalent Aptamers Blocking SARS-CoV-2 Infection" Molecules 28, no. 12: 4645. https://doi.org/10.3390/molecules28124645
APA StyleRahman, M. S., Han, M. J., Kim, S. W., Kang, S. M., Kim, B. R., Kim, H., Lee, C. J., Noh, J. E., Kim, H., Lee, J.-O., & Jang, S. K. (2023). Structure-Guided Development of Bivalent Aptamers Blocking SARS-CoV-2 Infection. Molecules, 28(12), 4645. https://doi.org/10.3390/molecules28124645