

Synthesis, Crystal Structure, DFT, and Anticancer Activity of Some Imine-Type Compounds via Routine Schiff Base Reaction: An Example of Unexpected Cyclization to Oxazine Derivative

 , , , and
, , , and
Abstract
:1. Introduction
2. Results and Discussion
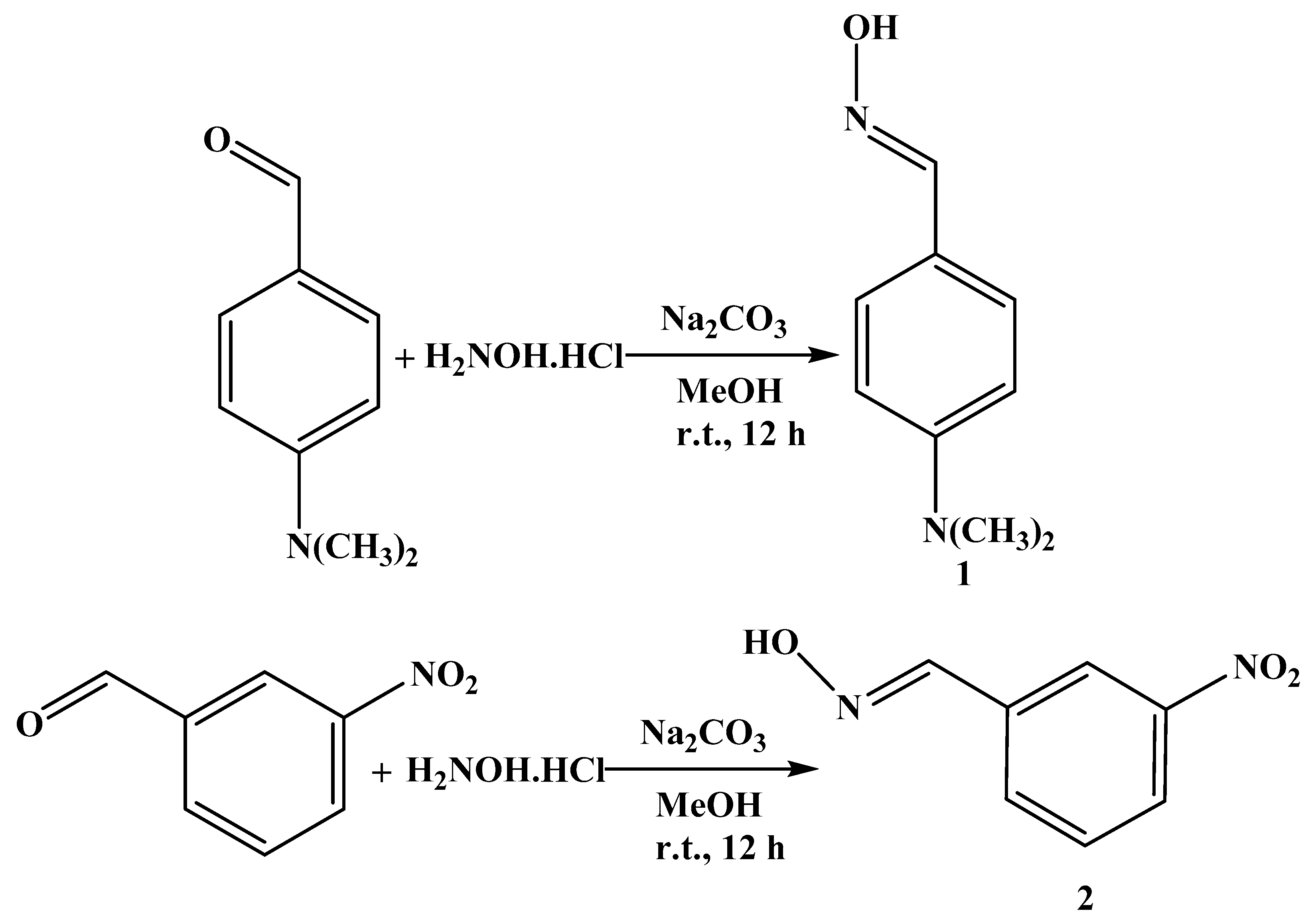
2.1. Synthesis and Characterization of Aryl Aldoximes 1 and 2
2.2. Synthesis and Characterization of Compounds 3 and 4
2.3. X-ray Structure Description
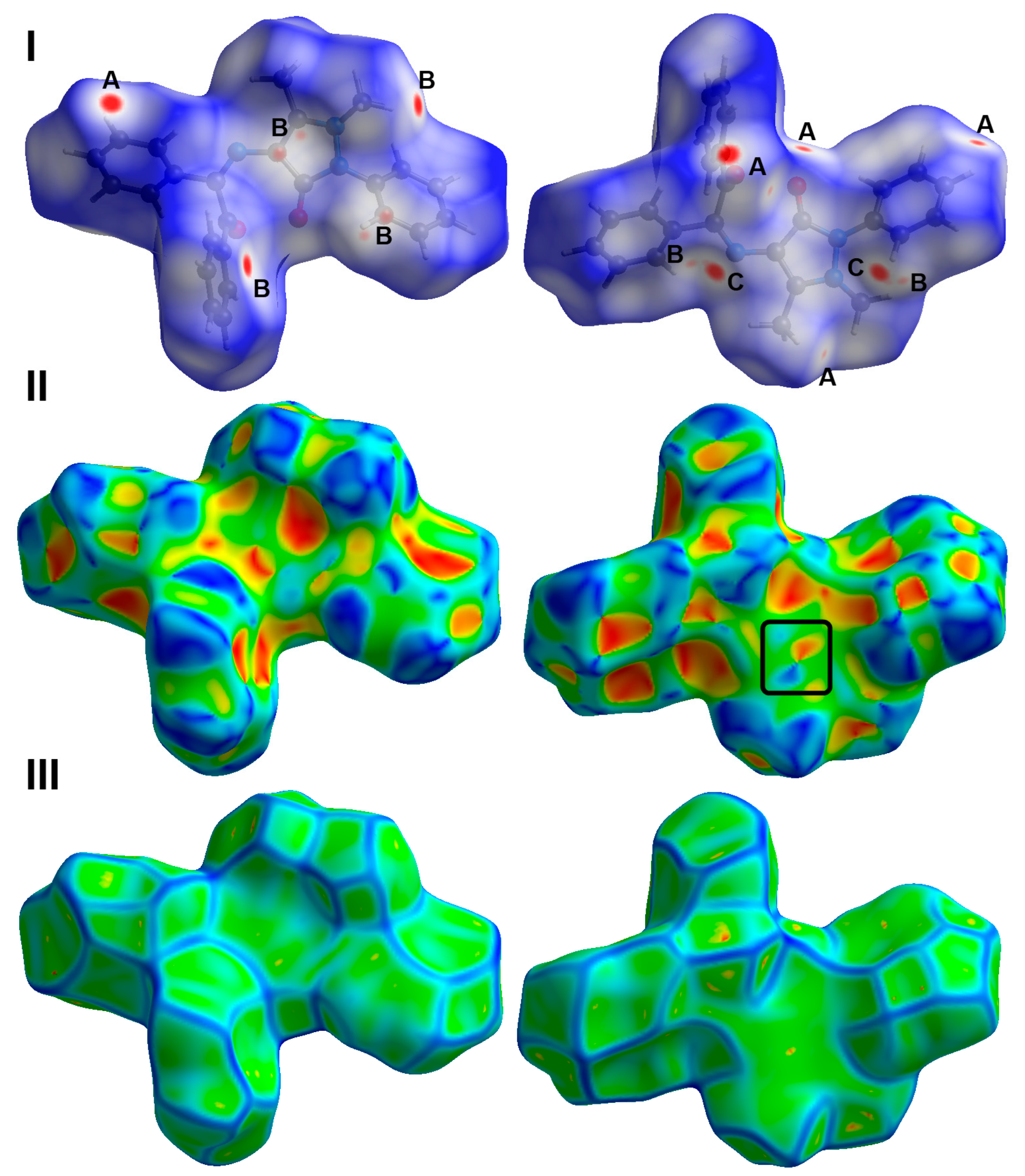
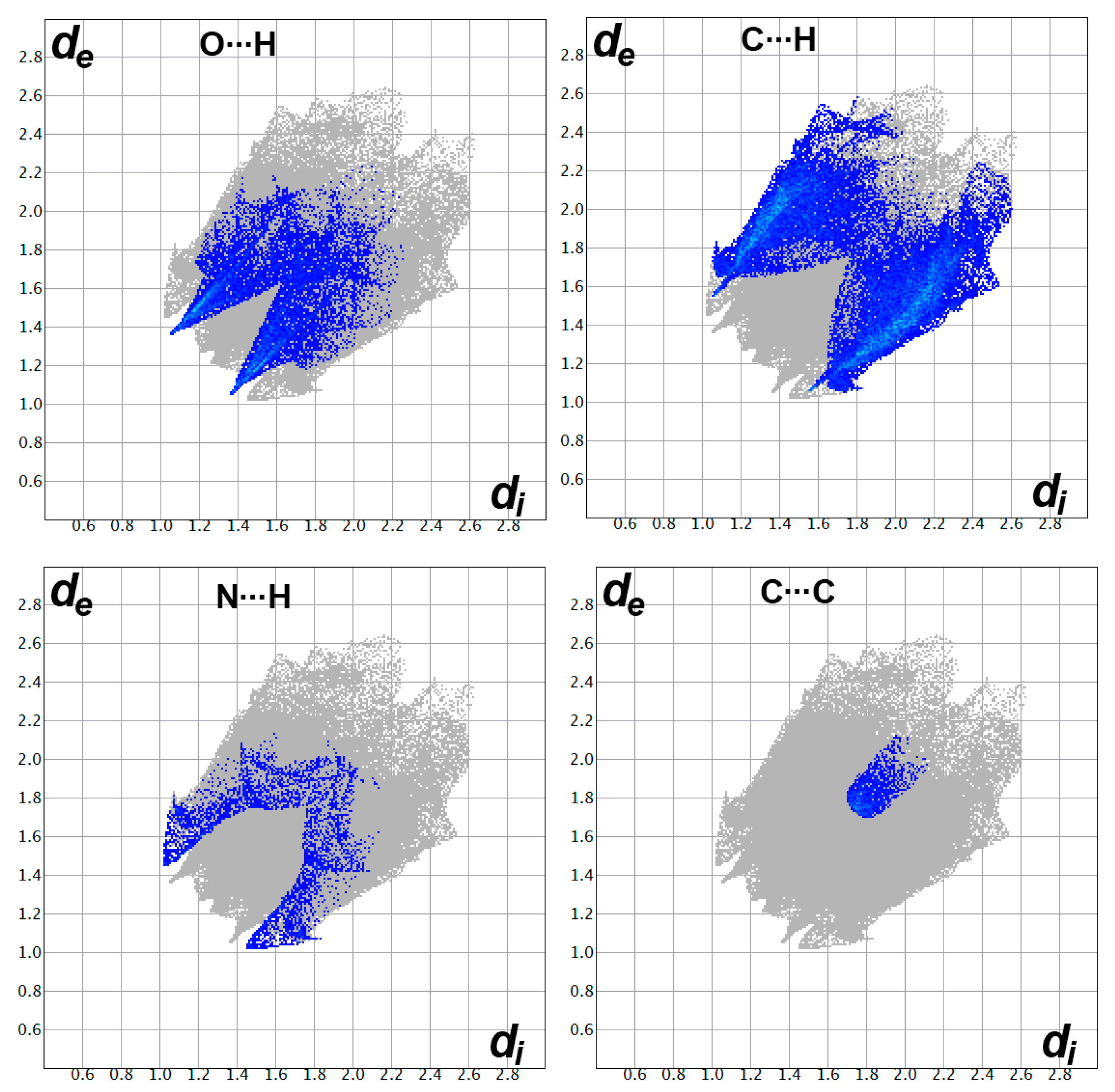
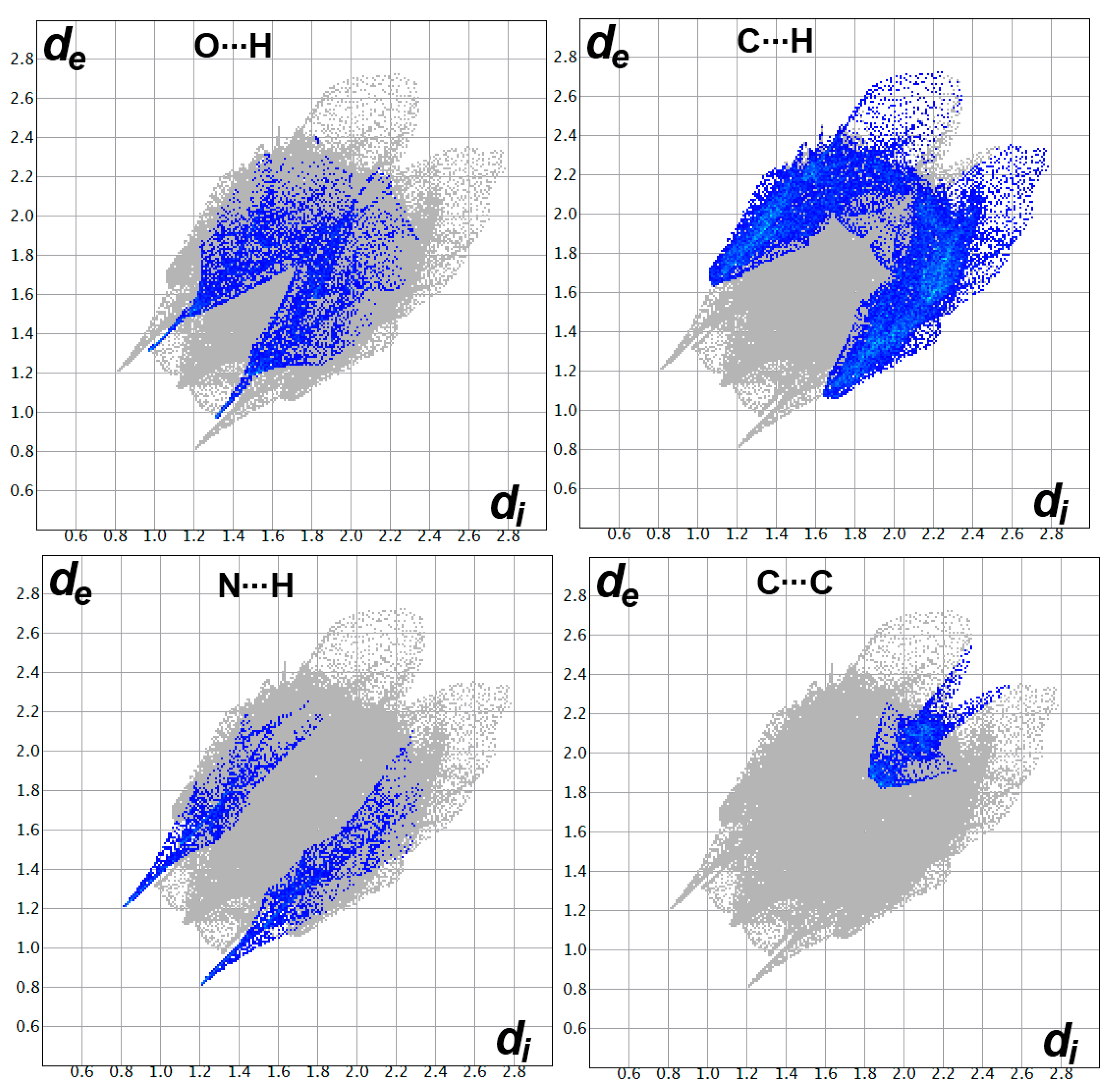
2.4. Analysis of Molecular Packing
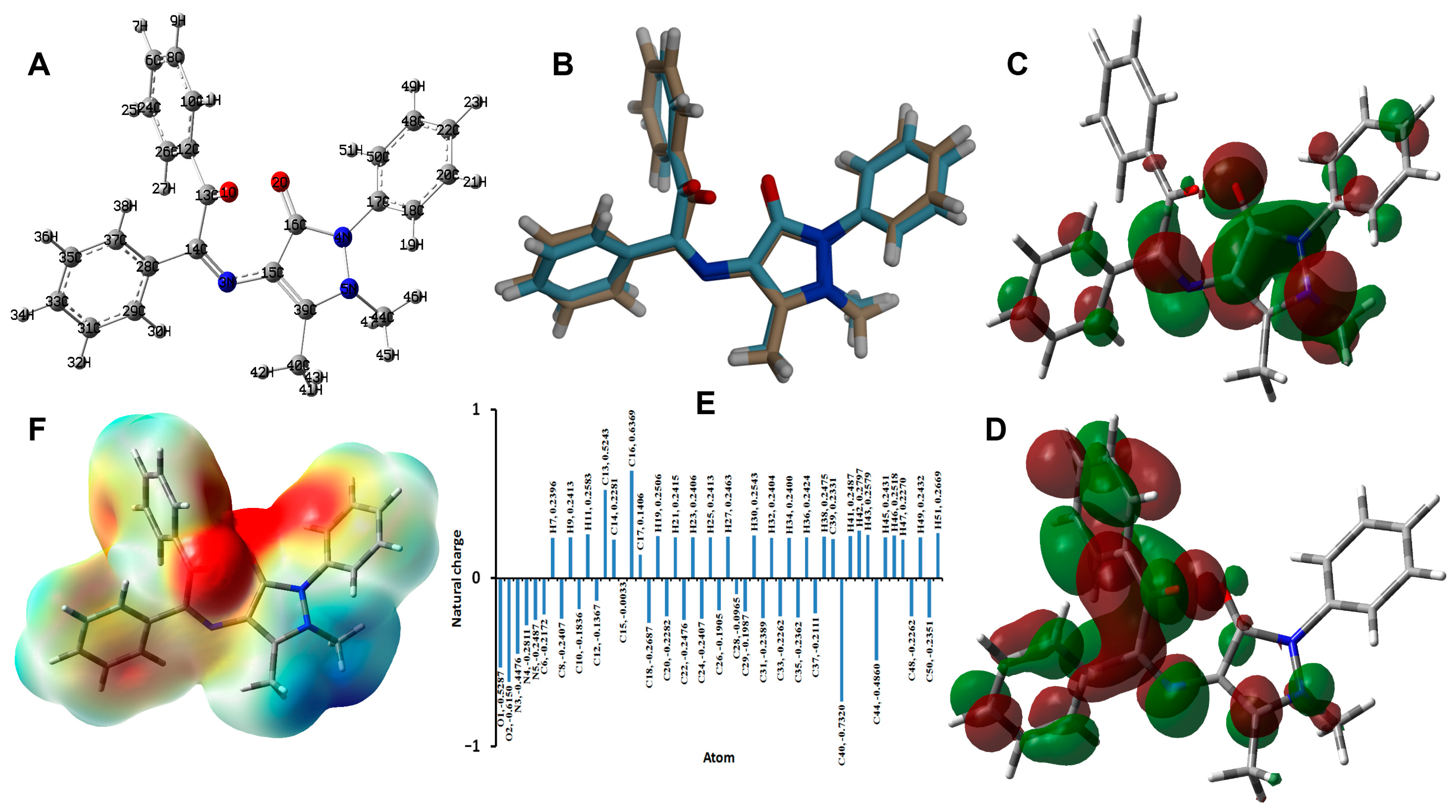
2.5. DFT Studies
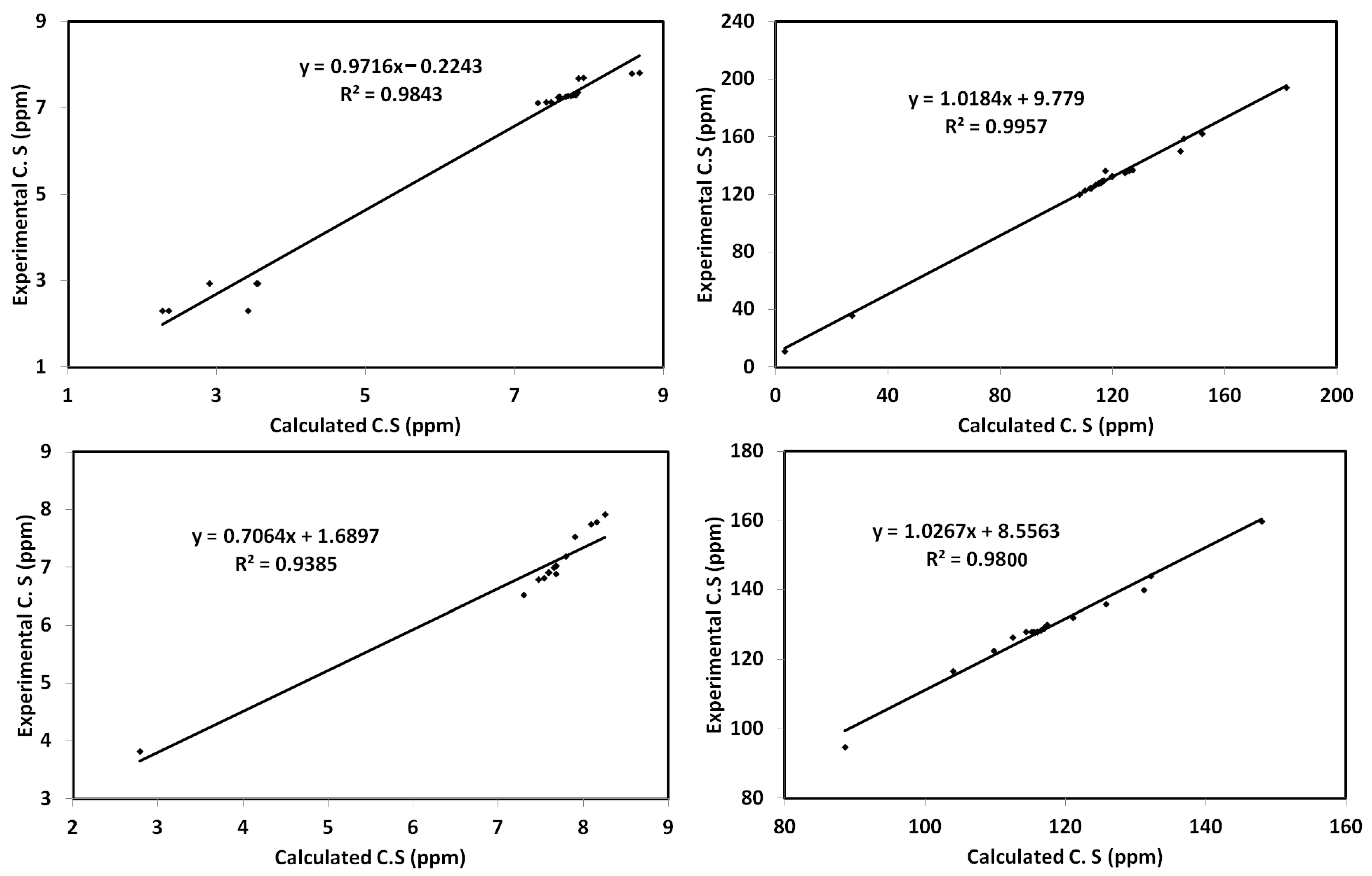
2.6. Calculated NMR Spectra
2.7. Anticancer Activity of Compounds 1–4
3. Experimental Section
3.1. Chemistry
3.1.1. General Methods
3.1.2. Synthesis of p-Dimethylaminobenzaldoxime 1 and m-Nitrobenzaldoxime 2
3.1.3. Synthesis of (4E)-4-(2-oxo-1,2-Diphenylethylideneamino)-1,2-dihydro-1,5-dimethyl-2-phenylpyrazol-3-one 3 and 2,3-diphenyl-2H-benzo[b][1,4]oxazin-2-ol 4
3.1.4. X-ray Structure Determinations
3.1.5. Methods and Calculations
Hirshfeld Surface Analysis
3.1.6. Computational Methods
3.2. Biological Experiments
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Sandier, S.R.; Karo, W. Organic Functional Group Preparations, 2nd ed.; Academic Press: San Diego, CA, USA, 1989; pp. 431–476. [Google Scholar]
- Greene, T.W.; Wuts, P.G.M. Protective Groups in Organic Synthesis, 3rd ed.; Wiley: Toronto, ON, Canada, 1999; pp. 355–358. [Google Scholar]
- Kassa, J. Review of oximes in the antidotal treatment of poisoning by organophosphorus nerve agents. J. Toxicol. Clin. Toxicol. 2002, 40, 803–816. [Google Scholar] [CrossRef]
- Ratković, A.; Pavlović, K.; Barić, D.; Marinić, Ž.; Grgičević, I.; Škorić, I. Modeling and synthesis of novel oxime derivatives as potential cholinesterase inhibitors. J. Mol. Struct. 2020, 1200, 127149. [Google Scholar] [CrossRef]
- da Silva, J.A.V.; Modesto-Costa, L.; de Koning, M.C.; Borges, I., Jr.; França, T.C.C. Theoretical NMR and conformational analysis of solvated oximes for organophosphates-inhibited acetylcholinesterase reactivation. J. Mol. Struct. 2018, 1152, 311–320. [Google Scholar] [CrossRef]
- Ritz, J.; Fuchs, H.; Kieczka, H.; Moran, W.C. Ullmann’s Encyclopedia of Industrial Chemistry; Wiley-VCH: Weinheim, Germany, 2012. [Google Scholar]
- Dewan, S.K.; Singh, R.; Kumar, A. One pot synthesis of nitriles from aldehydes and hydroxylamine hydrochloride using sodium sulphate (anhyd) and sodium bicarbonate in dry media under microwave irradiation. Arkivoc 2006, 2, 41–44. [Google Scholar]
- Dave, P.R.; Forshar, F. Facile preparation of 3,7-diazabicyclo[3.3.0]octane and 3,7,10-triheterocyclic[3.3.3]propellane ring systems from 1,5- diazacyclooctane-3,7-derivatives. J. Org. Chem. 1996, 61, 8897–8903. [Google Scholar] [CrossRef] [PubMed]
- Ballistreni, F.P.; Barbuzzi, E.; Tomaselli, G.A.; Toscano, R.M. Useful oxidation procedure of oximes to nitro compounds with Benz-Mo in acetonitrile. Syn. Lett. 1996, 11, 1093–1094. [Google Scholar]
- Smith, P.A.S.; Gloyer, S.E. Oxidation of dibenzylhydroxylamines to nitrones. Effects of structure and oxidizing agent on composition of the products. J. Org. Chem. 1975, 40, 2508–2512. [Google Scholar] [CrossRef]
- Negi, S.; Matsukura, M.; Mizuno, M.; Miyake, K.; Minami, N. Synthesis of (2R)-1-(4-Chloro-2-pyridyl)-2-(2-pyridyl)ethylamine: A selective oxime reduction and crystallization-induced asymmetric transformation. Synthesis 1996, 8, 991–996. [Google Scholar] [CrossRef]
- Narasaka, K. Synthesis of azaheterocycles from oxime derivatives. Pure Appl. Chem. 2003, 75, 19–28. [Google Scholar] [CrossRef] [Green Version]
- Whitesell, J.K.; Whitesell, M.A. Alkylation of ketones and aldehydes via their nitrogen derivatives. Synthesis 1983, 7, 517–536. [Google Scholar] [CrossRef]
- Ramalingan, C.; Park, T. Mercury-catalyzed rearrangement of ketoximes into amides and lactams in acetonitrile. J. Org. Chem. 2007, 72, 4536–4538. [Google Scholar] [CrossRef]
- Furuya, Y.; Ishihara, K.; Yamamoto, H. Cyanuric chloride as a mild and active Beckmann rearrangement catalyst. J. Am. Chem. Soc. 2005, 127, 11240–11241. [Google Scholar] [CrossRef] [PubMed]
- Song, B.A.; Liu, X.H.; Yang, S.; Hu, D.Y.; Jin, L.H.; Zhang, Y.T. Recent advance in synthesis and biological activity of oxime derivatives. Chin. J. Org. Chem. 2005, 25, 507–525. [Google Scholar]
- Kukushkin, V.Y.; Pombeiro, A.J.L. Oxime and oximate metal complexes: Unconventional synthesis and reactivity. Coord. Chem. Rev. 1999, 181, 147–175. [Google Scholar] [CrossRef]
- Kopylovich, M.N.; Lasri, J.; da Silva, M.F.C.G.; Pombeiro, A.J.L. Single-pot template transformations of cyanopyridines on a PdII centre: Syntheses of ketoimine and 2,4-dipyridyl-1,3,5-triazapentadiene palladium(II) complexes and their catalytic activity for microwave-assisted Suzuki–Miyaura and Heck reactions. Dalton Trans. 2009, 16, 3074–3084. [Google Scholar] [CrossRef] [PubMed]
- Lasri, J.; da Silva, M.F.C.G.; Charmier, M.A.J.; Pombeiro, A.J.L. Optically active mixed unsymmetric imine platinum(II) complexes—Utilization of the liberated imines for further syntheses of mixed imine-diazadiene complexes and of (E)-cyanoalkenes. Eur. J. Inorg. Chem. 2008, 2008, 3668–3677. [Google Scholar] [CrossRef]
- Lasri, J.; Charmier, M.A.J.; da Silva, M.F.C.G.; Pombeiro, A.J.L. Mixed unsymmetric oxadiazoline and/or imine platinum(II) complexes. Dalton Trans. 2007, 30, 3259–3266. [Google Scholar] [CrossRef]
- Kopylovich, M.N.; Kukushkin, V.Y.; da Silva, M.F.C.G.; Haukka, M.; da Silva, J.J.R.F.; Pombeiro, A.J.L. Conversion of alkanenitriles to amidines and carboxylic acids mediated by a cobalt(II)–ketoxime system. J. Chem. Soc. Perkin Trans. 2001, 1, 1569–1573. [Google Scholar] [CrossRef]
- Kopylovich, M.N.; Kukushkin, V.Y.; Haukka, M.; da Silva, J.J.R.F.; Pombeiro, A.J.L. Zinc(II)/ketoxime system as a simple and efficient catalyst for hydrolysis of organonitriles. Inorg. Chem. 2002, 41, 4798–4804. [Google Scholar] [CrossRef]
- Kopylovich, M.N.; Kukushkin, V.Y.; Haukka, M.; Luzyanin, K.V.; Pombeiro, A.J.L. An efficient synthesis of phthalocyanines based on an unprecedented double-addition of oximes to phthalonitriles. J. Am. Chem. Soc. 2004, 126, 15040–15041. [Google Scholar] [CrossRef]
- Kopylovich, M.N.; Haukka, M.; Kirillov, A.M.; Kukushkin, V.Y.; Pombeiro, A.J.L. Unsymmetrical NiII–imidoylamidine complexes derived from a novel oxime-mediated single-pot reaction of nitriles. Chem. Eur. J. 2007, 13, 786–791. [Google Scholar] [CrossRef] [PubMed]
- Lasri, J.; Elsherbiny, A.S.; Eltayeb, N.E.; Haukka, M.; El-Hefnawy, M.E. Synthesis and characterization of ferrocene-based Schiff base and ferrocenecarboxaldehyde oxime and their adsorptive removal of methyl blue from aqueous solution. J. Organomet. Chem. 2018, 866, 21–26. [Google Scholar] [CrossRef]
- Lasri, J.; Chulvi, K.; Eltayeb, N.E. Crystal structures of (E)-1-naphthaldehyde oxime and (E)-phenanthrene-9-carbaldehyde oxime. Acta Cryst. 2018, E74, 332–336. [Google Scholar] [CrossRef] [Green Version]
- Lasri, J.; Soliman, S.M.; Elsilk, S.E.; Haukka, M.; El-Faham, A. Synthesis, crystal structure, DFT and biological activity of E-pyrene-1-carbaldehyde oxime and E-2-naphthaldehyde oxime. J. Mol. Struct. 2020, 1207, 127848. [Google Scholar] [CrossRef]
- Abdel-Latif, E. Versatile synthesis of N,S-heterocycles containing the antipyrine moiety. Phosphorus Sulfur Silicon Relat. Elem. 2006, 181, 125–139. [Google Scholar] [CrossRef]
- Pedro, M.P.; Santos, A.M.; Antunes, J.N.; Eduarda, F.; Vieira, A.J.S.C. Scavenging activity of aminoantipyrines against hydroxyl radical. Eur. J. Med. Chem. 2010, 45, 2258–2264. [Google Scholar]
- Rizvi, M.A.; Dangat, Y.; Yaseen, Z.; Gupta, V.; Khan, K.Z. Synthesis, Crystal structure and in vitro DNA binding studies of combretastatin A-4 analogue. Croat. Chem. Acta 2015, 88, 289–296. [Google Scholar] [CrossRef]
- Wadkins, R.M.; Hyatt, J.L.; Wei, X.; Yoon, K.J.P.; Wierdl, M.; Edwards, C.C.; Morton, C.L.; Obenauer, J.C.; Damodaran, K.; Beroza, P.; et al. Identification and characterization of novel benzil (diphenylethane-1,2-dione) analogues as inhibitors of mammalian carboxylesterases. J. Med. Chem. 2005, 48, 2906–2915. [Google Scholar] [CrossRef]
- Schmidpeter, A.; Winmaier, J.H. Synthesis of oxazaphospholinobenzooxaazaphospholines and –oxadiazaphospholines compounds having pentacoordinated phosphorus as bridgehead of a bicyclo[3.3.0]octane skeleton. Angew. Chem. Int. Ed. 1975, 14, 489–490. [Google Scholar] [CrossRef]
- Singh, V.P.; Singh, R.V.; Tandon, J.P. Stereochemical and biochemical aspects of some organoboron complexes of sulphur donor ligands. J. Inorg. Biochem. 1990, 39, 237–245. [Google Scholar] [CrossRef]
- Marjani, K.; Asgarian, J.; Mousavi, M. Synthesis and structure determination of 2,3-diphenyl-2H-1,4-benzoxazin-2-ol. J. Chem. Res. 2007, 9, 548–551. [Google Scholar] [CrossRef]
- Liang, Q.; Wang, Q. 4-[(1-hydroxy-2-naphthyl)methyleneamino]-1,5-dimethyl-2-phenyl-1H-pyrazol-3(2H)-one. Acta Cryst. 2010, E66, o1968–o1969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eltayeb, N.E.; Şen, F.; Lasri, J.; Hussien, M.A.; Elsilk, S.E.; Babgi, B.A.; Gökce, H.; Sert, Y. Hirshfeld surface analysis, spectroscopic, biological studies and molecular docking of (4E)-4-((naphthalen-2-yl)methyleneamino)-1,2-dihydro-2,3-dimethyl-1-phenylpyrazol-5-one. J. Mol. Strut. 2020, 1202, 127315. [Google Scholar] [CrossRef]
- Boraei, A.T.A.; Soliman, S.M.; Haukka, M.; Barakat, A. X-ray structure, Hirshfeld analysis and DFT studies of two new hits of triazolyl-indole bearing alkylsulfanyl moieties. J. Mol. Struct. 2021, 1225, 129302. [Google Scholar] [CrossRef]
- Alshahrani, S.; Soliman, S.M.; Alamary, A.S.; Al-Majid, A.M.; Haukka, M.; Yousuf, S.; Barakat, A. Synthesis of enaminones-based benzo[d]imidazole scaffold: Characterization and molecular insight structure. Crystals 2020, 10, 955. [Google Scholar] [CrossRef]
- Altowyan, M.S.; Sultan, M.A.; Soliman, S.M.; Yousuf, S.; Ali, I.; Shawish, I.; Barakat, A. Synthesis, single crystal X-ray, Hirshfeld and DFT studies of 1,8-dichloro-9,10-dihydro-9,10-ethanoanthracene-11-carboxylic acid. Crystals 2021, 11, 1161. [Google Scholar] [CrossRef]
- Foresman, J.B.; Frisch, Æ. Exploring Chemistry with Electronic Structure Methods, 2nd ed.; Gaussian: Pittsburgh, PA, USA, 1996. [Google Scholar]
- Chang, R. Chemistry, 7th ed.; McGraw-Hill: New York, NY, USA, 2001. [Google Scholar]
- Kosar, B.; Albayrak, C. Spectroscopic investigations and quantum chemical computational study of (E)-4-methoxy-2-[(p-tolylimino)methyl]phenol. Spectrochim. Acta 2011, 78, 160–167. [Google Scholar] [CrossRef]
- Koopmans, T.A. Ordering of wave functions and eigenenergies to the individual electrons of an atom. Physica 1933, 1, 104–113. [Google Scholar] [CrossRef]
- Parr, R.G.; Yang, W. Density-Functional Theory of Atoms and Molecules; Oxford University Press: New York, NY, USA, 1989. [Google Scholar]
- Parr, R.G.; Szentpaly, L.V.; Liu, S. Electrophilicity index. J. Am. Chem. Soc. 1999, 121, 1922–1924. [Google Scholar] [CrossRef]
- Bruker AXS. APEX3—Software Suite for Crystallographic Programs; Bruker AXS, Inc.: Madison, WI, USA, 2016. [Google Scholar]
- Sheldrick, G.M. SADABS—Bruker Nonius Scaling and Absorption Correction; Bruker AXS, Inc.: Madison, WI, USA, 2016. [Google Scholar]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. 2015, C71, 3–8. [Google Scholar]
- Turner, M.J.; McKinnon, J.J.; Wolff, S.K.; Grimwood, D.J.; Spackman, P.R.; Jayatilaka, D.; Spackman, M.A. Crystal Explorer17; University of Western Australia: Crawley, Australia, 2017; Available online: http://hirshfeldsurface.net (accessed on 30 April 2017).
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. GAUSSIAN 09; Revision A02; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- GaussView; Version 4.1; Dennington, R., II; Keith, T.; Millam, J. (Eds.) Semichem Inc.: Shawnee Mission, KS, USA, 2007. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Stephens, P.J.; Devlin, F.J.; Chabalowski, C.F.; Frisch, M.J. Ab initio calculation of vibrational absorption and circular dichroism spectra using density functional force fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Ruud, K.; Helgaker, T.; Bak, K.L.; Jørgensen, P.; Jensen, H.J.A. Hartree-Fock limit magnetizabilities from London orbitals. J. Chem. Phys. 1993, 99, 3847–3859. [Google Scholar] [CrossRef] [Green Version]
- Reed, A.E.; Curtiss, L.A.; Weinhold, F. Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
- Cheeseman, J.R.; Trucks, G.W.; Keith, T.A.; Frisch, M.J. A comparison of models for calculating nuclear magnetic resonance shielding tensors. J. Chem. Phys. 1996, 104, 5497–5509. [Google Scholar] [CrossRef]
- Marten, B.; Kim, K.; Cortis, C.; Friesner, R.A.; Murphy, R.B.; Ringnalda, M.N.; Sitkoff, D.; Honig, B. New model for calculation of solvation free energies: Correction of self-consistent reaction field continuum dielectric theory for short-range hydrogen-bonding effects. J. Phys. Chem. 1996, 100, 11775–11788. [Google Scholar] [CrossRef]
- Plumb, J.A. Cell sensitivity assays: The MTT assay. In Cancer Cell Culture; Springer: Berlin/Heidelberg, Germany, 2004; pp. 165–169. [Google Scholar]
- Ali, E.M.; Elashkar, A.A.; El-Kassas, H.Y.; Salim, E.I. Methotrexate loaded on magnetite iron nanoparticles coated with chitosan: Biosynthesis, characterization, and impact on human breast cancer MCF-7 cell line. Int. J. Biol. Macromol. 2018, 120, 1170–1180. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Contact | Distance | Contact | Distance | Contact | Distance |
|---|---|---|---|---|---|
| 3 | 4 | ||||
| O1…H2 | 2.420 | C15…H19 | 2.748 | O2…H20 | 2.284 |
| O1…H25B | 2.557 | C6…H23 | 2.741 | N1…H1 | 2.028 |
| O2…H21 | 2.489 | C7…H23 | 2.775 | C1…H3 | 2.759 |
| N1…H23 | 2.482 | C16…C19 | 3.350 | C6…H3 | 2.718 |
| C11…H24B | 2.608 | ||||
| Compound | |||||
|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | Cisplatin | |
| HepG2 | 14.51–18.51 * | 24.67–30.96 * | 37.85–62.42 * | 51.39–73.26 * | 1.767–2.988 * |
| 16.39 ** | 27.64 ** | 48.61 ** | 61.36 ** | 2.297 ** | |
| MCF-7 | 19.67–25.79 * | 40.00–60.03 * | 40.15–58.49 * | 45.77–61.82 * | 3.781–6.837 * |
| 22.52 ** | 49.01 ** | 48.46 ** | 53.19 ** | 5.085 ** | |
| Compound | 3 | 4 |
|---|---|---|
| CCDC | 2245159 | 2245160 |
| Formula | C25H21N3O2 | C20H15NO2 |
| Formula Weight | 395.45 | 301.33 |
| Crystal System | Monoclinic | Orthorhombic |
| Space Group | P21/c | Pbca |
| a, b, c [Å] | 8.253 (3) | 8.202 (4) |
| 28.476 (12) | 12.001 (5) | |
| 8.776 (4) | 31.351 (13) | |
| alpha, beta, gamma [°] | 90 | |
| 96.841 (13) | ||
| 90 | ||
| V [Å3] | 2047.8 (15) | 3086 (2) |
| Z | 4 | 8 |
| D(calc) [g/cm3] | 1.283 | 1.297 |
| Mu(MoKa) [/mm ] | 0.083 | 0.084 |
| F(000) | 832 | 1264 |
| Crystal Size [mm] | 0.07 × 0.15 × 0.22 | 0.15 × 0.19 × 0.22 |
| Data Collection | ||
| Temperature (K) | 293 | 289 |
| Radiation [Å] | MoKa | MoKa |
| 0.71073 | 0.71073 | |
| Theta Min-Max [°] | 2.4, 25.4 | 2.6, 25.4 |
| Dataset | −9: 9; −34: 34; −10: 10 | −9: 9; −13: 14; −37: 37 |
| Tot., Uniq. Data, R(int) | 18671, 3750, 0.069 | 30981, 2825, 0.138 |
| Observed Data [I > 2.0 sigma(I)] | 2758 | 1795 |
| Refinement | ||
| Nref, Npar | 3750, 274 | 2825, 211 |
| R, wR2, S | 0.0849, 0.2321, 1.08 | 0.0510, 0.1118, 1.03 |
| w = ^2^(FO^2^) + (0.0699P)^2^ + 4.3819P] WHERE P = (FO^2^+2FC^2^)/3′ | w = ^2^(FO^2^) + (0.0537P)^2^ + 0.1289P] WHERE P = (FO^2^+2FC^2^)/3′ | |
| Min. and Max. Resd. Dens. [e/Å3] | −0.33, 0.32 | −0.21, 0.16 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lasri, J.; Eltayeb, N.E.; Soliman, S.M.; Ali, E.M.M.; Alhayyani, S.; Akhdhar, A. Synthesis, Crystal Structure, DFT, and Anticancer Activity of Some Imine-Type Compounds via Routine Schiff Base Reaction: An Example of Unexpected Cyclization to Oxazine Derivative. Molecules 2023, 28, 4766. https://doi.org/10.3390/molecules28124766
Lasri J, Eltayeb NE, Soliman SM, Ali EMM, Alhayyani S, Akhdhar A. Synthesis, Crystal Structure, DFT, and Anticancer Activity of Some Imine-Type Compounds via Routine Schiff Base Reaction: An Example of Unexpected Cyclization to Oxazine Derivative. Molecules. 2023; 28(12):4766. https://doi.org/10.3390/molecules28124766
Chicago/Turabian StyleLasri, Jamal, Naser E. Eltayeb, Saied M. Soliman, Ehab M. M. Ali, Sultan Alhayyani, and Abdullah Akhdhar. 2023. "Synthesis, Crystal Structure, DFT, and Anticancer Activity of Some Imine-Type Compounds via Routine Schiff Base Reaction: An Example of Unexpected Cyclization to Oxazine Derivative" Molecules 28, no. 12: 4766. https://doi.org/10.3390/molecules28124766