
Interaction between Cu and Thiols of Biological and Environmental Importance: Case Study Using Combined Spectrophotometric/Bathocuproine Sulfonate Disodium Salt Hydrate (BCS) Assay

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
2.1. Interaction of Cu and GSH
2.2. Interaction of Cu and L-cys
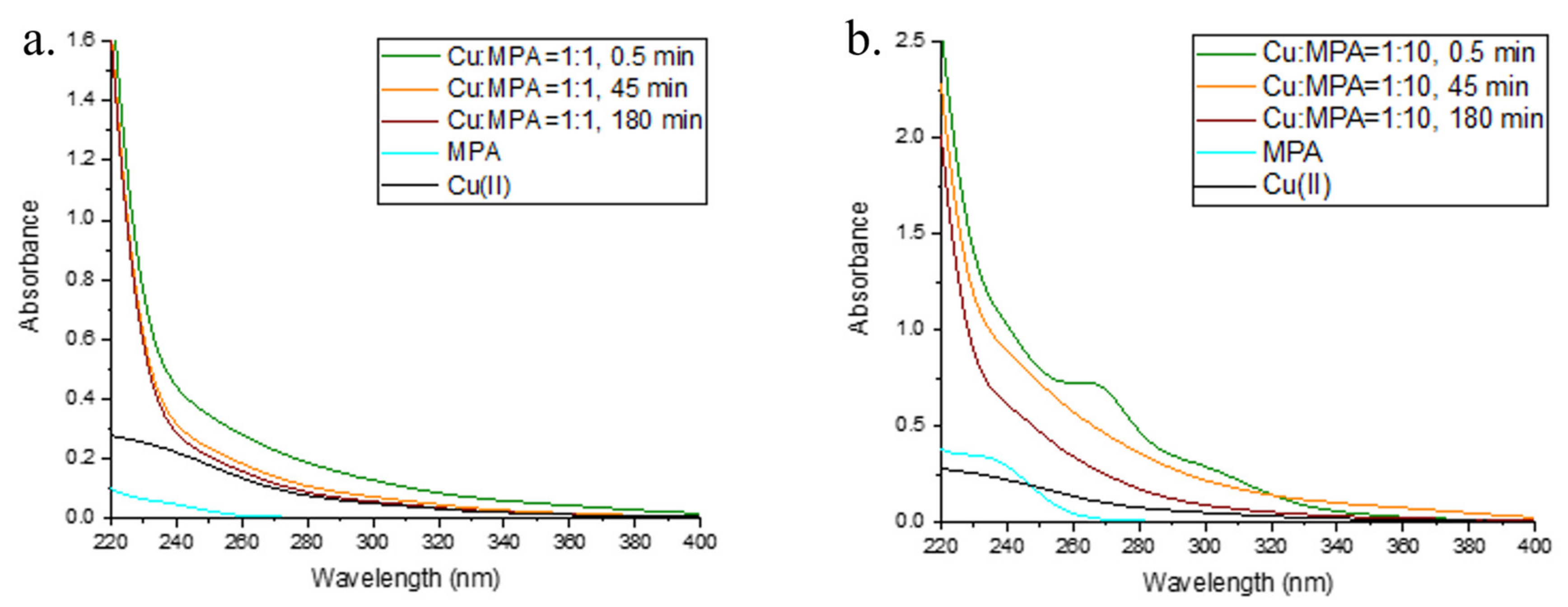
2.3. Interaction of Cu and 3-MPA
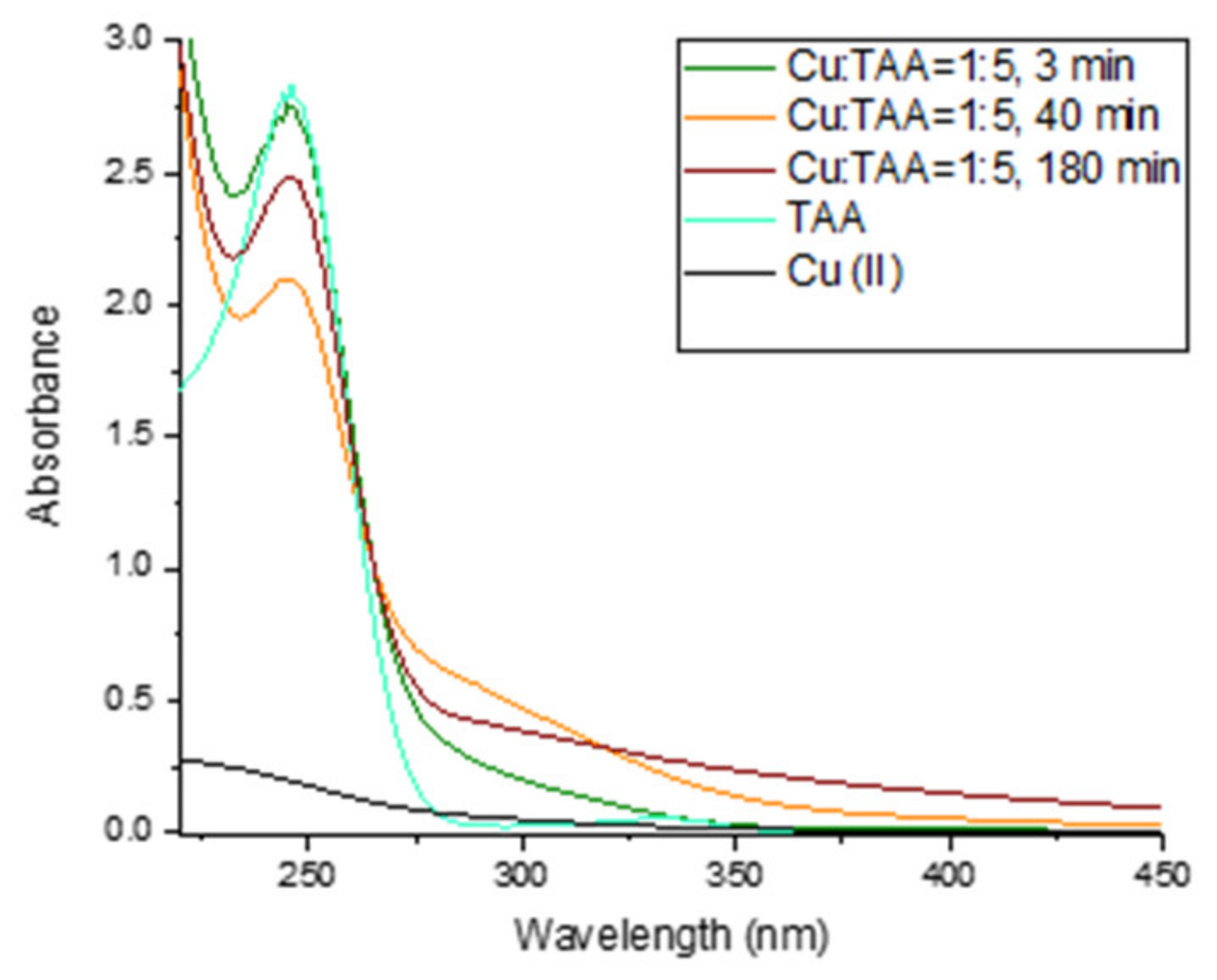
2.4. Interaction of Cu with TAA
3. Materials and Methods
3.1. Materials
3.2. Procedure
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Linder, M.C. Introduction and overview of copper as an element essential for life. In Biochemistry of Copper, 1st ed.; Frieden, E., Ed.; Springer: Boston, MA, USA, 1991; Volume 10, pp. 1–13. [Google Scholar] [CrossRef]
- Rensing, C.; McDevitt, S.F. The copper metallome in prokaryotic cells. Met. Ions Life. Sci. 2013, 12, 417–450. [Google Scholar] [CrossRef] [PubMed]
- Peña, M.M.; Lee, J.; Thiele, D.J. A delicate balance: Homeostatic control of copper uptake and distribution. J. Nutr. 1999, 129, 1251–1260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rucker, R.B.; Kosonen, T.; Clegg, M.S.; Mitchell, A.E.; Uriu-Hare, J.Y. Copper, lysyl oxidase, and extracelullar matrix protein cross linking. Am. J. Clin. Nutr. 1998, 67, 996–1002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solano, F. On the metal cofactor in the tyrosinase family. Int. J. Mol. Sci. 2018, 19, 633. [Google Scholar] [CrossRef] [Green Version]
- Raposo, B.; Rodríguez, C.; Martínez-González, J.; Badimon, L. High levels of homocysteine inhibit lysyl oxidase (LOX) and downregulate LOX expression in vascular endothelial cells. Atherosclerosis 2004, 177, 1–8. [Google Scholar] [CrossRef]
- Park, Y.D.; Lyou, Y.J.; Hahn, H.S.; Hahn, M.J.; Yang, J.M. Complex inhibition of tyrosinase by thiol-composed Cu2+ Chelators: A Clue for Designing Whitening Agents. J. Biomol. Struct. Dyn. 2006, 24, 131–138. [Google Scholar] [CrossRef]
- Bakavayev, S.; Chetrit, N.; Zvagelsky, T.; Mansour, R.; Vyazmensky, M.; Barak, Z.; Israelson, A.; Engel, S. Cu/Zn-superoxide dismutase and wild-type like fALS SOD1 mutants produce cytotoxic quantities of H2O2 via cysteine-dependent redox short-circuit. Sci. Rep. 2019, 9, 10826. [Google Scholar] [CrossRef] [Green Version]
- Klotz, L.O.; Krö, K.D.; Buchczyk, D.P.; Sies, H. Role of copper, zinc, selenium and tellurium in the cellular defense against oxidative and nitrosative stress. J. Nutr. 2003, 133, 1448S–1451S. [Google Scholar] [CrossRef] [Green Version]
- Bremner, I. Manifestations of Cu excess. Am. J. Clin. Nutr. 1998, 67, 1069–1073. [Google Scholar] [CrossRef] [Green Version]
- Hodgson, E.K.; Fridovich, I. The interaction of bovine erythrocyte superoxyde dismutase with hydrogen peroxide: Inactivation of the enzyme. Biochemistry 1975, 14, 5294–5299. [Google Scholar] [CrossRef]
- Winterbourn, C.C.; Peskin, A.V.; Parsons-Mair, H.N. Thiol oxidase activity of copper, zinc superoxide dismutase. J. Biol. Chem. 2002, 277, 1906–1911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaetke, L.M.; Chow-Johnson, H.S.; Chow, C.K. Copper: Toxicological relevance and mechanisms. Arch. Toxicol. 2014, 88, 1929–1938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaetke, L.M.; Chow, C.K. Copper toxicity, oxidative stress, and antioxidant nutrient. Toxicology 2003, 169, 147–163. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Tewari, R.K.; Sharma, P.N. Modulation of copper toxicity-induced oxidative damage by excess supply of iron in maize plants. Plant. Cell. Rep. 2008, 27, 399–409. [Google Scholar] [CrossRef] [PubMed]
- Valko, M.; Morris, H.; Cronin, M.T.D. Metals, toxicity and oxidative stress. Curr. Med. Chem. 2005, 12, 1161–1208. [Google Scholar] [CrossRef] [Green Version]
- Murakami, K.; Tsubouchi, R.; Fukayama, M.; Yoshino, M. Copper-dependent inhibition and oxidative inactivation with affinity cleavage of yeast glutathione reductase. Biometals 2014, 27, 551–558. [Google Scholar] [CrossRef]
- Xiao, Y.; Chen, D.; Zhang, X.; Cui, X.; Fan, J.; Bi, C.; Dou, Q.P. Molecular study on copper-mediated tumor proteasome inhibition and cell death. Int. J. Oncol. 2010, 37, 81–87. [Google Scholar] [CrossRef] [Green Version]
- Prohaska, J.R. Role of copper transporters in copper homeostasis. Am. J. Clin. Nutr. 2008, 88, 826–829. [Google Scholar] [CrossRef] [Green Version]
- Shenberger, Y.; Marciano, O.; Gottlieb, H.E.; Ruthstein, S. Insights into the N-terminal Cu(II) and Cu(I) binding sites of the human copper transporter CTR1. J. Coord. Chem. 2018, 71, 1985–2002. [Google Scholar] [CrossRef] [Green Version]
- Rubino, J.T.; Chenkin, M.P.; Keller, M.; Riggs-Gelasco, P.; Franz, K.J. A comparison of methionine, histidine and cysteine in copper(I)-binding peptides reveals differences relevant to copper uptake by organisms in diverse environments. Metallomics 2011, 3, 61–73. [Google Scholar] [CrossRef]
- Hatori, Y.; Lutsenko, S. An expanding range of functions for the copper chaperone/antioxidant protein atox1. Antioxid. Redox Signal. 2013, 19, 945–957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatori, Y.; Clasen, S.; Hasan, N.M.; Barry, A.N.; Lutsenko, S. Functional partnership of the copper export machinery and glutathione balance in human cells. J. Biol. Chem. 2012, 287, 26678–26687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heaton, D.N.; George, G.N.; Garrison, G.; Winge, D.R. The mitochondrial copper metallochaperone Cox17 exists as an oligomeric, polycopper complex. Biochemistry 2001, 40, 743–751. [Google Scholar] [CrossRef] [PubMed]
- Banci, L.; Bertini, I.; Ciofi-Baffoni, S.; Hadjiloi, T.; Martinelli, M.; Palumaa, P. Mitochondrial copper(I) transfer from cox17 to sco1 is coupled to electron transfer. Proc. Natl. Acad. Sci. USA 2008, 105, 6803–6808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moffet, J.W.; Zika, R.G.; Brand, L.E. Distribution and potential sources and sinks of copper chelators in the Sargasso sea. Deep Sea Res. 1990, 37, 27–36. [Google Scholar] [CrossRef]
- Moffett, J.W.; Brand, L.E. Production of strong, extracellular Cu chelators by marine cyanobacteria in response to Cu stress. Limnol. Oceanogr. 1996, 41, 388–395. [Google Scholar] [CrossRef]
- Whitby, H.; Posacka, A.M.; Maldonado, M.T.; van den Berg, C.M.G. Copper-binding ligands in the NE Pacific. Mar. Chem. 2018, 204, 36–48. [Google Scholar] [CrossRef]
- Tang, D.; Shafer, M.M.; Karner, D.A.; Armstrong, D.E. Response of nonprotein thiols to copper stress and extracellular release of glutathione in the diatom Thalassiosira Weissflogii. Limnol. Oceanogr. 2005, 50, 516–525. [Google Scholar] [CrossRef]
- Valent, I.; Bednárová, L.; Schreiber, I.; Bujdák, J.; Valachová, K.; Šoltés, L. Reaction of N-Acetylcysteine with Cu2+: Appearance of intermediates with high free radical scavenging activity: Implications for anti-/pro-oxidant properties of thiols. Int. J. Mol. Sci. 2022, 23, 6199. [Google Scholar] [CrossRef]
- Smith, R.C.; Reed, V.D.; Hill, W.E. Oxidation of thiols by copper(II). Phosphorus Sulfur Silicon Relat. Elem. 1994, 90, 147–154. [Google Scholar] [CrossRef]
- Anderson, M.E. Glutathione: An overview of biosynthesis and modulation. Chem. Biol. Interact. 1998, 111, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Freedman, J.H.; Ciriolo, M.R.; Peisach, J. The role of glutathione in copper metabolism and toxicity. J. Biol. Chem. 1989, 264, 5598–5605. [Google Scholar] [CrossRef] [PubMed]
- Maryon, E.B.; Molloy, S.A.; Kaplan, J.H. Cellular glutathione plays a key role in copper uptake mediated by human copper transporter 1. Am. J. Physiol. 2013, 304, 768–779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasanuzzaman, M.; Nahar, K.; Anee, T.I.; Fujita, M. Glutathione in plants: Biosynthesis and physiological role in environmental stress tolerance. Physiol. Mol. Biol. Plants 2017, 23, 249–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dupont, C.L.; Ahner, B.A. Effects of copper, cadmium, zinc on the production and exudation of thiols by Emiliania Huxleyi. Limnol. Oceanogr. 2005, 50, 508–515. [Google Scholar] [CrossRef] [Green Version]
- Leal, M.F.C.; Vasconcelos, T.D.M.S.; van den Berg, C.M.G. Copper-induced release of complexing ligands similar to thiols by Emiliania Huxleyi in seawater cultures. Limnol. Oceanogr. 1999, 44, 1750–1762. [Google Scholar] [CrossRef] [Green Version]
- Hu, H.; Mylon, S.E.; Benoit, G. Distribution of the thiols glutathione and 3-mercaptopropionic acid in Connecticut Lakes. Limnol. Oceanogr. 2006, 51, 2763–2774. [Google Scholar] [CrossRef] [Green Version]
- Tang, D.; Shafer, M.; Karner, D.A.; Overdier, J.; Armstrong, D.E. Factors affecting the presence of dissolved glutathione in estuarine waters. Environ. Sci. Technol. 2004, 38, 4247–4253. [Google Scholar] [CrossRef]
- Liem-Nguyen, V.; Bouchet, S.; Björn, E. Determination of sub-nanomolar levels of low molecular mass thiols in natural waters by liquid chromatography tandem mass spectrometry after derivatization with p-(hydroxymercuri) benzoate and online preconcentration. Anal. Chem. 2015, 87, 1089–1096. [Google Scholar] [CrossRef]
- Swarr, G.J.; Kading, T.; Lamborg, C.H.; Hammerschmidt, C.R.; Bowman, K.L. Dissolved low-molecular weight thiol concentrations from the U.S. GEOTRACES North Atlantic Ocean zonal transect. Deep Sea Res. 2016, 116, 77–87. [Google Scholar] [CrossRef] [Green Version]
- Van Den Berg, C.M.G.; Househam, B.C.; Riley, J.P. Determination of cystine and cysteine in seawater using cathodic stripping voltammetry in the presence of Cu(II). J. Electroanal. Chem. Interfacial Electrochem. 1988, 239, 137–148. [Google Scholar] [CrossRef]
- Takagi, H.; Ohtsu, I. L-cysteine metabolism and fermentation in microorganisms. In Advances in Biochemical Engineering/Biotechnology, 1st ed.; Yokota, A., Ikeda, M., Eds.; Springer: Tokio, Japan, 2017; Volume 159, pp. 129–151. [Google Scholar] [CrossRef]
- Poole, L.B. The basics of thiols and cysteines in redox biology and chemistry. Free Radic. Biol. Med. 2015, 80, 148–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pushie, M.J.; Zhang, L.; Pickering, I.J.; George, G.N. The fictile coordination chemistry of cuprous-thiolate sites in copper chaperones. Biochim. Biophys. Acta Bioenerg. 2012, 1817, 938–947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiene, R.P.; Taylor, B.F. Biotransformations of organosulphur compounds in sediments via 3-mercaptopropionate. Nature 1988, 332, 148–150. [Google Scholar] [CrossRef]
- Allen, K.D.; White, R.H. Occurrence and biosynthesis of 3-mercaptopropionic acid in Methanocaldococcus Jannaschii. FEMS Microbiol. Lett. 2016, 363, 217. [Google Scholar] [CrossRef] [Green Version]
- Hill, J. Sulfur and the Origins of Life. Master’s Thesis, University of Canterbury, Christchurch, New Zealand, 8 December 2000. [Google Scholar]
- Chandru, K.; Gilbert, A.; Butch, C.; Aono, M.; Cleaves, H.J. The abiotic chemistry of thiolated acetate derivatives and the origin of life. Sci. Rep. 2016, 6, 29883. [Google Scholar] [CrossRef] [Green Version]
- Sanden, S.A.; Yi, R.; Hara, M.; McGlynn, S.E. Simultaneous synthesis of thioesters and iron-sulfur clusters in water: Two universal components of energy metabolism. Chem. Comm. 2020, 56, 11989–11992. [Google Scholar] [CrossRef]
- Battin, E.E.; Brumaghim, J.L. Metal specificity in DNA damage prevention by sulfur antioxidants. J. Inorg. Biochem. 2008, 102, 2036–2042. [Google Scholar] [CrossRef]
- Yin, S.N.; Liu, Y.; Zhou, C.; Yang, S. Glutathione-mediated Cu(I)/Cu(II) Complexes: Valence-dependent effects on clearance and in vivo imaging application. Nanomaterials 2017, 7, 132. [Google Scholar] [CrossRef] [Green Version]
- Aliaga, M.E.; López-Alarcón, C.; Bridi, R.; Speisky, H. Redox-implications associated with the formation of complexes between copper ions and reduced or oxidized glutathione. J. Inorg. Biochem. 2016, 154, 78–88. [Google Scholar] [CrossRef]
- Kachur, A.V.; Koch, C.J.; Biaglow, J.E. Mechanism of copper-catalyzed oxidation of glutathione. Free Radic. Res. 1998, 28, 259–269. [Google Scholar] [CrossRef]
- Moffet, J.W.; Zika, R.G. Oxidation kinetics of Cu(I) in seawater: Implications for its existance in the marine environment. Mar. Chem. 1983, 13, 239–251. [Google Scholar] [CrossRef]
- Ngamchuea, K.; Batchelor-Mcauley, C.; Compton, R.G. The copper(II)-catalyzed oxidation of glutathione. Eur. J. Chem. 2016, 22, 15937–15944. [Google Scholar] [CrossRef] [PubMed]
- Corazza, A.; Harvey, I.; Sadler, P.J. LH,13C-NMR and X-Ray absorption studies of copper(I) glutathione complexes. Eur. J. Biochem. 1996, 236, 697–705. [Google Scholar] [CrossRef] [PubMed]
- Aliaga, M.E.; Carrasco-Pozo, C.; López-Alarcón, C.; Speisky, H. The Cu(I)-glutathione complex: Factors affecting its formation and capacity to generate reactive oxygen species. Trans. Met. Chem. 2010, 35, 321–329. [Google Scholar] [CrossRef]
- Pecci, L.; Montefoschi, G.; Musci, G.; Cavallini, D. Novel findings on the copper catalysed oxidation of cysteine. J. Amino Acids 1997, 13, 355–367. [Google Scholar] [CrossRef]
- Rigo, A.; Corazza, A.; Luisa Di Paolo, M.; Rossetto, M.; Ugolini, R.; Scarpa, M. Interaction of copper with cysteine: Stability of cuprous complexes and catalytic role of cupric ions in anaerobic thiol oxidation. J. Inorg. Biochem. 2004, 98, 1495–1501. [Google Scholar] [CrossRef]
- Cavallini, D.; De Marco, C.; Dupre, S.; Rotilio, G. The copper catalyzed oxidation of cysteine to cystine. Arch. Biochem. Biophys. 1969, 130, 354–361. [Google Scholar] [CrossRef]
- Kolthoff, I.M.; Stricks, W. Polarographic investigations of reactions in aqueous solutions containing copper and cysteine (cystine). J. Am. Chem. Soc. 1951, 73, 1728–1733. [Google Scholar] [CrossRef]
- Flitman, R.; Frieden, E. Cuprous ion formation in cupric ion catalyzed oxidations. J. Am. Chem. Soc. 1957, 79, 5198–5203. [Google Scholar] [CrossRef]
- Knežević, L.; Bura-Nakić, E. Investigation of thiol compounds (L-cysteine, thioacetic acid and ethanethiol) with V(V) and V(IV) using combined spectroscopy and chromatography. J. Inorg. Biochem. 2023, 242, 112158. [Google Scholar] [CrossRef] [PubMed]
- Fausto, R.; Batista de Carvalho, L.A.E.; Teixeira-Dias, J.J.C. Molecular structure and properties of thioacetic acid. J. Mol. Struct. 1990, 207, 67–83. [Google Scholar] [CrossRef]
- Moffet, J.W.; Zika, R.G.; Petasne, R.G. Evaluation of bathocuproine for the spectrophotometric determination of copper (I) in copper redox studies with applications in studies of natural waters. Anal. Chim. Acta 1985, 175, 171–179. [Google Scholar] [CrossRef]
- Buerge-Weirich, D.; Sulzberger, B. Formation of Cu(I) in estuarine and marine waters: Application of a new solid-phase extraction method to measure Cu(I). Environ. Sci. Technol. 2004, 38, 1843–1848. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Crmarić, D.; Bura-Nakić, E. Interaction between Cu and Thiols of Biological and Environmental Importance: Case Study Using Combined Spectrophotometric/Bathocuproine Sulfonate Disodium Salt Hydrate (BCS) Assay. Molecules 2023, 28, 5065. https://doi.org/10.3390/molecules28135065
Crmarić D, Bura-Nakić E. Interaction between Cu and Thiols of Biological and Environmental Importance: Case Study Using Combined Spectrophotometric/Bathocuproine Sulfonate Disodium Salt Hydrate (BCS) Assay. Molecules. 2023; 28(13):5065. https://doi.org/10.3390/molecules28135065
Chicago/Turabian StyleCrmarić, Dora, and Elvira Bura-Nakić. 2023. "Interaction between Cu and Thiols of Biological and Environmental Importance: Case Study Using Combined Spectrophotometric/Bathocuproine Sulfonate Disodium Salt Hydrate (BCS) Assay" Molecules 28, no. 13: 5065. https://doi.org/10.3390/molecules28135065