

The Oxygen Evolution Reaction at MoS2 Edge Sites: The Role of a Solvent Environment in DFT-Based Molecular Simulations

Abstract
:1. Introduction
2. Methodologies
2.1. The Implicit Solvent Model
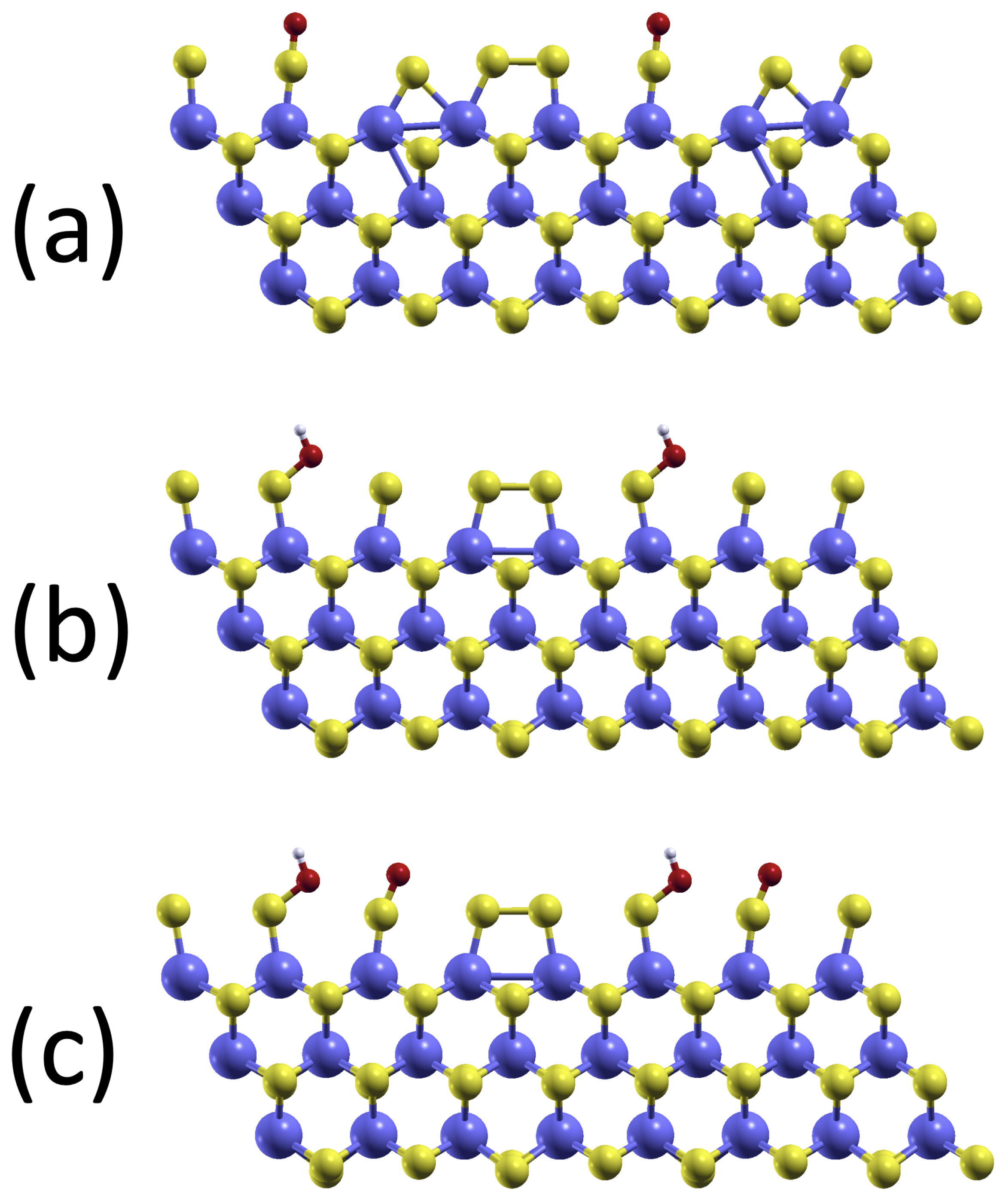
2.2. Model System of the MoS2 Edge
2.3. Water Splitting and PCETs
2.4. Density Functional Theory Calculations
3. Results and Discussion
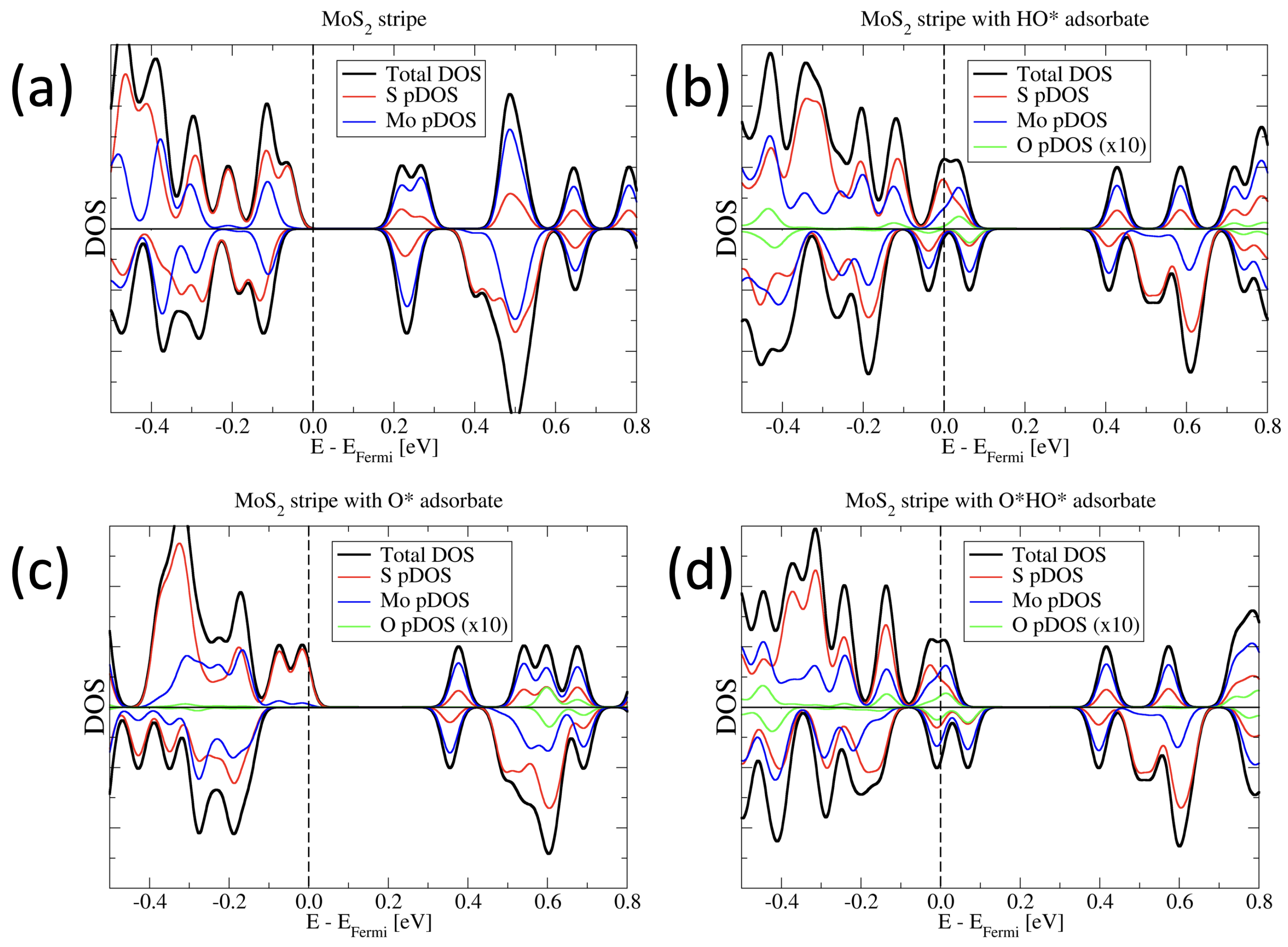
3.1. Electronic and Magnetic Structure
3.2. Energetics of the OER
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hota, P.; Das, A.; Maiti, D.K. A short review on generation of green fuel hydrogen through water splitting. Int. J. Hydrogen Energy 2023, 48, 523–541. [Google Scholar] [CrossRef]
- Zhang, H.; Wang, J.; Zhao, X.; Yang, J.; Bu sinnah, Z.A. Modeling a hydrogen-based sustainable multi-carrier energy system using a multi-objective optimization considering embedded joint chance constraints. Energy 2023, 278, 127643. [Google Scholar] [CrossRef]
- Solati, N.; Karakaya, C.; Kaya, S. Advancing the Understanding of the Structure–Activity–Durability Relation of 2D MoS 2 for the Hydrogen Evolution Reaction. ACS Catal. 2023, 13, 342–354. [Google Scholar] [CrossRef]
- de Oliveira, C.C.; Autreto, P.A.d.S. Optimized 2D nanostructures for catalysis of hydrogen evolution reactions. MRS Adv. 2023, 8, 307–310. [Google Scholar] [CrossRef]
- Wang, F.; Shifa, T.A.; Zhan, X.; Huang, Y.; Liu, K.; Cheng, Z.; Jiang, C.; He, J. Recent advances in transition-metal dichalcogenide based nanomaterials for water splitting. Nanoscale 2015, 7, 19764–19788. [Google Scholar] [CrossRef] [PubMed]
- Manzeli, S.; Ovchinnikov, D.; Pasquier, D.; Yazyev, O.V.; Kis, A. 2D transition metal dichalcogenides. Nat. Rev. Mater. 2017, 2, 17033. [Google Scholar] [CrossRef]
- Chhowalla, M.; Shin, H.S.; Eda, G.; Li, L.J.; Loh, K.P.; Zhang, H. The chemistry of two-dimensional layered transition metal dichalcogenide nanosheets. Nat. Chem. 2013, 5, 263–275. [Google Scholar] [CrossRef]
- Politano, A.; Chiarello, G.; Kuo, C.N.; Lue, C.S.; Edla, R.; Torelli, P.; Pellegrini, V.; Boukhvalov, D.W. Tailoring the Surface Chemical Reactivity of Transition-Metal Dichalcogenide PtTe2 Crystals. Adv. Funct. Mater. 2018, 28, 1706504. [Google Scholar] [CrossRef]
- Ye, Z.; Tan, C.; Huang, X.; Ouyang, Y.; Yang, L.; Wang, Z.; Dong, M. Emerging MoS2 Wafer-Scale Technique for Integrated Circuits. Nano-Micro Lett. 2023, 15, 38. [Google Scholar] [CrossRef]
- Li, X.; Wei, Y.; Wang, Z.; Kong, Y.; Su, Y.; Lu, G.; Mei, Z.; Su, Y.; Zhang, G.; Xiao, J.; et al. One-dimensional semimetal contacts to two-dimensional semiconductors. Nat. Commun. 2023, 14, 111. [Google Scholar] [CrossRef]
- Liu, L.; Kumar, S.B.; Ouyang, Y.; Guo, J. Performance Limits of Monolayer Transition Metal Dichalcogenide Transistors. IEEE Trans. Electron Devices 2011, 58, 3042–3047. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A.; Faella, E.; Durante, O.; Giubileo, F.; Pelella, A.; Viscardi, L.; Intonti, K.; Sleziona, S.; Schleberger, M.; Di Bartolomeo, A. Optoelectronic memory in 2D MoS2 field effect transistor. J. Phys. Chem. Solids 2023, 179, 111406. [Google Scholar] [CrossRef]
- Li, X.; Wang, R.; Wu, Q.; Yu, Y.; Gao, T.; Yao, T.; Wang, X.; Han, J.; Song, B. Synergistically Designed Dual Interfaces to Enhance the Electrochemical Performance of MoO2/MoS2 in Na- and Li-Ion Batteries. Small 2023, 19, 2206940. [Google Scholar] [CrossRef]
- Bai, Z.; Xu, H.; Li, G.; Yang, B.; Yao, J.; Guo, K.; Wang, N. MoS2 Nanosheets Decorated with Fe3O4 Nanoparticles for Highly Efficient Solar Steam Generation and Water Treatment. Molecules 2023, 28, 1719. [Google Scholar] [CrossRef] [PubMed]
- Guo, M.; Yuan, B.; Sui, Y.; Xiao, Y.; Dong, J.; Yang, L.; Bai, L.; Yang, H.; Wei, D.; Wang, W.; et al. Rational design of molybdenum sulfide/tungsten oxide solar absorber with enhanced photocatalytic degradation toward dye wastewater purification. J. Colloid Interface Sci. 2023, 631, 33–43. [Google Scholar] [CrossRef]
- Li, G.L.; Miao, Y.Y.; Qiao, X.Y.; Wang, T.Y.; Deng, F. Engineering edge sites based on NiS2/MoS2/CNTs heterojunction catalyst for overall water splitting. Appl. Surf. Sci. 2023, 615, 156309. [Google Scholar] [CrossRef]
- Chen, B.; Hu, P.; Yang, F.; Hua, X.; Yang, F.F.; Zhu, F.; Sun, R.; Hao, K.; Wang, K.; Yin, Z. In Situ Porousized MoS2 Nano Islands Enhance HER/OER Bifunctional Electrocatalysis. Small 2023, 19, 2207177. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, Y.; Ling, C.; Chen, Q.; Wang, Z.; Shi, L.; Wang, J. Activating Inert Basal Planes of MoS2 for Hydrogen Evolution Reaction through the Formation of Different Intrinsic Defects. Chem. Mater. 2016, 28, 4390–4396. [Google Scholar] [CrossRef]
- German, E.; Gebauer, R. Why are MoS2 monolayers not a good catalyst for the oxygen evolution reaction? Appl. Surf. Sci. 2020, 528, 146591. [Google Scholar] [CrossRef]
- Pesci, F.M.; Sokolikova, M.S.; Grotta, C.; Sherrell, P.C.; Reale, F.; Sharda, K.; Ni, N.; Palczynski, P.; Mattevi, C. MoS2/WS2 Heterojunction for Photoelectrochemical Water Oxidation. ACS Catal. 2017, 7, 4990–4998. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Tang, M.T.; Cao, A.; Chan, K.; Nørskov, J.K. Insights into the Hydrogen Evolution Reaction on 2D Transition-Metal Dichalcogenides. J. Phys. Chem. C 2022, 126, 5151–5158. [Google Scholar] [CrossRef]
- Hohenberg, P.; Kohn, W. Inhomogeneous Electron Gas. Phys. Rev. 1964, 136, B864–B871. [Google Scholar] [CrossRef] [Green Version]
- Kohn, W.; Sham, L.J. Self-Consistent Equations Including Exchange and Correlation Effects. Phys. Rev. 1965, 140, A1133–A1138. [Google Scholar] [CrossRef] [Green Version]
- Klamt, A.; Schüürmann, G. COSMO: A new approach to dielectric screening in solvents with explicit expressions for the screening energy and its gradient. J. Chem. Soc. Perkin Trans. 1993, 2, 799–805. [Google Scholar] [CrossRef]
- Tomasi, J.; Persico, M. Molecular Interactions in Solution: An Overview of Methods Based on Continuous Distributions of the Solvent. Chem. Rev. 1994, 94, 2027–2094. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum mechanical continuum solvation models. Chem. Rev. 2005, 105, 2999–3093. [Google Scholar] [CrossRef] [PubMed]
- Onsager, L. Electric Moments of Molecules in Liquids. J. Am. Chem. Soc. 1936, 58, 1486–1493. [Google Scholar] [CrossRef]
- Corni, S.; Tomasi, J. Enhanced response properties of a chromophore physisorbed on a metal particle. J. Chem. Phys. 2001, 114, 3739–3751. [Google Scholar] [CrossRef]
- Fattebert, J.L.; Gygi, F. Density functional theory for efficientab initio molecular dynamics simulations in solution. J. Comput. Chem. 2002, 23, 662–666. [Google Scholar] [CrossRef] [PubMed]
- Fattebert, J.L.; Gygi, F. First-principles molecular dynamics simulations in a continuum solvent. Int. J. Quantum Chem. 2003, 93, 139–147. [Google Scholar] [CrossRef]
- Andreussi, O.; Dabo, I.; Marzari, N. Revised self-consistent continuum solvation in electronic-structure calculations. J. Chem. Phys. 2012, 136, 064102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giannozzi, P.; Andreussi, O.; Brumme, T.; Bunau, O.; Buongiorno Nardelli, M.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Cococcioni, M.; et al. Advanced capabilities for materials modelling with Quantum ESPRESSO. J. Phys. Condens. Matter 2017, 29, 465901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Deng, J.J.; Yao, W.W.; Gurti, J.I.; Li, W.; Wang, W.J.; Yao, J.X.; Ding, X.L. Non-stoichiometric molybdenum sulfide clusters and their reactions with the hydrogen molecule. Phys. Chem. Chem. Phys. 2021, 23, 347–355. [Google Scholar] [CrossRef]
- Wang, Y.Y.; Deng, J.J.; Wang, X.; Che, J.T.; Ding, X.L. Small stoichiometric (MoS2) n clusters with the 1T phase. Phys. Chem. Chem. Phys. 2018, 20, 6365–6373. [Google Scholar] [CrossRef] [PubMed]
- Ulman, K.; Nguyen, M.T.; Seriani, N.; Gebauer, R. Passivation of surface states of α-Fe2O3(0001) surface by deposition of Ga2O3 overlayers: A density functional theory study. J. Chem. Phys. 2016, 144, 094701. [Google Scholar] [CrossRef]
- Nguyen, M.T.; Farnesi Camellone, M.; Gebauer, R. On the electronic, structural, and thermodynamic properties of Au supported on α-MoS2 surfaces and their interaction with CO. J. Chem. Phys. 2015, 143, 034704. [Google Scholar] [CrossRef] [PubMed]
- Byskov, L.S.; Nørskov, J.K.; Clausen, B.S.; Topsøe, H. Edge termination of MoS2 and CoMos catalyst particles. Catal. Lett. 2000, 64, 95–99. [Google Scholar] [CrossRef]
- Wang, H.; Tsai, C.; Kong, D.; Chan, K.; Abild-Pedersen, F.; Nørskov, J.K.; Cui, Y. Transition-metal doped edge sites in vertically aligned MoS2 catalysts for enhanced hydrogen evolution. Nano Res. 2015, 8, 566–575. [Google Scholar] [CrossRef]
- Kim, T.; Lee, T.H.; Park, S.Y.; Eom, T.H.; Cho, I.; Kim, Y.; Kim, C.; Lee, S.A.; Choi, M.J.; Suh, J.M.; et al. Drastic Gas Sensing Selectivity in 2-Dimensional MoS2 Nanoflakes by Noble Metal Decoration. ACS Nano 2023, 17, 4404–4413. [Google Scholar] [CrossRef]
- Norskov, J.K.; Rossmeisl, J.; Logadottir, A.; Lindqvist, L.; Kitchin, J.R.; Bligaard, T.; Jónsson, H. Origin of the Overpotential for Oxygen Reduction at a Fuel-Cell Cathode. J. Phys. Chem. B 2004, 108, 17886–17892. [Google Scholar] [CrossRef]
- Gebauer, R.; Serra, S.; Chiarotti, G.; Scandolo, S.; Baroni, S.; Tosatti, E. Noncolinear spin polarization from frustrated antiferromagnetism: A possible scenario for molecular oxygen at high pressure. Phys. Rev. B 2000, 61, 6145–6149. [Google Scholar] [CrossRef]
- Groß, A. Challenges in the modeling of elementary steps in electrocatalysis. Curr. Opin. Electrochem. 2023, 37, 101170. [Google Scholar] [CrossRef]
- Chan, K.; Norskov, J.K. Electrochemical Barriers Made Simple. J. Phys. Chem. Lett. 2015, 6, 2663–2668. [Google Scholar] [CrossRef]
- Righi, G.; Fabris, S.; Piccinin, S. Oxygen Evolution Reaction on the Fe3O4(001) Surface: Theoretical Insights into the Role of Terminal and Bridging Oxygen Atoms. J. Phys. Chem. C 2021, 125, 18752–18761. [Google Scholar] [CrossRef]
- Prandini, G.; Marrazzo, A.; Castelli, I.E.; Mounet, N.; Marzari, N. Precision and efficiency in solid-state pseudopotential calculations. npj Comput. Mater. 2018, 4, 72. [Google Scholar] [CrossRef] [Green Version]
- Lejaeghere, K.; Bihlmayer, G.; Bjorkman, T.; Blaha, P.; Blugel, S.; Blum, V.; Caliste, D.; Castelli, I.E.; Clark, S.J.; Dal Corso, A.; et al. Reproducibility in density functional theory calculations of solids. Science 2016, 351, aad3000. [Google Scholar] [CrossRef] [Green Version]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Marzari, N.; Vanderbilt, D.; De Vita, A.; Payne, M.C. Thermal Contraction and Disordering of the Al(110) Surface. Phys. Rev. Lett. 1999, 82, 3296–3299. [Google Scholar] [CrossRef] [Green Version]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [Green Version]
- Zhao, W.N.; Liu, Z.P. Mechanism and active site of photocatalytic water splitting on titania in aqueous surroundings. Chem. Sci. 2014, 5, 2256–2264. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Solvation | Intermediate | d(S−O) | d(O−H) | d(S−O(H)) |
|---|---|---|---|---|
| O*HO | 1.481 | 0.979 | 1.704 | |
| Vacuum | HO | - | 0.980 | 1.702 |
| O | 1.480 | - | - | |
| O*HO | 1.485 | 0.986 | 1.661 | |
| Water | HO | - | 0.986 | 1.661 |
| O | 1.482 | - | - |
| Solvation | Intermediate | |
|---|---|---|
| O*HO | −2.375 | |
| Vacuum | HO | −1.651 |
| O | −1.225 | |
| O*HO | −2.003 | |
| Water | HO | −1.530 |
| O | −0.946 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
German, E.; Gebauer, R. The Oxygen Evolution Reaction at MoS2 Edge Sites: The Role of a Solvent Environment in DFT-Based Molecular Simulations. Molecules 2023, 28, 5182. https://doi.org/10.3390/molecules28135182
German E, Gebauer R. The Oxygen Evolution Reaction at MoS2 Edge Sites: The Role of a Solvent Environment in DFT-Based Molecular Simulations. Molecules. 2023; 28(13):5182. https://doi.org/10.3390/molecules28135182
Chicago/Turabian StyleGerman, Estefania, and Ralph Gebauer. 2023. "The Oxygen Evolution Reaction at MoS2 Edge Sites: The Role of a Solvent Environment in DFT-Based Molecular Simulations" Molecules 28, no. 13: 5182. https://doi.org/10.3390/molecules28135182