

Potency of Xanthone Derivatives from Garcinia mangostana L. for COVID-19 Treatment through Angiotensin-Converting Enzyme 2 and Main Protease Blockade: A Computational Study

Abstract
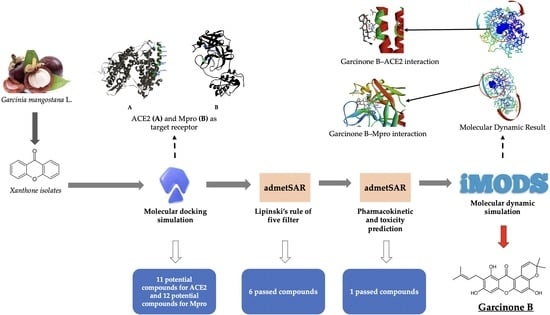
:
1. Introduction
2. Results
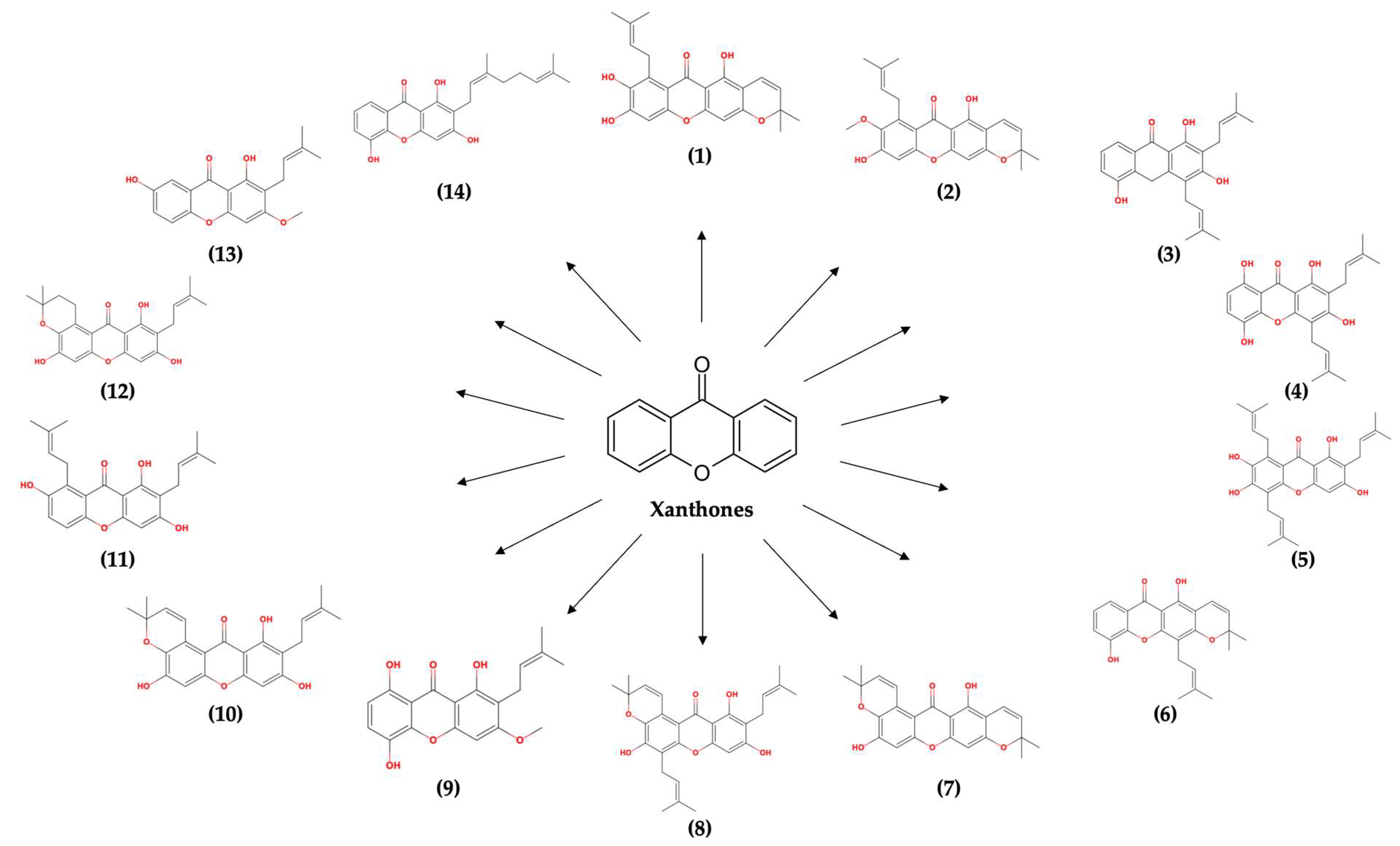
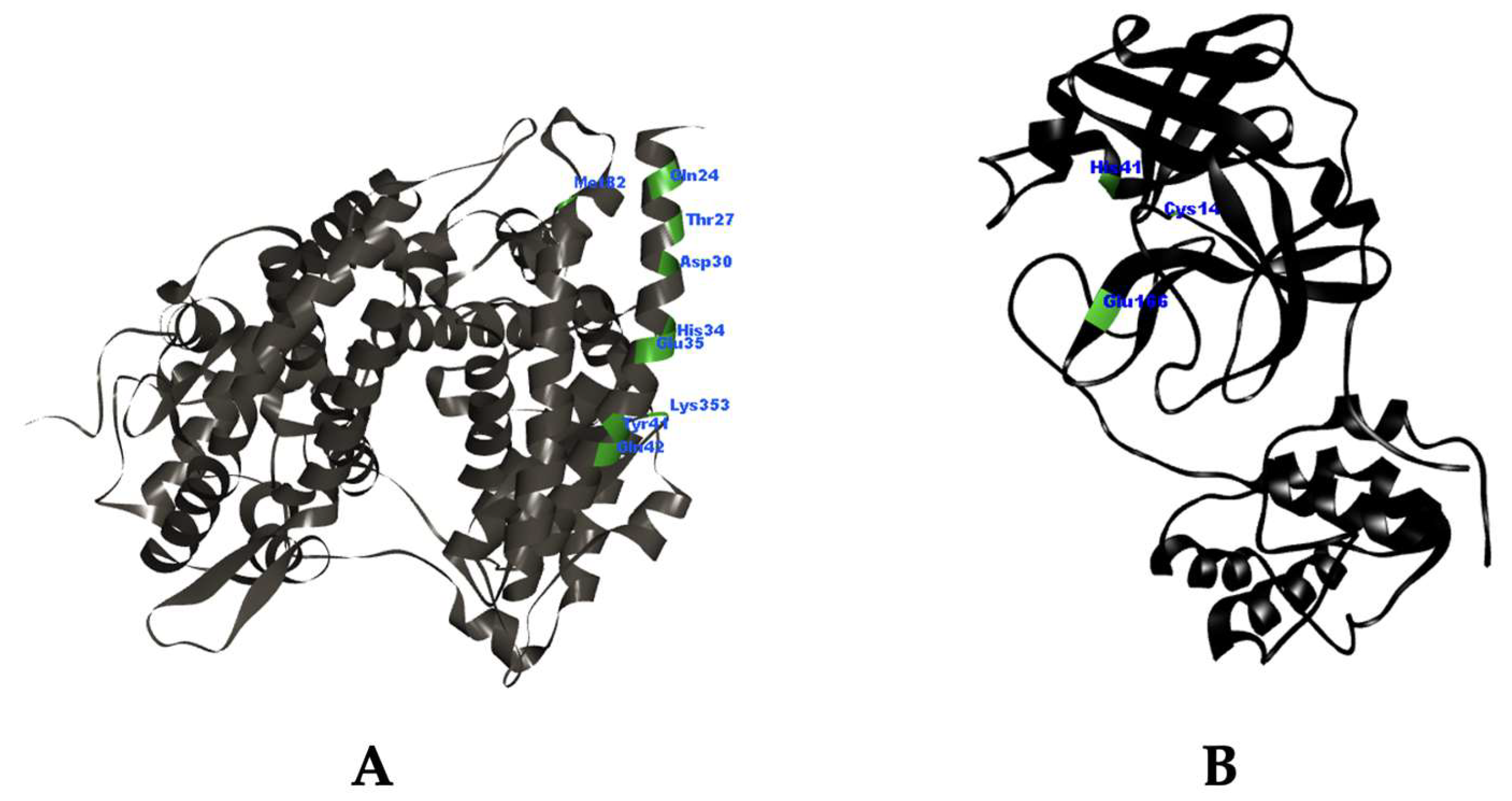
2.1. Molecular Docking Simulation
2.2. Lipinski’s Rule of Five Filtration
2.3. Pharmacokinetic Profile and Toxicity Prediction
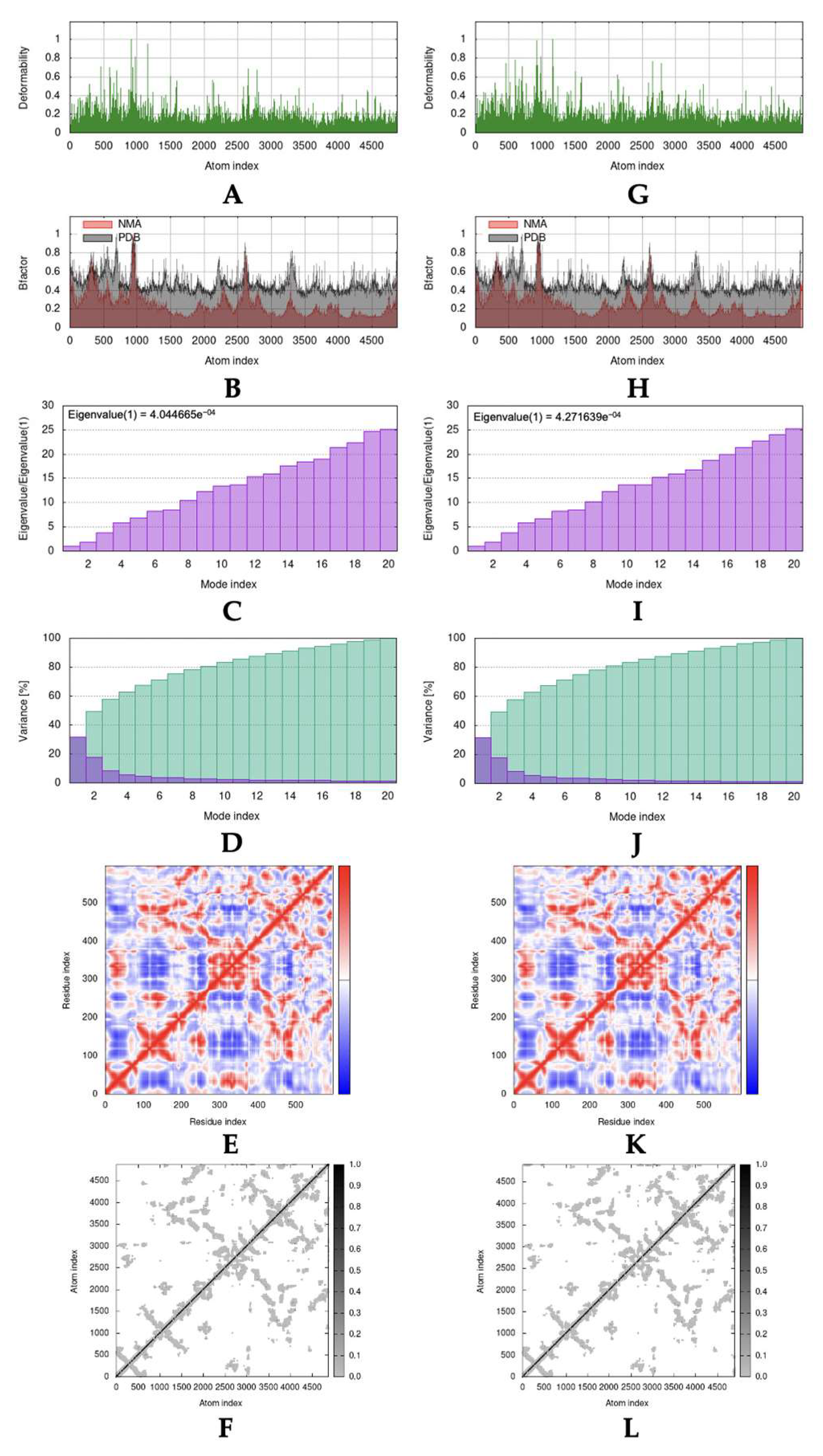
2.4. Molecular Dynamic Simulation
3. Discussion
3.1. Molecular Docking Simulation
3.2. Lipinski’s Rule of Five Filtration
3.3. Pharmacokinetic Profile and Toxicity Prediction
3.4. Molecular Dynamic Simulation
4. Materials and Methods
4.1. Molecular Docking Study
4.1.1. Ligand Preparation
4.1.2. Receptor Preparation
4.1.3. Molecular Docking Simulation
4.2. Lipinski’s Rule of Five Filtration
4.3. Pharmacokinetic Profile and Toxicity Prediction
4.4. Molecular Dynamic Simulation
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Cleofas, J.V.; Oducado, R.M.F. COVID-19 Death Occurrences, Pandemic Fatigue, and Well-Being. J. Loss Trauma 2022, 27, 679–682. [Google Scholar] [CrossRef]
- Kashte, S.; Gulbake, A.; El-Amin, S.F.; Gupta, A. COVID-19 Vaccines: Rapid Development, Implications, Challenges and Future Prospects. Hum. Cell. 2021. [CrossRef]
- Hadj Hassine, I. COVID-19 Vaccines and Variants of Concern: A Review. Rev. Med. Virol. 2022, 32, e2313. [Google Scholar] [CrossRef]
- Seyed Hosseini, E.; Riahi Kashani, N.; Nikzad, H.; Azadbakht, J.; Hassani Bafrani, H.; Haddad Kashani, H. The Novel Coronavirus Disease-2019 (COVID-19): Mechanism of Action, Detection and Recent Therapeutic Strategies. Virology 2020, 551, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Rabascall, C.X.; Lou, B.X.; Navetta-Modrov, B.; Hahn, S.S. Effective Use of Monoclonal Antibodies for Treatment of Persistent COVID-19 Infection in a Patient on Rituximab. BMJ Case Rep. 2021, 14, e243469. [Google Scholar] [CrossRef]
- Issa, H.; Eid, A.H.; Berry, B.; Takhviji, V.; Khosravi, A.; Mantash, S.; Nehme, R.; Hallal, R.; Karaki, H.; Dhayni, K.; et al. Combination of Angiotensin (1-7) Agonists and Convalescent Plasma as a New Strategy to Overcome Angiotensin Converting Enzyme 2 (ACE2) Inhibition for the Treatment of COVID-19. Front. Med. 2021, 8, 620990. [Google Scholar] [CrossRef]
- Kassegn, A.; Endris, E. Review on Socio-Economic Impacts of ‘Triple Threats’ of COVID-19, Desert Locusts, and Floods in East Africa: Evidence from Ethiopia. Cogent Soc. Sci. 2021, 7, 1–28. [Google Scholar] [CrossRef]
- Singh, A.K.; Singh, A.; Singh, R.; Misra, A. Remdesivir in COVID-19: A Critical Review of Pharmacology, Pre-Clinical and Clinical Studies. Diabetes Metab. Syndr. Clin. Res. Rev. 2020, 14, 641–648. [Google Scholar] [CrossRef]
- Pardo, J.; Shukla, A.M.; Chamarthi, G.; Gupte, A. The Journey of Remdesivir: From Ebola to COVID-19. Drugs Context 2020, 9, 1–9. [Google Scholar] [CrossRef]
- Saha, A.; Sharma, A.R.; Bhattacharya, M.; Sharma, G.; Lee, S.S.; Chakraborty, C. Probable Molecular Mechanism of Remdesivir for the Treatment of COVID-19: Need to Know More. Arch. Med. Res. 2020, 51, 585–586. [Google Scholar] [CrossRef]
- Khan, F.I.; Kang, T.; Ali, H.; Lai, D. Remdesivir Strongly Binds to RNA-Dependent RNA Polymerase, Membrane Protein, and Main Protease of SARS-CoV-2: Indication from Molecular Modeling and Simulations. Front. Pharmacol. 2021, 12, 710778. [Google Scholar] [CrossRef]
- Nguyen, H.L.; Thai, N.Q.; Truong, D.T.; Li, M.S. Remdesivir Strongly Binds to Both RNA-Dependent RNA Polymerase and Main Protease of SARS-COV-2: Evidence from Molecular Simulations. J. Phys. Chem. B 2020, 124, 11337–11348. [Google Scholar] [CrossRef] [PubMed]
- Hussein, M.I.H.; Albashir, A.A.D.; Elawad, O.A.M.A.; Homeida, A. Malaria and COVID-19: Unmasking Their Ties. Malar. J. 2020, 19, 457. [Google Scholar] [CrossRef] [PubMed]
- Lestari, K.; Sitorus, T.; Instiaty, S.M.; Levita, J. Molecular Docking of Quinine, Chloroquine and Hydroxychloroquine to Angiotensin Converting Enzyme 2 (ACE2) Receptor for Discovering New Potential COVID-19 Antidote. J. Adv. Pharm. Educ. Res. 2020, 10, 1–4. [Google Scholar]
- Khelfaoui, H.; Harkati, D.; Saleh, B.A. Molecular Docking, Molecular Dynamics Simulations and Reactivity, Studies on Approved Drugs Library Targeting ACE2 and SARS-CoV-2 Binding with ACE2. J. Biomol. Struct. Dyn. 2020, 39, 7246–7262. [Google Scholar] [CrossRef]
- Wang, N.; Han, S.; Liu, R.; Meng, L.; He, H.; Zhang, Y.; Wang, C.; Lv, Y.; Wang, J.; Li, X.; et al. Chloroquine and Hydroxychloroquine as ACE2 Blockers to Inhibit Viropexis of 2019-NCoV Spike Pseudotyped Virus. Phytomedicine 2020, 79, 153333. [Google Scholar] [CrossRef]
- Bignardi, P.R.; Vengrus, C.S.; Aquino, B.M.; Cerci Neto, A. Use of Hydroxychloroquine and Chloroquine in Patients with COVID-19: A Meta-Analysis of Randomized Clinical Trials. Pathog. Glob. Health 2021, 115, 139–150. [Google Scholar] [CrossRef]
- Ebina-Shibuya, R.; Namkoong, H.; Horita, N.; Kato, H.; Hara, Y.; Kobayashi, N.; Kaneko, T. Hydroxychloroquine and Chloroquine for Treatment of Coronavirus Disease 19 (COVID-19): A Systematic Review and Meta-Analysis of Randomized and Non-Randomized Controlled Trials. J. Thorac. Dis. 2021, 13, 202–212. [Google Scholar] [CrossRef]
- Doyno, C.; Sobieraj, D.M.; Baker, W.L. Toxicity of Chloroquine and Hydroxychloroquine Following Therapeutic Use or Overdose. Clin. Toxicol. 2020, 59, 12–23. [Google Scholar] [CrossRef]
- Javorac, D.; Grahovac, L.; Manić, L.; Stojilković, N.; Anđelković, M.; Bulat, Z.; Đukić-Ćosić, D.; Curcic, M.; Djordjevic, A.B. An Overview of the Safety Assessment of Medicines Currently Used in the COVID-19 Disease Treatment. Food Chem. Toxicol. 2020, 144, 111639. [Google Scholar] [CrossRef]
- Farooq, S.; Ngaini, Z. Natural and Synthetic Drugs as Potential Treatment for Coronavirus Disease 2019 (COVID-2019). Chem. Afr. 2020, 4, 1–13. [Google Scholar] [CrossRef]
- Santhi, V.P.; Masilamani, P.; Sriramavaratharajan, V.; Murugan, R.; Gurav, S.S.; Sarasu, V.P.; Parthiban, S.; Ayyanar, M. Therapeutic Potential of Phytoconstituents of Edible Fruits in Combating Emerging Viral Infections. J. Food Biochem. 2021, 45, e13851. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Liu, Y.; Yang, Y.; Zhang, P.; Zhong, W.; Wang, Y.; Wang, Q.; Xu, Y.; Li, M.; Li, X.; et al. Analysis of Therapeutic Targets for SARS-CoV-2 and Discovery of Potential Drugs by Computational Methods. Acta Pharm. Sin. B 2020, 10, 766–788. [Google Scholar] [CrossRef]
- Al Adem, K.; Shanti, A.; Stefanini, C.; Lee, S. Inhibition of SARS-CoV-2 Entry into Host Cells Using Small Molecules. Pharmaceuticals 2020, 13, 447. [Google Scholar] [CrossRef]
- Pomalingo, D.R.; Suhandi, C.; Megantara, S.; Muchtaridi, M. The Optimization of α-Mangostin as a New Drug Candidate through Molecular Docking and Dynamic Simulations. Rasayan J. Chem. 2021, 14, 698–704. [Google Scholar] [CrossRef]
- Megantara, S.; Wathoni, N.; Mohammed, A.F.A.; Suhandi, C.; Ishmatullah, M.H.; Putri, M.F.F.D. In Silico Study: Combination of α-Mangostin and Chitosan Conjugated with Trastuzumab against Human Epidermal Growth Factor Receptor 2. Polymers 2022, 14, 2747. [Google Scholar] [CrossRef]
- Hidayat, S.; Ibrahim, F.; Pratama, K.; Muchtaridi, M. The Interaction of Alpha-Mangostin and Its Derivatives against Main Protease Enzyme in COVID-19 Using in Silico Methods. J. Adv. Pharm. Technol. Res. 2021, 12, 285–290. [Google Scholar] [CrossRef]
- Suharyani, I.; Muchtaridi, M.; Mohammed, A.F.A.; Elamin, K.M.; Wathoni, N.; Abdassah, M. α-Mangostin/γ-Cyclodextrin Inclusion Complex: Formation and Thermodynamic Study. Polymers 2021, 13, 2890. [Google Scholar] [CrossRef] [PubMed]
- Alyami, H.; Dahmash, E.; Alyami, F.; Dahmash, D.; Huynh, C.; Terry, D.; Mohammed, A.R. Dosage form Preference Consultation Study in Children and Young Adults: Paving the Way for Patient-Centred and Patient-Informed Dosage form Development. Eur. J. Hosp. Pharm. 2016, 24, 332–337. [Google Scholar] [CrossRef]
- Yang, R.; Li, P.; Li, N.; Zhang, Q.; Bai, X.; Wang, L.; Xiao, Y.; Sun, L.; Yang, Q.; Yan, J. Xanthones from the Pericarp of Garcinia Mangostana. Molecules 2017, 22, 683. [Google Scholar] [CrossRef] [Green Version]
- Kataria, R.; Khatkar, A. Molecular Docking, Synthesis, Kinetics Study, Structure-Activity Relationship and ADMET Analysis of Morin Analogous as Helicobacter Pylori Urease Inhibitors. BMC Chem. 2019, 13, 45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, D.P.; Penn-Nicholson, A.; Cho, M.W. Identification of Critical Determinants on ACE2 for SARS-CoV Entry and Development of a Potent Entry Inhibitor. Virology 2006, 350, 15–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giordano, D.; De Masi, L.; Argenio, M.A.; Facchiano, A. Structural Dissection of Viral Spike-protein Binding of SARS-CoV-2 and SARS-CoV-1 to the Human Angiotensin-converting Enzyme 2 (Ace2) as Cellular Receptor. Biomedicines 2021, 9, 1038. [Google Scholar] [CrossRef] [PubMed]
- Natarajan, P.M.; Umapathy, V.R.; Murali, A.; Swamikannu, B. Computational Simulations of Identified Marine-Derived Natural Bioactive Compounds as Potential Inhibitors of Oral Cancer. Future. Sci. OA 2022, 8, FSO782. [Google Scholar] [CrossRef]
- Adeoye, A.O.; Oso, B.J.; Olaoye, I.F.; Tijjani, H.; Adebayo, A.I. Repurposing of Chloroquine and Some Clinically Approved Antiviral Drugs as Effective Therapeutics to Prevent Cellular Entry and Replication of Coronavirus. J. Biomol. Struct. Dyn. 2020, 39, 3469–3479. [Google Scholar] [CrossRef]
- Abdelli, I.; Hassani, F.; Bekkel Brikci, S.; Ghalem, S. In Silico Study the Inhibition of Angiotensin Converting Enzyme 2 Receptor of COVID-19 by Ammoides Verticillata Components Harvested from Western Algeria. J. Biomol. Struct. Dyn. 2021, 39, 3263–3276. [Google Scholar] [CrossRef]
- López-Blanco, J.R.; Aliaga, J.I.; Quintana-Ortí, E.S.; Chacón, P. IMODS: Internal Coordinates Normal Mode Analysis Server. Nucleic Acids Res. 2014, 42, W271–W276. [Google Scholar] [CrossRef]
- Samavati, L.; Uhal, B.D. ACE2, Much More Than Just a Receptor for SARS-CoV-2. Front. Cell. Infect. Microbiol. 2020, 10, 317. [Google Scholar] [CrossRef]
- Silva, M.G.; Falcoff, N.L.; Corradi, G.R.; Di Camillo, N.; Seguel, R.F.; Tabaj, G.C.; Guman, G.R.; de Matteo, E.; Nuñez, M.; Gironacci, M.M. Effect of Age on Human ACE2 and ACE2-Expressing Alveolar Type II Cells Levels. Pediatr. Res. 2022, 93, 948–952. [Google Scholar] [CrossRef]
- Zulli, A.; Burrell, L.M.; Buxton, B.F.; Hare, D.L. ACE2 and AT4R Are Present in Diseased Human Blood Vessels. Eur. J. Histochem. 2008, 52, 39–44. [Google Scholar] [CrossRef] [Green Version]
- Luan, J.; Lu, Y.; Jin, X.; Zhang, L. Spike Protein Recognition of Mammalian ACE2 Predicts the Host Range and an Optimized ACE2 for SARS-CoV-2 Infection. Biochem. Biophys. Res. Commun. 2020, 526, 165–169. [Google Scholar] [CrossRef] [PubMed]
- Ali, A.; Vijayan, R. Dynamics of the ACE2–SARS-CoV-2/SARS-CoV Spike Protein Interface Reveal Unique Mechanisms. Sci. Rep. 2020, 10, 14214. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, I.; Pawara, R.; Surana, S.; Patel, H. The Repurposed ACE2 Inhibitors: SARS-CoV-2 Entry Blockers of COVID-19. Top. Curr. Chem. 2021, 379, 40. [Google Scholar] [CrossRef] [PubMed]
- Chaouat, A.E.; Brizic, I.; Kucan Brlic, P.; Atari, N.; Kliker, L.; Alfi, O.; Mandelboim, M.; Wolf, D.; Tafish, L.; Kol, I.; et al. Anti-Human ACE2 Antibody Neutralizes and Inhibits Virus Production of SARS-CoV-2 Variants of Concern. iScience 2022, 25, 104935. [Google Scholar] [CrossRef]
- Hu, Q.; Xiong, Y.; Zhu, G.H.; Zhang, Y.N.; Zhang, Y.W.; Huang, P.; Ge, G.B. The SARS-CoV-2 Main Protease (Mpro): Structure, Function, and Emerging Therapies for COVID-19. MedComm 2022, 3, e151. [Google Scholar] [CrossRef]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; et al. Structure of the SARS-CoV-2 Spike Receptor-Binding Domain Bound to the ACE2 Receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef] [Green Version]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of Mpro from SARS-CoV-2 and Discovery of Its Inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef] [Green Version]
- Halim, S.A.; Waqas, M.; Khan, A.; Al-Harrasi, A. In Silico Prediction of Novel Inhibitors of SARS-Cov-2 Main Protease through Structure-Based Virtual Screening and Molecular Dynamic Simulation. Pharmaceuticals 2021, 14, 896. [Google Scholar] [CrossRef]
- Du, X.; Li, Y.; Xia, Y.L.; Ai, S.M.; Liang, J.; Sang, P.; Ji, X.L.; Liu, S.Q. Insights into Protein–Ligand Interactions: Mechanisms, Models, and Methods. Int. J. Mol. Sci. 2016, 17, 144. [Google Scholar] [CrossRef] [Green Version]
- Torres, P.H.M.; Sodero, A.C.R.; Jofily, P.; Silva, F.P., Jr. Key Topics in Molecular Docking for Drug Design. Int. J. Mol. Sci. 2019, 20, 4574. [Google Scholar] [CrossRef] [Green Version]
- Lipinski, C.A. Drug-like Properties and the Causes of Poor Solubility and Poor Permeability. J. Pharmacol. Toxicol. Methods 2000, 44, 235–249. [Google Scholar] [CrossRef]
- Benet, L.Z.; Hosey, C.M.; Ursu, O.; Oprea, T.I. BDDCS, the Rule of 5 and Drugability. Adv. Drug Deliv. Rev. 2016, 101, 89–98. [Google Scholar] [CrossRef] [Green Version]
- Cheng, F.; Li, W.; Liu, G.; Tang, Y. In Silico ADMET Prediction: Recent Advances, Current Challenges and Future Trends. Curr. Top. Med. Chem. 2013, 13, 1273–1289. [Google Scholar] [CrossRef] [PubMed]
- Yazdanian, M.; Glynn, S.L.; Wright, J.L.; Hawi, A. Correlating Partitioning and Caco-2 Cell Permeability of Structurally Diverse Small Molecular Weight Compounds. Pharm. Res. 1998, 15, 1490–1494. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Fan, Y.; Wang, Y.; Lu, Y.; Yin, Z. Characterization of Plasma Protein Binding Dissociation with Online SPE-HPLC. Sci. Rep. 2015, 5, 14866. [Google Scholar] [CrossRef] [Green Version]
- Ma, X.L.; Chen, C.; Yang, J. Predictive Model of Blood-Brain Barrier Penetration of Organic Compounds. Acta Pharmacol. Sin. 2005, 26, 500–512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deodhar, M.; Al Rihani, S.B.; Arwood, M.J.; Darakjian, L.; Dow, P.; Turgeon, J.; Michaud, V. Mechanisms of Cyp450 Inhibition: Understanding Drug-Drug Interactions Due to Mechanism-Based Inhibition in Clinical Practice. Pharmaceutics 2020, 12, 846. [Google Scholar] [CrossRef]
- Yamakuni, T.; Aoki, K.; Nakatani, K.; Kondo, N.; Oku, H.; Ishiguro, K.; Ohizumi, Y. Garcinone B Reduces Prostaglandin E2 Release and NF-ΚB-Mediated Transcription in C6 Rat Glioma Cells. Neurosci. Lett. 2006, 394, 206–210. [Google Scholar] [CrossRef]
- Sims, J.T.; Krishnan, V.; Chang, C.Y.; Engle, S.M.; Casalini, G.; Rodgers, G.H.; Bivi, N.; Nickoloff, B.J.; Konrad, R.J.; de Bono, S.; et al. Characterization of the Cytokine Storm Reflects Hyperinflammatory Endothelial Dysfunction in COVID-19. J. Allergy Clin. Immunol. 2021, 147, 107–111. [Google Scholar] [CrossRef] [PubMed]
- Madanagopal, P.; Ramprabhu, N.; Jagadeesan, R. In Silico Prediction and Structure-Based Multitargeted Molecular Docking Analysis of Selected Bioactive Compounds against Mucormycosis. Bull. Natl. Res. Cent. 2022, 46, 24. [Google Scholar] [CrossRef]
- Brown, T. ChemDraw. Sci. Teach. 2014, 81, 67. [Google Scholar]
- Shivanika, C.; Deepak Kumar, S.; Ragunathan, V.; Tiwari, P.; Sumitha, A.; Brindha Devi, P. Molecular Docking, Validation, Dynamics Simulations, and Pharmacokinetic Prediction of Natural Compounds against the SARS-CoV-2 Main-Protease. J. Biomol. Struct. Dyn. 2022, 40, 585–611. [Google Scholar] [CrossRef]
- BIOVIA. Dassault Systèmes BIOVIA, Discovery Studio Modeling Environment, Release 2017. In Dassault Systèmes; BIOVIA: San Diego, CA, USA, 2017. [Google Scholar]
- Xue, Q.; Liu, X.; Russell, P.; Li, J.; Pan, W.; Fu, J.; Zhang, A. Evaluation of the Binding Performance of Flavonoids to Estrogen Receptor Alpha by Autodock, Autodock Vina and Surflex-Dock. Ecotoxicol. Environ. Saf. 2022, 233, 113323. [Google Scholar] [CrossRef]
- Sethi, A.; Sanam, S.; Munagalasetty, S.; Jayanthi, S.; Alvala, M. Understanding the Role of Galectin Inhibitors as Potential Candidates for SARS-CoV-2 Spike Protein: In Silico Studies. RSC Adv. 2020, 10, 29873–29884. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Lou, C.; Sun, L.; Li, J.; Cai, Y.; Wang, Z.; Li, W.; Liu, G.; Tang, Y. AdmetSAR 2.0: Web-Service for Prediction and Optimization of Chemical ADMET Properties. Bioinformatics 2019, 35, 1067–1069. [Google Scholar] [CrossRef] [PubMed]
- Moon, A.; Khan, D.; Gajbhiye, P.; Jariya, M. Insilico Prediction of Toxicity of Ligands Utilizing Admetsar. Int. J. Pharma Bio Sci. 2017, 8, 674–677. [Google Scholar] [CrossRef]
- Pokharkar, O.; Lakshmanan, H.; Zyryanov, G.; Tsurkan, M. In Silico Evaluation of Antifungal Compounds from Marine Sponges against COVID-19-Associated Mucormycosis. Mar. Drugs 2022, 20, 215. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | ΔG (kcal/mol) | Ki (μM) | Amino Acid Residues Interaction | ||
|---|---|---|---|---|---|
| Hydrogen Bond | Hydrophobic | Others | |||
| Chloroquine | −4.1 | 991.4 | Lys31 | π-π stacked: His34; Alkyl: His34; π-alkyl: Lys31, His34 | N/A |
| Remdesivir | −4.38 | 611.23 | Lys31, Glu35 | π-alkyl: His34 | N/A |
| α-Mangostin | −4.27 | 746.2 | Asp30 | π-π stacked: His34; π-alkyl: Lys31, His34 | π-anion: Asp30 |
| 7-O-demethyl mangostanin | −5.17 | 162.05 | Asp30, Lys31, Glu35 | π-π stacked: His34; Amide-π stacked: His34; π-alkyl: Lys31, His34 | π-anion: Asp30 |
| Mangostanin | −4.35 | 653.11 | Lys31 | π-π stacked: His34; Alkyl: His34; π-alkyl: Lys31, His34 | π-anion: Asp30 |
| 8-Deoxygartanin | −5.13 | 173.07 | Lys31, Glu35 | π-π stacked: His34; Alkyl: Lys31 | π-sigma: His34 |
| Gartanin | −5.13 | 174.42 | N/A | π-π stacked: His34; Amide-π stacked: His34; π-alkyl: Lys31, His34 | van der Waals: Glu35 |
| Garcinone E | −3.65 | 2130 | Thr27 | π-alkyl: Lys31 | N/A |
| Trapezifolixanthone | −6.01 | 39.34 | Asp30, Glu35 | π-π stacked: His34; Amide-π stacked: His34; Alkyl: His34; π-alkyl: Lys31, His34 | Salt bridge: Lys31; π-sigma: His34 |
| Padiaxanthone | −5.75 | 61.41 | Asp30, Lys31 | π-π stacked: His34; Amide-π stacked: His34; Alkyl: His34; π-alkyl: Lys31, His34 | π-anion: Asp30; van der Waals: Glu35 |
| Tovophyllin A | −4.08 | 1020 | Asp30, Lys31 | π-π stacked: His34; Alkyl: His34; π-alkyl: Lys31, His34 | N/A |
| 1,5,8-Trihydroxy-3-methoxy-2-prenylxanthone | −5.17 | 161.09 | N/A | π-π stacked: His34; Alkyl: His34; π-alkyl: Lys31, His34 | N/A |
| Garcinone B | −5.21 | 152.73 | Asp30, Glu35 | π-π stacked: His34; Alkyl: His34; π-alkyl: Lys31, His34 | N/A |
| 1,7-Dihydroxy-2-(3-methylbut-2-enyl)-3-methoxyxanthone | −4.61 | 418 | Lys31, Glu35 | Alkyl: His34; π-alkyl: Lys31, His34 | π-sigma: His34 |
| Mangostenone D | −5.33 | 122.96 | Thr27 | π-π stacked: His34; Alkyl: His34; π-alkyl: Lys31, His34 | π-anion: Asp30 |
| Mangostinone | −6.81 | 10.19 | Glu35 | π-π stacked: His34; Alkyl: His34; π-alkyl: Lys31, His34 | Attractive charge: Lys31 |
| 1,7-Dihydroxy-2-(3-methylbut-2-enyl)-3-methoxyxanthone | −4.85 | 279.52 | Lys31 | Amide-π stacked: His34; Alkyl: His34; π-alkyl: Lys31, His34 | van der Waals: Glu35 |
| Compound | ΔG (kcal/mol) | Ki (μM) | Amino Acid Residues Interaction | ||
|---|---|---|---|---|---|
| Hydrogen Bond | Hydrophobic | Others | |||
| Chloroquine | −7.11 | 6.10 | His164 | Alkyl: His163, His172, Arg188 | π-sigma: His41; π-sulfur: Cys145 |
| Remdesivir | −6.50 | 17.33 | Glu166, Thr190, Gln192 | Alkyl: Pro168 | N/A |
| α-Mangostin | −8.31 | 0.805 | Cys145, Glu166, Thr190, Gln192 | Alkyl: His163, His172; π-alkyl: Met165 | π-π T-shaped: His41 |
| 7-O-demethyl mangostanin | −8.97 | 0.268 | Thr190, Gln192 | Alkyl: Cys145, His163; π-alkyl: Met165 | π-π T-shaped: His41; π-sigma: Gln189 |
| Mangostanin | −7.92 | 1.57 | Glu166, Arg188 | Alkyl: His41, Met49, Cys145, His163, Pro168; π-alkyl: Met165 | N/A |
| 8-Deoxygartanin | −8.87 | 0.318 | Met49, Glu166 | Alkyl: His163, His172; π-alkyl: Cys145 | N/A |
| Gartanin | −9.13 | 0.203 | His163, Glu166, Thr190 | Alkyl: Pro168; π-alkyl: Met165 | N/A |
| Garcinone E | −9.61 | 0.091 | His164, Met165, Arg188 | Alkyl: Cys44, Met49, Cys145, His163 | π-π T-shaped: His41 |
| Trapezifolixanthone | −9.34 | 0.143 | Cys145, His164, Thr190, Gln192 | Alkyl: His41, Met49; π-alkyl: Met165 | π-lone pair: Glu166 |
| Padiaxanthone | −10.23 | 0.032 | Gly143, Glu166 | Alkyl: Met49, His163, His172; π-alkyl: Cys145 | π-sigma: His41 |
| Tovophyllin A | −9.24 | 0.170 | Met165, Arg188, Thr190, Gln192 | Alkyl: His41, Met49, Pro52, His163; π-alkyl: Pro168 | π-lone pair: Glu166 |
| 1,5,8-Trihydroxy-3-methoxy-2-prenylxanthone | −9.25 | 0.166 | N/A | Alkyl: Pro52, Tyr54; π-alkyl: Met49, Met165 | N/A |
| Garcinone B | −9.59 | 0.094 | Leu141, Gly143, Ser144 | Alkyl: Leu27, Cys145, His163; π-alkyl: His41, Met49, Met165 | N/A |
| 1,7-Dihydroxy-2-(3-methylbut-2-enyl)-3-methoxyxanthone | −7.24 | 4.90 | Cys145, His164, Glu166 | Alkyl: Met165 | π-sulfur: Cys145 |
| Mangostenone D | −8.56 | 0.528 | Thr190 | Alkyl: Cys145, His163, Pro168 | π-sulfur: Met165 |
| Mangostinone | −10.30 | 0.028 | Cys145, His164, Thr190 | Alkyl: His41, Met49, Arg188; π-alkyl: Met165 | π-lone pair: Glu166 |
| 1,7-Dihydroxy-2-(3-methylbut-2-enyl)-3-methoxyxanthone | −9.98 | 0.048 | Cys145, His164, Glu166 | Alkyl: Leu27; π-alkyl: Met165 | π-cation: His163; π-sigma: His41 |
| Compound | Molecular Weight (g/mol) | Hydrogen Bond Donor | Hydrogen Bond Acceptor | LogP | Violation |
|---|---|---|---|---|---|
| 7-O-demethyl mangostanin | 380.44 | 3 | 5 | 5.08 | 1 (LogP > 5) |
| Mangostanin | 342.35 | 3 | 6 | 3.58 | 0 |
| 8-Deoxygartanin | 326.35 | 2 | 5 | 3.87 | 0 |
| Gartanin | 394.42 | 3 | 6 | 4.76 | 0 |
| Garcinone E | 380.44 | 3 | 5 | 5.08 | 1 (LogP > 5) |
| Trapezifolixanthone | 394.42 | 3 | 6 | 4.76 | 0 |
| Padiaxanthone | 464.56 | 4 | 6 | 6.29 | 1 (LogP > 5) |
| Tovophyllin A | 396.44 | 4 | 6 | 4.79 | 0 |
| 1,5,8-Trihydroxy-3-methoxy-2-prenylxanthone | 408.45 | 2 | 6 | 5.06 | 1 (LogP > 5) |
| Garcinone B | 396.44 | 3 | 6 | 4.68 | 0 |
| 1,7-Dihydroxy-2-(3-methylbut-2-enyl)-3-methoxyxanthone | 380.44 | 3 | 5 | 5.30 | 1 (LogP > 5) |
| Mangostenone D | 392.41 | 2 | 6 | 4.73 | 0 |
| Mangostinone | 462.54 | 3 | 6 | 6.26 | 1 (LogP > 5) |
| 1,7-Dihydroxy-2-(3-methylbut-2-enyl)-3-methoxyxanthone | 378.42 | 2 | 5 | 5.05 | 1 (LogP > 5) |
| Compound | Absorption | Distribution | Metabolism *) | Toxicity *) | ||||
|---|---|---|---|---|---|---|---|---|
| HIA | Caco-2 | PPB | BBB | Inhibitor | Substrate | Carcinogenicity | Ames Test (Mutagenicity) | |
| 7-O-demethyl mangostanin | 0.9895 | 0.5775 | 0.525 | 0.915 | + | − | + | − |
| Mangostanin | 0.9886 | 0.4906 | 0.500 | 0.86 | + | − | + | − |
| 8-Deoxygartanin | 0.9919 | 0.7836 | 0.500 | 0.933 | + | − | + | − |
| Gartanin | 0.9732 | 0.5446 | 0.625 | 0.733 | + | − | + | − |
| Garcinone E | 0.9855 | 0.4877 | 0.575 | 0.961 | + | − | + | − |
| Trapezifolixanthone | 0.9846 | 0.5797 | 0.600 | 0.779 | + | − | + | − |
| Padiaxanthone | 0.9465 | 0.7296 | 0.650 | 0.756 | + | − | + | − |
| Tovophyllin A | 0.9855 | 0.6397 | 0.575 | 0.769 | + | − | + | − |
| 1,5,8-Trihydroxy-3-methoxy-2-prenylxanthone | 0.9743 | 0.7549 | 0.525 | 0.719 | + | − | + | − |
| Garcinone B | 0.9632 | 0.6002 | 0.55 | 0.708 | + | − | − | − |
| 1,7-Dihydroxy-2-(3-methylbut-2-enyl)-3-methoxyxanthone | 0.9776 | 0.8016 | 0.525 | 0.946 | + | − | + | − |
| Mangostenone D | 0.9838 | 0.5834 | 0.675 | 0.823 | + | − | + | − |
| Mangostinone | 0.9846 | 0.6456 | 0.6 | 0.737 | + | − | + | − |
| 1,7-Dihydroxy-2-(3-methylbut-2-enyl)-3-methoxyxanthone | 0.9905 | 0.592 | 0.575 | 0.921 | + | − | + | − |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Suhandi, C.; Alfathonah, S.S.; Hasanah, A.N. Potency of Xanthone Derivatives from Garcinia mangostana L. for COVID-19 Treatment through Angiotensin-Converting Enzyme 2 and Main Protease Blockade: A Computational Study. Molecules 2023, 28, 5187. https://doi.org/10.3390/molecules28135187
Suhandi C, Alfathonah SS, Hasanah AN. Potency of Xanthone Derivatives from Garcinia mangostana L. for COVID-19 Treatment through Angiotensin-Converting Enzyme 2 and Main Protease Blockade: A Computational Study. Molecules. 2023; 28(13):5187. https://doi.org/10.3390/molecules28135187
Chicago/Turabian StyleSuhandi, Cecep, Siti Sarah Alfathonah, and Aliya Nur Hasanah. 2023. "Potency of Xanthone Derivatives from Garcinia mangostana L. for COVID-19 Treatment through Angiotensin-Converting Enzyme 2 and Main Protease Blockade: A Computational Study" Molecules 28, no. 13: 5187. https://doi.org/10.3390/molecules28135187
APA StyleSuhandi, C., Alfathonah, S. S., & Hasanah, A. N. (2023). Potency of Xanthone Derivatives from Garcinia mangostana L. for COVID-19 Treatment through Angiotensin-Converting Enzyme 2 and Main Protease Blockade: A Computational Study. Molecules, 28(13), 5187. https://doi.org/10.3390/molecules28135187