
Gram-Scale Total Synthesis of TAB with Cardioprotective Activity and the Structure-Activity Relationship of Its Analogs

 , ,
, ,
Abstract
:1. Introduction
2. Results
2.1. Total Synthesis of TAB
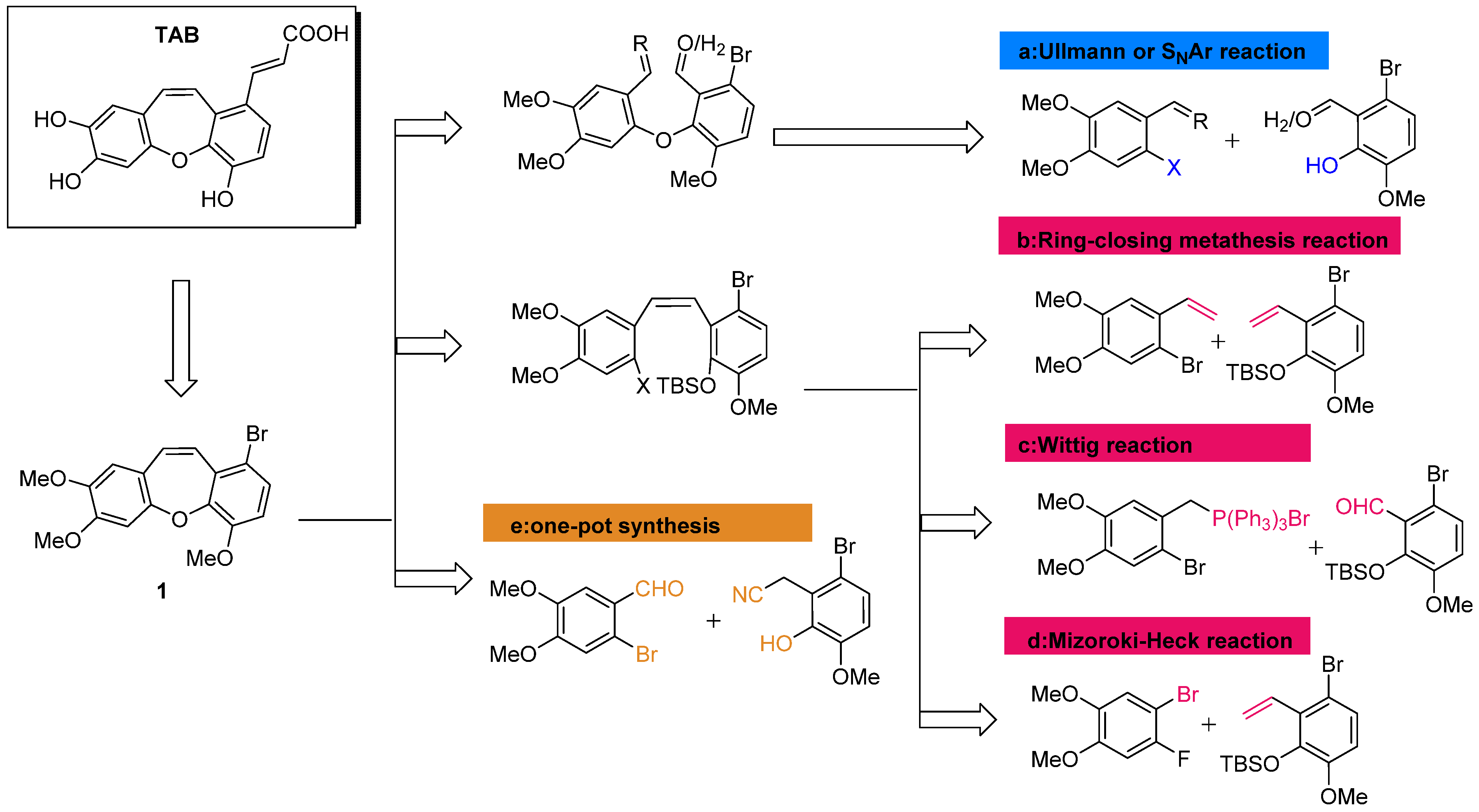
2.1.1. Retrosynthesis of TAB
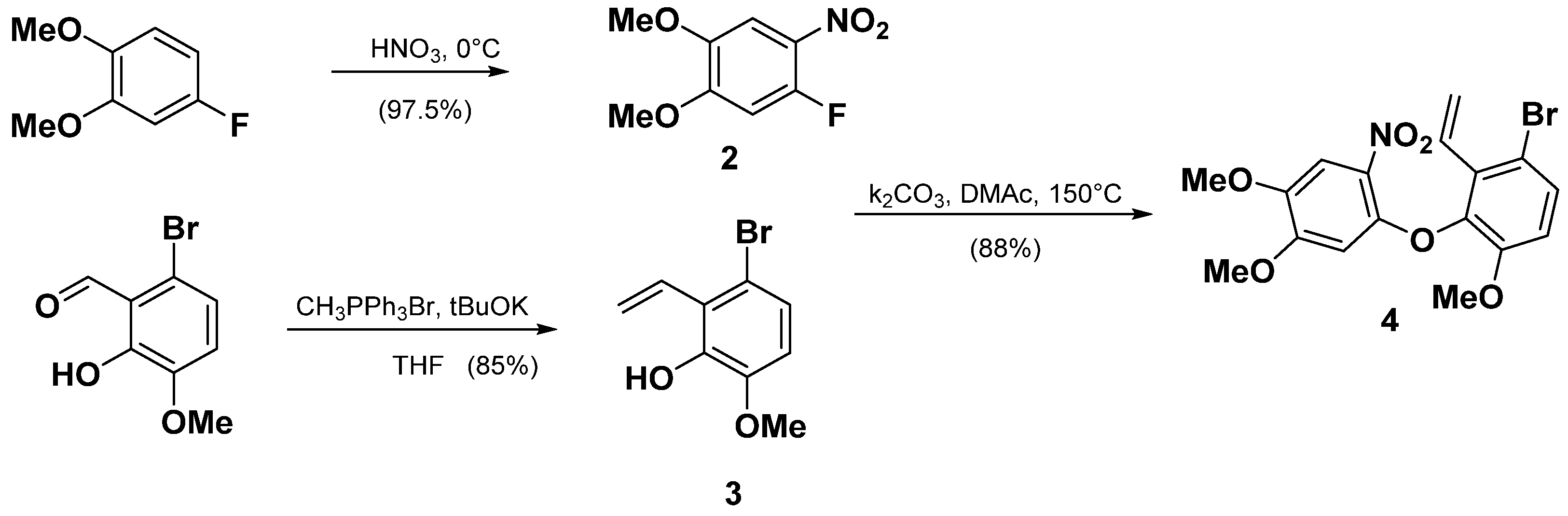
2.1.2. Synthesis of TAB
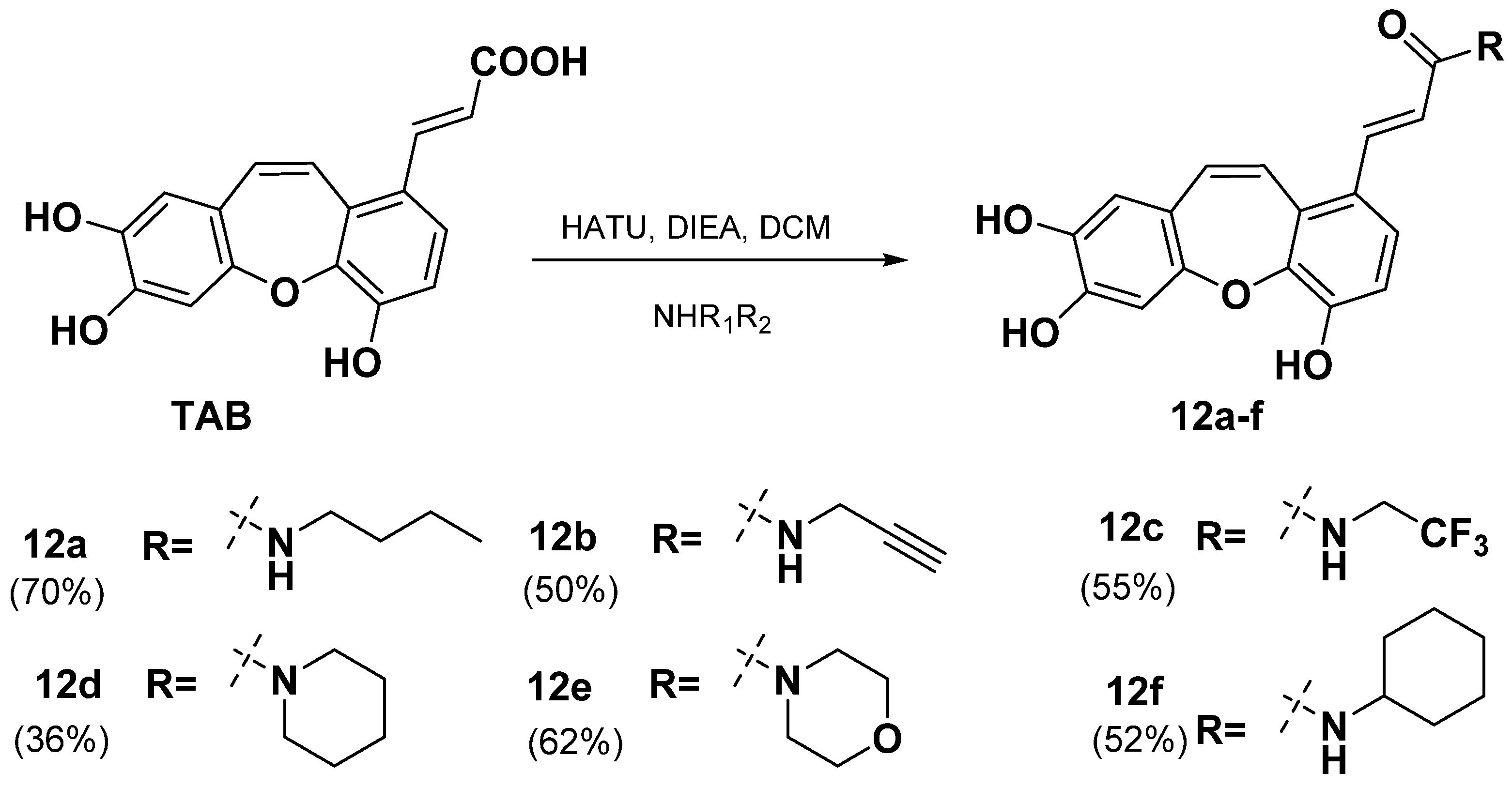
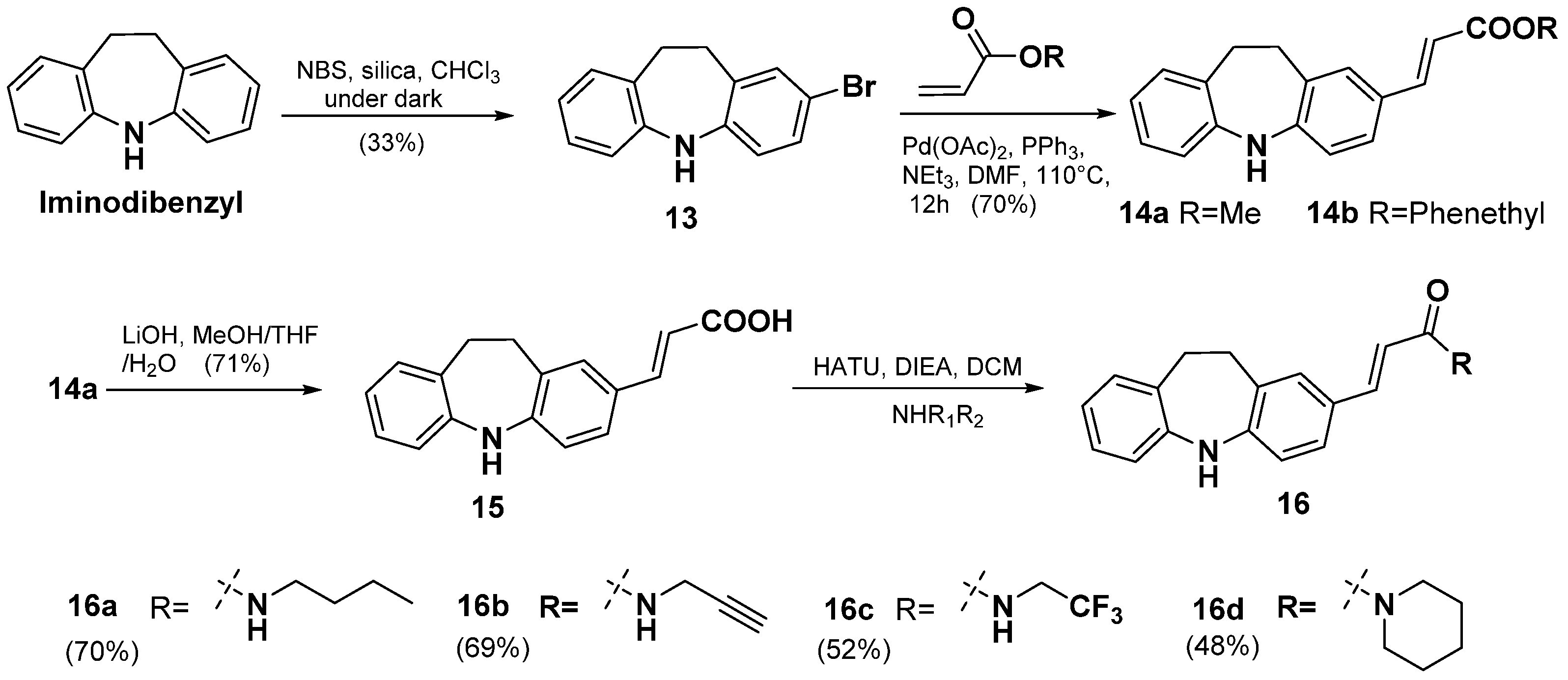
2.2. Synthesis of TAB Derivatives
2.3. Cardioprotective Activity
2.4. Structure-Activity Relationship
3. Discussion
4. Materials and Methods
4.1. General
4.2. Synthesis and Characterization Data
- 1-fluoro-4,5-dimethoxy-2-nitrobenzene (2)
- 3-bromo-6-methoxy-2-vinylphenol (3)
- 1-bromo-3-(4,5-dimethoxy-2-nitrophenoxy)-4-methoxy-2-vinylbenzene (4)
- 2-(3-bromo-6-methoxy-2-vinylphenoxy)-4,5-dimethoxyaniline (5)
- 1-bromo-3-(2-iodo-4,5-dimethoxyphenoxy)-4-methoxy-2-vinylbenzene (6) and 1-bromo-9-(iodomethyl)-4,6,7-trimethoxy-9H-xanthene (7)
- 1-bromo-4,6,7-trimethoxy-9-methylene-9H-xanthene (8)
- (1-bromo-4,6,7-trimethoxy-9H-xanthen-9-yl)methanol (9)
- 1-bromo-4,7,8-trimethoxydibenzo[b,f]oxepine (1)
- Methyl (E)-3-(4,7,8-trimethoxydibenzo[b,f]oxepin-1-yl)acrylate (10)
- (E)-3-(4,7,8-trimethoxydibenzo[b,f]oxepin-1-yl)acrylic acid (11)
- Tournefolic acid B (TAB)
- (E)-N-butyl-3-(4,7,8-trihydroxydibenzo[b,f]oxepin-1-yl)acrylamide (12a)
- (E)-N-(prop-2-yn-1-yl)-3-(4,7,8-trihydroxydibenzo[b,f]oxepin-1-yl)acrylamide (12b)
- (E)-N-(2,2,2-trifluoroethyl)-3-(4,7,8-trihydroxydibenzo[b,f]oxepin-1-yl)acrylamide (12c)
- (E)-1-(piperidin-1-yl)-3-(4,7,8-trihydroxydibenzo[b,f]oxepin-1-yl)prop-2-en-1-one (12d)
- (E)-1-morpholino-3-(4,7,8-trihydroxydibenzo[b,f]oxepin-1-yl)prop-2-en-1-one (12e)
- (E)-N-cyclohexyl-3-(4,7,8-trihydroxydibenzo[b,f]oxepin-1-yl)acrylamide (12f)
- 2-bromo-10,11-dihydro-5H-dibenzo[b,f]azepine (13)
- Methyl (E)-3-(10,11-dihydro-5H-dibenzo[b,f]azepin-2-yl)acrylate (14a) and phenethyl (E)-3-(10,11-dihydro-5H-dibenzo[b,f]azepin-2-yl)acrylate (14b)
- (E)-3-(10,11-dihydro-5H-dibenzo[b,f]azepin-2-yl)acrylic acid (15)
- (E)-N-butyl-3-(10,11-dihydro-5H-dibenzo[b,f]azepin-2-yl)acrylamide (16a)
- (E)-3-(10,11-dihydro-5H-dibenzo[b,f]azepin-2-yl)-N-(prop-2-yn-1-yl)acrylamide (16b)
- (E)-3-(10,11-dihydro-5H-dibenzo[b,f]azepin-2-yl)-N-(2,2,2-trifluoroethyl)acrylamide (16c)
- (E)-3-(10,11-dihydro-5H-dibenzo[b,f]azepin-2-yl)-1-(piperidin-1-yl)prop-2-en-1-one (16d)
4.3. Cardioprotective Activity Assays
4.3.1. Cell Culture and Treatment
4.3.2. Hypoxia/Reoxygenation Protocol
4.3.3. Cell Viability Assay
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Golshiri, K.; Ataabadi, E.A.; Fernandez, P.; Danser, J.; Roks, A.J. The importance of the nitric oxide-cGMP pathway in age-related cardiovascular disease: Focus on phosphodiesterase-1 and soluble guanylate cyclase. Basic Clin. Pharmacol. Toxicol. 2020, 127, 67–80. [Google Scholar]
- Johoson, J.A.; Cavallari, L.H. Pharmacogenetics and cardiovascular diseasee implications for personalized medicine. Pharmacol. Rev. 2013, 65, 987–1009. [Google Scholar] [CrossRef] [PubMed]
- Yellon, D.M.; Hausenloy, D.J. Myocardial reperfusion injury. N. Engl. J. Med. 2007, 357, 1121–1135. [Google Scholar] [CrossRef] [PubMed]
- Kurian, G.A.; Rajagopal, R.; Vedantham, S.; Rajesh, M. The Role of Oxidative Stress in Myocardial Ischemia and Reperfusion Injury and Remodeling: Revisited. Oxidative Med. Cell. Longev. 2016, 2014, 1656450. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Engelman, R.M.; Wei, Z.; Bagchi, D.; Rousou, J.A.; Das, D.K. Attenuation of myocardial reperfusion injury by reducing intracellular calcium overloading with dihydropyridines. Biochem. Pharmacol. 1993, 45, 1333–1341. [Google Scholar] [CrossRef]
- Liu, Y.; Yang, H.; Liu, L.X.; Yan, W.; Guo, H.J.; Li, W.J.; Tian, C.; Li, H.H.; Wang, H.X. NOD2 contributes to myocardial ischemia/reperfusion injury by regulating cardiomyocyte apoptosis and inflammation. Life Sci. 2016, 149, 10–17. [Google Scholar] [CrossRef]
- McCully, J.D.; Wakiyama, H.; Hsieh, Y.J.; Jones, M.; Levitsky, S. Differential contribution of necrosis and apoptosis in myocardial ischemia-reperfusion injury. AJP Heart Circ. Physiol. 2004, 286, 1923–1935. [Google Scholar] [CrossRef]
- Kong, R.; Gao, Y.; Sun, B.; Chen, H.; Wang, G.; Wang, X.; Zhu, H.; Pan, S.; Xue, D.; Jiang, H. The strategy of combined ischemia preconditioning and salvianolic acid-B pretreatment to prevent hepatic ischemia-reperfusion injury in rats. Dig. Dis. Sci. 2009, 54, 2568–2576. [Google Scholar] [CrossRef]
- Chen, J.; Wong, H.S.; Leung, H.Y.; Leong, P.K.; Chan, W.M.; Chen, N.; Ko, K.M. An ursolic acid-enriched extract of Cynomorium songaricum protects against carbon tetrachloride hepatotoxicity and gentamicin nephrotoxicity in rats possibly through a mitochondrial pathway: A comparison with ursolic acid. J. Funct. Foods 2014, 7, 330–341. [Google Scholar] [CrossRef]
- Lu, Y.; Kan, H.; Wang, Y.; Wang, D.; Wang, X.; Gao, J.; Zhu, L. Asiatic acid ameliorates hepatic ischemia/reperfusion injury in rats via mitochondria-targeted protective mechanism. Toxicol. Appl. Pharm. 2018, 338, 214–223. [Google Scholar] [CrossRef]
- Wang, M.; Zhang, J.; Zhang, J.; Sun, K.; Li, Q.; Kuang, B.; Wang, M.M.Z.; Hou, S.; Gong, H. Methyl eugenol attenuates liver ischemia reperfusion injury via activating PI3K/Akt signaling. Int. Immunopharmacol. 2021, 99, 108023. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Wu, S.; Jiang, Y.; Wang, L.; Li, G.; Qing, Y.; Liu, J.; Zhang, D. Inhibition of autophagy by geniposide protects against myocardial ischemia/reperfusion injury. Int. Immunopharmacol. 2020, 85, 106609. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Zhang, Y.; Liu, M.; Han, X.; Zhang, J.; Zhang, X.; Chu, L. Protective effect of quercetin against myocardial ischemia as a Ca2+ channel inhibitor: Involvement of inhibiting contractility and Ca2+ influx via L-type Ca2+ channels. Arch. Pharmacal Res. 2020, 43, 808–820. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Bian, L.; Fu, X.; Ai, Q.; Sui, Y.; Zhang, A.; Gao, H.; Zhong, L.L.; Lu, D. Gastrodin pretreatment alleviates rat brain injury caused by cerebral ischemic-reperfusion. Brain Res. 2019, 1712, 207–216. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Sun, X.; Xu, X.; Yu, Y.L.; Zhu, Y.D. Application of Tournefolic acid B in the Preparation for the Treatment and Prevention of Ischemic Heart Disease. CN Patent 106562950A, 19 April 2017. [Google Scholar]
- Yu, Y.; Xing, N.; Xu, X.; Zhu, Y.; Wang, S.; Sun, G.; Sun, X. Tournefolic acid B, derived from Clinopodium chinense (Benth.) Kuntze, protects against myocardial ischemia/reperfusion injury by inhibiting endoplasmic reticulum stress-regulated apoptosis via PI3K/AKT pathways. Phytomedicine 2018, 52, 178–186. [Google Scholar] [CrossRef]
- Lin, Y.; Chang, Y.Y.; Kuo, Y.; Shiao, M. Anti-Lipid-Peroxidative Principles from Tournefortia sarmentosa. J. Nat. Prod. 2002, 65, 745–747. [Google Scholar] [CrossRef]
- Chi, C.W.; Lin, Y.L.; Wang, Y.H.; Chen, C.F.; Wang, C.N.; Shiao, Y.J. Tournefolic acid B attenuates amyloid β protein-mediated toxicity by abrogating the calcium overload in mitochondria and retarding the caspase 8-truncated Bid-cytochrome c pathway in rat cortical neurons. Eur. J. Pharmacol. 2008, 586, 35–43. [Google Scholar] [CrossRef]
- Wang, C.N.; Pan, H.C.; Lin, Y.L.; Chi, C.W.; Shiao, Y. Ester Derivatives of Tournefolic Acid B Attenuate N-Methyl-Daspartate-Mediated Excitotoxicity in Rat Cortical Neurons. Mol Pharmacol. 2006, 69, 950–959. [Google Scholar] [CrossRef]
- Kittakoop, P.; Nopichai, S.; Thongon, N.; Charoenchai, P.; Thebtaranonth, Y. Bauhinoxepins A and B: New Antimycobacterial Dibenzo[b,f]oxepins from Bauhinia saccocalyx. Helv. Chim. Acta 2004, 87, 175–179. [Google Scholar] [CrossRef]
- Qian, T.X.; Li, L.N. Isosalvianolic acid C, a depside possessing a dibenzooxepin skeleton. Phytochemistry 1992, 31, 1068–1070. [Google Scholar]
- Lin, Y.; Wang, J.; He, L.; Hou, Y.; Fu, J.; Wei, D.; Jia, Q.; Lv, Y.; Wang, C.; Han, S. Isosalvianolic acid C-induced pseudo-allergic reactions via the mast cell specific receptor MRGPRX2. Int. Immunopharmacol. 2019, 71, 22–31. [Google Scholar] [CrossRef]
- Mori, M.; Nakamura, Y.; Shirai, Y.; Seto, Y.; Nakamura, H.; Makita, H.; Imasato, Y. Prolongation of antipyretic action and reduction of gastric ulcerogenicity in the rat by controlled-release granules of bermoprofen, a new nonsteroidal anti-inflammatory drug. J. Pharm. Sci. 1991, 80, 876–880. [Google Scholar] [CrossRef] [PubMed]
- Fagan, P.J.; Hauptman, E.; Shapiro, R.; Casalnuovo, A. Using Intelligent/Random Library Screening to Design Focused Libraries for the Optimization of Homogeneous Catalysts: Ullmann Ether Formation. J. Am. Chem. Soc. 2000, 122, 5043–5051. [Google Scholar] [CrossRef]
- Yeager, G.; Schissel, D. An Umpoled synthon Approach to the synthesis of 2-Aryloxyphenols. Synthesis 1995, 1995, 28–30. [Google Scholar] [CrossRef]
- Krawczyk, H.; Mielecki, D.; Szczecinski, P.; Grzesiuk, E. Synthesis of derivatives of methoxydibenzo[b, f]oxepine in the presence of sodium azide. Tetrahedron 2016, 72, 3877–3884. [Google Scholar] [CrossRef]
- Jepsen, T.H.; Larsen, M.; Jørgensen, M.; Nielsen, M.B. Three-Step synthesis of (Thio)xanthene and Dibenzothiepine/Diebenzoxepine by an Intramolecular Mizoroki-Heck Reaction of Diaryl (Thio)Ethers. Synlett 2012, 23, 418–422. [Google Scholar]
- Arnold, L.A.; Luo, W.; Guy, R.K. Synthesis of Medium Ring Heterocycles Using an Intramolecular Heck Reaction. Org. Lett. 2004, 6, 3005–3007. [Google Scholar] [CrossRef]
- Comber, M.; Sargent, M. The Synthesis of Pacharin: A Dibenzoxepine from the Heartwood of Bauhinia racemose Lamk. J. Chem. Soc. Perkin Trans. 1 1990, 5, 1371–1373. [Google Scholar] [CrossRef]
- Matsuda, T.; Sato, S. Synthesis of Dibenzoheteropines of Group 13-16 Elements via Ring-Closing Metathesis. J. Org. Chem. 2013, 78, 3329–3335. [Google Scholar] [CrossRef]
- Choi, Y.L.; Lim, H.S.; Lim, H.J.; Heo, J.N. One-Pot Transition-Metal-Free Synthesis of Dibenzo[b,f]oxepins from 2-Halobenzaldehydes. Org. Lett. 2012, 14, 5102–5105. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, Y.; He, Q.; Xie, Y.; Yang, C. Copper-Assisted/Copper-Free Synthesis of Functionalized Dibenzo[b, f]oxepins and Their Analogs via a One-Pot Tandem Reaction. Helv. Chim. Acta 2013, 96, 296–308. [Google Scholar] [CrossRef]
- Carlos, L.; Castedo, L. A new synthesis of 10, 11- dihydrodlbenzo[b, f]oxepin-10-ones: Key intermediates to cularine alkaoids. Tetrahedron Lett. 1989, 30, 6927–6928. [Google Scholar]
- Stopks, T.; Marzo, L.; Daniliuc, C.G. Oxidative C-H Bond Functionalization and Ring Expansion with TMSCHN2: A Copper(I)-Catalyzed Approach to Dibenzoxepines and Dibenzoazepines. Angew. Chem. Int. Ed. 2015, 54, 5049–5053. [Google Scholar] [CrossRef] [PubMed]
- Molinspiration v2018.10. Available online: https://www.molinspiration.com/ (accessed on 20 June 2023).
- Snow, A.; Weigele, M.; Larsen, L.; Nguyen, B.; Lake, T.; Castillo, G.; Sanders, V.; Lorimer, S.; Larsen, D.; Coffen, D.L.; et al. Preparation of Substituted N-aryl Benzamides and Related Compounds for Treatment of Amyloid Diseases and Synuclein Opathies and as Imaging Agents. U.S. Patent 20110104055, 5 May 2011. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compd. | Cell Viability (% of Control) | clogP a | |||||
|---|---|---|---|---|---|---|---|
| 0 μM | 0.5 μM | 1 μM | 2 μM | 4 μM | 8 μM | ||
| TAB | 56.0 ± 0.7 | 60.2 ± 0.4 | 77.2 ± 1.1 | 68.6 ± 0.9 | 65.7 ± 0.6 | 65.5 ± 1.2 | 2.69 |
| 10 | 56.5 ± 1.3 | 58.2 ± 1.3 | 56.8 ± 0.9 | 62.5 ± 1.6 | 62.0 ± 0.8 | 59.5 ± 1.4 | 4.20 |
| 11 | 56.3 ± 0.8 | 60.5 ± 0.7 | 60.4 ± 0.6 | 61.7 ± 1.2 | 67.4 ± 0.5 | 64.3 ± 0.6 | 3.58 |
| 12a | 56.6 ± 1.5 | 72.5 ± 0.5 | 75.7 ± 1.2 | 77.3 ± 1.5 | 74.6 ± 0.9 | 75.3 ± 1.1 | 3.99 |
| 12b | 56.4 ± 0.5 | 68.6 ± 1.5 | 70.7 ± 1.4 | 73.8 ± 0.9 | 77.3 ± 1.1 | 70.6 ± 0.7 | 2.71 |
| 12c | 56.7 ± 0.9 | 73.6 ± 0.5 | 78.5 ± 0.8 | 82.6 ± 1.1 | 80.4 ± 1.3 | 79.5 ± 0.8 | 3.48 |
| 12d | 56.5 ± 0.5 | 71.0 ± 0.6 | 75.6 ± 1.1 | 74.6 ± 0.8 | 72.5 ± 1.6 | 59.6 ± 1.7 | 3.70 |
| 12e | 56.1 ± 0.7 | 69.4 ± 1.2 | 74.1 ± 1.5 | 75.9 ± 1.2 | 73.7 ± 0.7 | 65.2 ± 0.3 | 2.64 |
| 12f | 56.7 ± 0.3 | 63.3 ± 1.3 | 68.5 ± 0.6 | 68.3 ± 1.0 | 66.2 ± 1.6 | 60.9 ± 1.2 | 4.46 |
| 14a | 56.9 ± 1.1 | 55.6 ± 1.0 | 57.4 ± 1.1 | 57.1 ± 1.4 | 59.7 ± 0.4 | 61.5 ± 1.1 | 4.62 |
| 14b | 56.4 ± 1.4 | 58.4 ± 1.6 | 56.1 ± 1.2 | 57.4 ± 1.7 | 58.5 ± 1.8 | 62.5 ± 0.8 | 6.41 |
| 15 | 56.0 ± 1.2 | 60.3 ± 1.0 | 64.8 ± 0.5 | 71.8 ± 1.4 | 69.3 ± 1.1 | 63.9 ± 0.9 | 4.00 |
| 16a | 55.8 ± 0.5 | 65.1 ± 0.6 | 65.7 ± 1.3 | 72.6 ± 0.3 | 70.8 ± 1.2 | 69.4 ± 0.5 | 5.30 |
| 16b | 56.4 ± 0.8 | 67.3 ± 0.6 | 75.0 ± 1.2 | 72.5 ± 1.7 | 68.2 ± 0.9 | 66.3 ± 0.6 | 4.02 |
| 16c | 56.2 ± 1.6 | 64.6 ± 1.5 | 72.4 ± 1.0 | 75.3 ± 1.2 | 69.3 ± 0.3 | 63.3 ± 0.5 | 4.79 |
| 16d | 56.7 ± 0.5 | 69.5 ± 1.5 | 71.4 ± 0.6 | 72.9 ± 1.1 | 65.4 ± 0.8 | 67.2 ± 1.0 | 5.01 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, Z.; Sun, Z.; Wu, D.; Yi, F.; Wu, H.; Ma, G.; Xu, X. Gram-Scale Total Synthesis of TAB with Cardioprotective Activity and the Structure-Activity Relationship of Its Analogs. Molecules 2023, 28, 5197. https://doi.org/10.3390/molecules28135197
Sun Z, Sun Z, Wu D, Yi F, Wu H, Ma G, Xu X. Gram-Scale Total Synthesis of TAB with Cardioprotective Activity and the Structure-Activity Relationship of Its Analogs. Molecules. 2023; 28(13):5197. https://doi.org/10.3390/molecules28135197
Chicago/Turabian StyleSun, Zhonghao, Zhaocui Sun, Daoshun Wu, Fan Yi, Haifeng Wu, Guoxu Ma, and Xudong Xu. 2023. "Gram-Scale Total Synthesis of TAB with Cardioprotective Activity and the Structure-Activity Relationship of Its Analogs" Molecules 28, no. 13: 5197. https://doi.org/10.3390/molecules28135197
APA StyleSun, Z., Sun, Z., Wu, D., Yi, F., Wu, H., Ma, G., & Xu, X. (2023). Gram-Scale Total Synthesis of TAB with Cardioprotective Activity and the Structure-Activity Relationship of Its Analogs. Molecules, 28(13), 5197. https://doi.org/10.3390/molecules28135197